

Abstract
Aberrant overexpression of ERG induced by the TMPRSS2-ERG gene fusion is likely involved in the development of prostate cancer. Synthetic pyrrole–imidazole (PI) polyamides recognize and attach to the minor groove of DNA with high affinity and specificity. In the present study, we designed a PI polyamide targeting TMPRSS2-ERG translocation breakpoints and assessed its effect on human prostate cancer cells. Our study identified that this PI polyamide repressed the cell and tumor growth of androgen-sensitive LNCaP prostate cancer cells. Targeting of these breakpoint sequences by PI polyamides could be a novel approach for the treatment of prostate cancer.
Keywords: Androgen receptor, fusion gene, prostate cancer, pyrrole–imidazole polyamide, TMPRSS2-ERG
The androgen receptor (AR) plays a key role in the physiological development of the normal prostate epithelium as well as in the onset and progression of prostate cancer.1 AR is a member of the nuclear receptor superfamily and functions as a ligand-dependent transcription factor.2 Upon activation by androgens, AR translocates into the nucleus and binds to androgen responsive elements (ARE).
Recently, fusion of the prostate-specific androgen-regulated TMPRSS2 gene to the E26 transformation-specific (ETS) family transcription factor gene ERG was reported as a common event in prostate cancer.3–7 ETS family members modulate many cellular functions, including proliferation, apoptosis, differentiation, tissue remodeling, migration, invasion and angiogenesis.8 Altered expression or properties of ETS transcription regulators affect the control of these processes and are involved in carcinogenesis and cancer progression. Several studies have demonstrated that ETS family gene re-arrangements are linked to clinicopathological indicators in prostate cancer.6,9,10 Because TMPRSS2, 5′-fusion partners, are upregulated by androgen, AR has been supposed to be important to regulate the fusion of genes in prostate cancer. A recent study shows that AR induces intronic binding sites in TMPRSS2 and ERG to facilitate specific chromosomal translocations by utilizing a common motif (TGT/AGGGA/T: break fusion site) for break/ligation in the human prostate cancer cell line LNCaP, which does not harbor TMPRSS2-ERG endogenously.11–14
Pyrrole–imidazole (PI) polyamides are small synthetic molecules that recognize and form non-covalent bonds to the minor groove of DNA, followed by inhibition of DNA–protein interactions with high affinity and sequence specificity.15–17 DNA recognition depends on a code of side-by-side pairing of pyrrole and imidazole in the hairpin polyamide, which binds to the minor groove. A pairing of imidazole opposite pyrrole targets for the G–C base pair, and pyrrole–pyrrole targets for both T–A and A–T base pairs. Recently, various types of sequence-specific PI polyamides have been developed to control gene expression.18–22 These investigations indicate that targeted PI polyamides could be potential gene silencers for the treatment of cancer. The aim of the present study is to investigate the effects of a PI polyamide targeting the TMPRSS2-ERG translocation break fusion site (fusion polyamide) in prostate cancer. We demonstrate that the fusion polyamide decreases the expression of TMPRSS2-ERG and ERG in LNCaP cells. In addition, we show that this polyamide represses the growth and migration of prostate cancer cells in vitro and in vivo.
Material and Methods
Cell culture, treatment with pyrrole–imidazole polyamide and antibody
The human prostate cancer cell lines LNCaP, VCaP and PC3 were purchased from the American Type Culture Collection (Rockville, MD, USA). LNCaP cells were maintained as previously described;23 VCaP and PC3 cells were cultured in DMEM supplemented with 10% FBS. PI polyamides were synthesized at Nihon University (Tokyo, Japan), as previously described.18 LNCaP cells were treated with negative control PI polyamide (negative control) and PI polyamide that targets break fusion sites (fusion polyamide) as previously described.18 A rabbit polyclonal anti-cleaved caspase-3 antibody was purchased from Cell Signaling Technology (Danvers, MA, USA).
DNA binding assay
FITC-labeled oligonucleotides were synthesized for gel mobility shift assays as described below.
TMPRSS2: 5′-FITC-TGTTAAGCTGAGGGTTGTGGGAGAGTGTTTTTCACTCTCCCACAACCCTCAGCTTAACA-3′.
ERG: 5′-FITC-TTCATGTTTGTGGGTGGGTGTATGTTTTTCATACACCCACCCACAAACATGAA-3′.
Both TMPRSS2 and ERG nucleotides contain the sequence TGT/AGGGA/T, which is the break fusion site in TMPRSS2 and ERG. Next, 1 μM of FITC-labeled oligonucleotides were dissolved in annealing buffer (20 mM Tris–HCl, 2 mM EDTA, 200 mM NaCl) and incubated at 100°C for 3 min. The solution was cooled down gradually to 30°C in the next 70 min to anneal the oligonucleotides in such a way that hairpin structures were formed. Then, 15 μL of annealed oligonucleotides and 5 μL of 2, 4 or 20 μM polyamides were mixed and incubated at 37°C for 1 h. The mixtures were separated by electrophoresis in 1× Tris-buffered EDTA on a 4–20% acrylamide gel and visualized with the luminescent image analyzer LAS-4000 (Fujifilm, Tokyo, Japan).
FISH
LNCaP cells were stimulated with vehicle or 100 nM dihydrotestosterone (DHT) in the presence of negative control or fusion polyamide for 24 h. Isolation of nuclear proteins and DNA-FISH were carried out according to methods previously described (Ourgenic, Tokushima, Japan).24 The probes were synthesized by Integrated DNA Technologies (Coralville, IA, USA) as listed bellow:
-
ERG
Biotin-GACTCCAGGAGCGCTCCCCAGAATCCCCTTCCTTAACCCAAACTCGAGCC.
-
TMPRSS2
FITC(FAM)-GATCTTTGGAGACCCGAGGAAAGCCGTGTTGACCAAAAGCAAGACAAATG.
Quantitative RT-PCR
Total RNA extraction, first-strand cDNA synthesis and quantitative RT-PCR (qRT-PCR) were performed as previously described.23 LNCaP cells were treated with 5 μM of negative control PI polyamide or 1 or 5 μM of fusion polyamide. Two days after treatment with DHT (100 nM), the mRNA expression levels of TMPRSS2-ERG and ERG were analyzed. As a negative control, cDNA samples prepared without reverse transcriptase were used. The primer sequences were as listed below:11,25
-
ERG
Forward: 5′- ACCGTTGGGATGAACTACGGCA-3′
Reverse: 5′- TGGAGATGTGAGAGAAGGATGTCG
-
TMPRSS2-ERG
Forward: 5′- AGCGCGGCAGGTTATTCCA-3′
Reverse: 5′- ATCATGTCCTTCAGTAAGCCA-3′.
Cell proliferation assay
The cell growth rate was measured using an MTS proliferation assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA), as previously described26 following the manufacturer's instructions. Briefly, 5000 cells/well were seeded in 96-well plates and cultured in phenol red-free RPMI 1640 (LNCaP cells) or phenol red-free DMEM (PC3 and VCaP cells) medium supplemented with 2.5% charcoal-stripped FBS for 24, 48 and 96 h. Each condition was tested in quadruplicate wells and each experiment was repeated at least twice.
Cell migration assay
The cell migration assay was performed as previously described.26 Briefly, 50 000 cells were suspended in 30 μL of RPMI 1640 (LNCaP cells) or DMEM (PC3 and VCaP cells) medium containing 10% FBS and added to the upper chamber. After incubation for 24 h in LNCaP and PC3 cells or 48 h in VCaP cells at 37°C in a humid 5% CO2 atmosphere, the cells on the lower surface of the filter were fixed in methanol for 30 min, then stained with Giemsa solution (Muto Pure Chemicals, Tokyo, Japan) for 30 s. The cells on the lower surface were counted in at least five fields at a magnification of ×200 under a microscope.
Analysis of the in vivo effects of polyamides
Three million LNCaP cells were injected subcutaneously into each side of 7-week-old male nude mice (n = 12). When the tumor size reached 100 mm3, fusion polyamide or negative control polyamide (6 mg/kg body weight) dissolved in dH2O were injected via the tail vein once a week for 4 weeks. The tumor size was measured every week until 1 week after the final injection, at which point the animals were killed and dissected. Tumor tissues were collected and kept both frozen and fixed in 10% formalin for analysis. Immunohistochemistry analysis was performed as previously described.25 The sections were incubated with the cleaved caspase-3 polyclonal antibody (1:100 dilution) overnight, followed by a 60-min incubation with Histofine Simple Stain MAX-PO (Nichirei, Tokyo, Japan).
Statistical analysis
Data are presented as mean ± SD or SEM. Statistical differences between the results of each group and its corresponding control were evaluated using Student's t-test. A P-value of <0.05 was considered significant.
Results
Binding of the polyamide to double-stranded DNA
The fusion polyamide was designed to bind to the break fusion site in TMPRSS2 and ERG. As a control, negative control polyamide, which does not bind to these sites, was used (Fig.1a,b). To determine the binding affinity and specificity of polyamide to target DNA, gel mobility shift assays were performed. Whereas oligonucleotides containing the break fusion site of TMPRSS or ERG showed mobility retardation when they were incubated with the fusion polyamide, nucleotides incubated with negative control polyamide did not show a clear mobility shift. The degree of mobility shift by the fusion polyamide was shown to be dose-dependent (Fig.1c). The distribution of FITC-labeled fusion polyamide and negative control polyamide in LNCaP cells are shown in Fig. S1. After 2 h of incubation of LNCaP cells with 5 μM of FITC-labeled polyamide, strong fluorescent signals were detected in nuclei.
Figure 1.
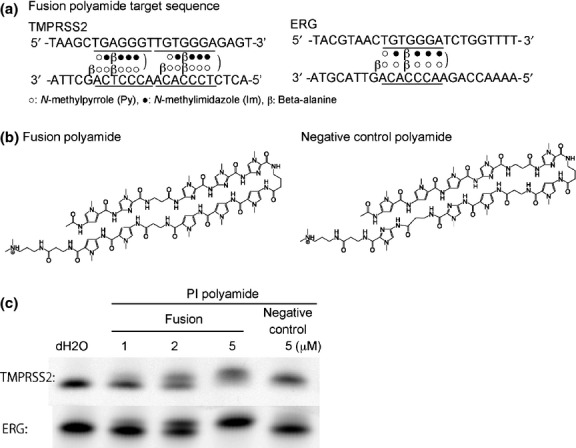
Target sequence and structure of synthetic pyrrole–imidazole (PI) polyamide that targets the break fusion sites in TMPRSS2 and ERG. (a) PI poly-amide targeting break fusion sites. The polyamide was designed to bind to the break fusion site in TMPRSS2 and ERG. (b) Structure of the fusion polyamide and negative control polyamide. They were synthesized by employing a solid-phase method and purified by high performance liquid chromatography (0.1% AcOH/CH3CN, 0–66% linear gradient, 0–20 min, 254 nm, through a Chemcobond 5-ODS-H column). (c) Gel mobility shift assay and distribution of fluorescein isothiocyanate (FITC)-labeled PI polyamide in vitro. FITC-labeled DNA corresponding to the break fusion sites in TMPRSS2 was synthesized and incubated with vehicle (water), fusion PI polyamide, or negative control PI polyamide for 1 h at 37°C and loaded onto a 20% polyacrylamide gel.
Pyrrole–imidazole polyamide targeting the break fusion site decreased fusion transcript and endogenous ERG
Because previous reports show that TMPRSS2-ERG transcripts are induced in LNCaP cells by stimulation of DHT (100 nM) for 24 h,11–14 we analyzed LNCaP cells treated with the same protocol. To determine whether the fusion polyamide affects DHT-dependent inter-chromosomal movement and TMPRSS2-ERG expression in LNCaP cells, we performed FISH analysis and measured TMPRSS2-ERG expression levels using qRT-PCR. After DHT treatment, the number of cells showing co-localization of TMPRSS2 and ERG was significantly increased in LNCaP cells cultured in the presence of 5 μM of negative control polyamide (Fig.2). This DHT-induced inter-chromosomal movement, however, was significantly decreased in cells cultured with 5 μM of fusion polyamide. Although the TMPRSS2-ERG transcript was significantly and constantly expressed in VCaP cells, which harbor this fusion gene, its expression was induced to a detectable level by the stimulation with DHT in LNCaP cells. The expression of the TMPRSS2-ERG transcript was significantly suppressed in the presence of 5 μM of the fusion polyamide compared with 5 μM of negative control polyamide in LNCaP cells. In contrast, the fusion polyamide did not affect its expression in VCaP cells (Fig.3a). Moreover, we tested whether the fusion polyamide could downregulate endogenous ERG gene expression. Both 1 and 5 μM of fusion polyamide substantially reduced mRNA expression levels of ERG in LNCaP cells (Fig.3b).
Figure 2.
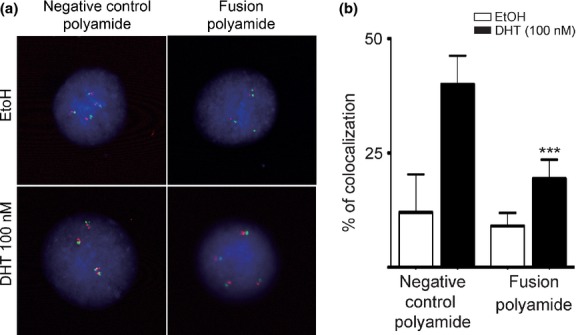
Presence of the fusion polyamide resulted in decreased androgen receptor-induced and chromosomal interactions of TMPRSS2 and ERG loci. (a, b) Following treatment of LNCaP cells with the fusion polyamide for 72 h, cells were stimulated with dihydrotestosterone (DHT) for 24 h, and FISH was performed with TMPRSS2 (green) and ERG (red) probes. (***P < 0.0001 versus negative control polyamide).
Figure 3.
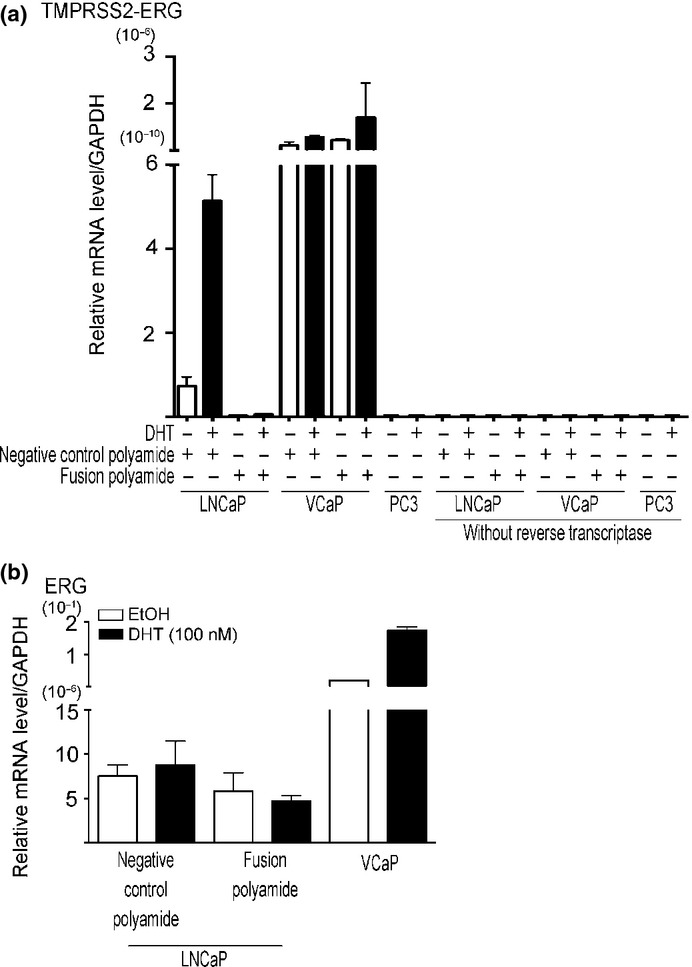
Efficacy of the pyrrole–imidazole (PI) polyamide targeting the break fusion site for fusion transcript and endogenous ERG expressions. (a, b) The presence of the fusion polyamide resulted in reduced expression of TMPRSS2-ERG and ERG expressions. LNCaP cells were treated with 5 μM of negative control PI polyamide or 5 μM of fusion polyamide (Fusion). Two days after treatment with DHT (100 nM), the mRNA expression levels of TMPRSS2-ERG and ERG were analyzed by quantitative RT-PCR. We used VCaP cells as positive and PC3 cells as a negative control for TMPRSS2-ERG expression.
Fusion polyamide repressed cell growth and cell migration
To assess the effect of the fusion polyamide on the viability of prostate cancer cells, we analyzed the cell proliferation activity by MTS assay.26 LNCaP cells treated with 1 and 5 μM fusion polyamide showed a significant decrease in cell proliferation after 96 h of DHT treatment compared to cells treated with negative control polyamide (P < 0.05; Fig.4). The MTS assay also revealed that the fusion polyamide had no significant effect on cell proliferation in AR-negative and TMPRSS2-ERG-negative prostate cancer cell line PC3 cells and VCaP cells (Fig.4). Next, we assessed the effects of the fusion polyamide on the migratory ability of LNCaP cells by conducting cell migration assays. Cell migration was significantly reduced in fusion polyamide-treated cells compared to negative control polyamide-treated cells (P < 0.0001, Fig.5). Neither PC3 nor VCaP cells showed significant differences in average number of migratory cells between fusion polyamide-treated cells and negative control polyamide-treated cells (Fig.5).
Figure 4.
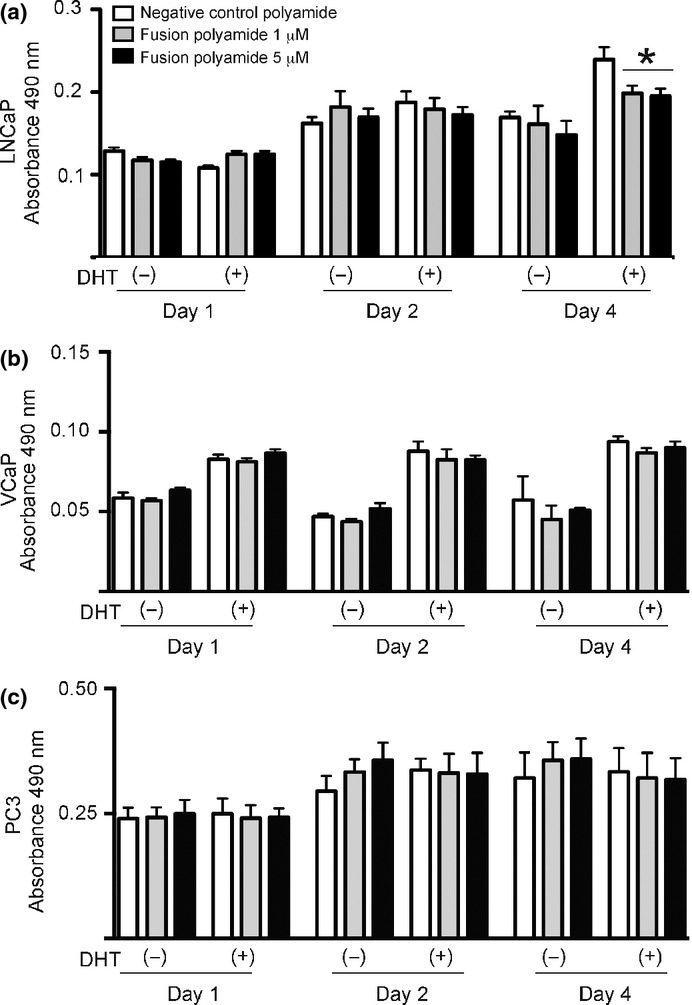
Effect of the fusion polyamide on cell growth. The fusion polyamide reduced (a) LNCaP cell growth in an androgen-dependent manner. (b) VCaP cells and (c) PC3 cells in phenol red-free DMEM and LNCaP cells in phenol red-free RPMI medium were treated for 48 h with 5 μM of negative control polyamide (negative control) or 1 or 5 μM of pyrrole–imidazole (PI) polyamide targeting the break fusion site (fusion polyamide). After stimulation with 100 nM dihydrotestosterone (DHT), an MTS assay was performed to assess the cell proliferation rate of PI polyamide-treated cells. Results are presented as mean and SD of triplicate assays (*P < 0.05).
Figure 5.
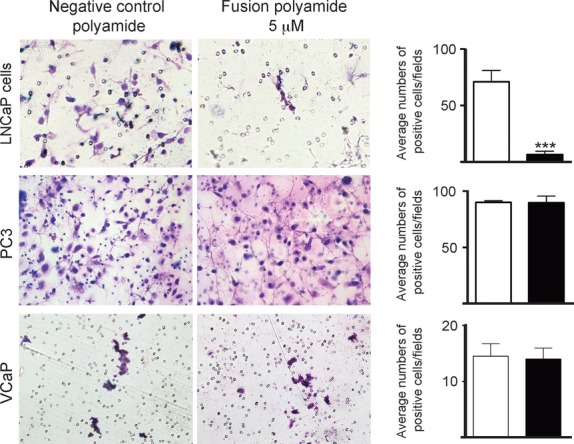
Effect of the fusion polyamide on cell migration. Effect of the fusion polyamide on cell migration. A cell migration assay was performed to analyze the motility of fusion polyamide-treated LNCaP, PC3 and VCaP cells and negative control polyamide-treated cells. Migrated cells were stained with Giemsa solution. Right panel shows average number of cells that migrated through the PET filter. Five representative fields in each well were quantif-ied to determine the number of migrated cells under a light microscope. Results are presented as the mean and SD of triplicate assays (***P < 0.0001).
Fusion polyamide repressed tumor growth in vivo
Athymic male mice bearing LNCaP cell-derived tumors were treated with the fusion polyamide or negative control polyamide. Tumor growth was prominent in mice treated with the negative control polyamide, but it was substantially reduced in mice treated with the fusion polyamide (Fig.6a,b). Moreover, the expression of cleaved caspase-3 tended to increase in LNCaP xenografts derived from mice treated with the fusion polyamide (Fig.6c).
Figure 6.
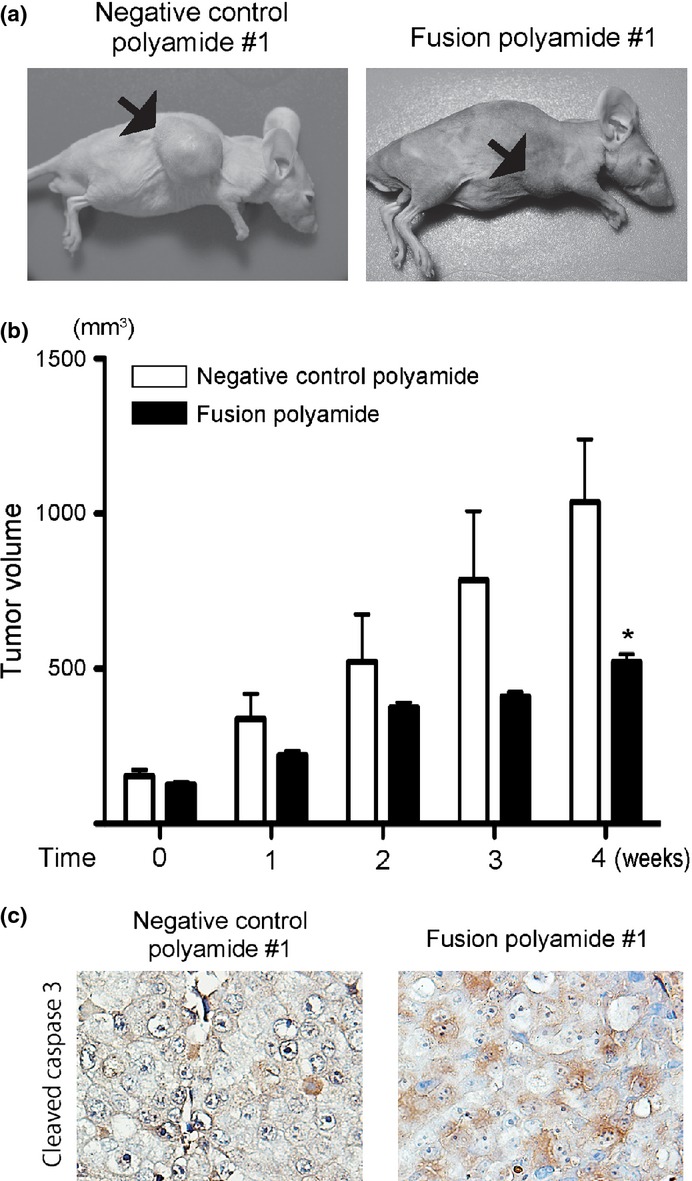
The fusion polyamide represses tumor formation of LNCaP cells in nude mice. (a, b) Seven-week-old male mice were implanted with 3 × 106 tumor cells and fusion polyamide or negative control polyamide was injected into the tail vein once weekly. (a) Photographs of mice bearing tumors after 4 weeks of treatment with pyrrole–imidazole (PI) polyamide. (b) Treatment with the fusion polyamide significantly reduced the tumor volume compared to treatment with the negative control polyamide. Line plots, means of tumor volume (V mm3) formed in mice; bars, SEM (n = 6 each), as determined by the formula: V = 0.5 × maximal diameter × middle diameter × minimal diameter. *P < 0.05 for fusion polyamide versus negative control polyamide. (c) Representative image of immunohistochemistry for cleaved caspase-3 in tumor xenograft tissues. (Magnification: ×400.)
Discussion
Activation of AR signaling mediated by androgens promotes cancer progression. Several studies have shown that AR expression is positively correlated with standard clinical and pathologic parameters, including the Gleason grade, clinical stage, lymph node status, extracapsular extension and seminal vesicle invasion.27,28 The reports of the fusion of TMPRSS2 with ETS family members in prostate cancer have opened a new field in prostate cancer research.3,5–7,29–31 Approximately 80% of prostate tumors harbor genomic fusions of TMPRSS2 and members of the ETS family of transcription factors. Among them, approximately 50% contain TMPRSS2-ERG fusions.32,33 Recently, urine testing to detect TMPRSS2-ERG has been reported to demonstrate significant correlation of the expression level of this fusion transcript with Gleason score, clinical stage and extracapsular extension of the tumor.34,35 In addition, the expression of TMPRSS2-ERG promoted by AR has been associated with a poor clinical outcome.36,37 Those findings indicate that evaluation of the expression level of TMPRSS2-ERG is valuable not only for diagnosis but also for predicting prognosis. Furthermore, it is also indicated that repressing the mechanism of TMPRSS2-ERG expression is crucial for the development of therapeutic approaches. In the present study, we attempted to develop a new compound that can repress the fusion gene formation and/or expression and to assess its effect on prostate cancer growth.
A previous report showed that the expression of fusion transcripts was inhibited by siRNA, which target specific components involved in homologous recombination of DNA.11 Shao et al. (2012) report that siRNA targeting most common isoforms of the TMPRSS2-ERG fusion transcript efficiently suppressed the growth of prostate cancer in vivo.38 Those data strongly suggest that inhibition of TMPRSS2-ERG fusion gene formation or expression could be a good therapeutic approach for prostate cancer. siRNA can effectively repress the expression of specific genes. However, there is a disadvantage in its application; that is, siRNA can be easily degraded by nucleases. One of the most important advantages of PI polyamide is that it is resistant to biological degradation (e.g. by nucleases and proteases). It can be taken up by cells and transported to nuclei without requiring any specific drug delivery system. Moreover, PI polyamide can inhibit DNA–protein interaction by binding to the minor groove of double-helical DNA with high affinity and sequence specificity.15,16 Another important advantage is that intravenous, subcutane-ous or peritoneal injections of PI polyamide have never indu-ced significant health injuries when tested in mice and rats.18,19,39,40 Raskatov et al.41 report that intra-peritoneal or subcutaneous administration of 120 nmol/mouse (4.5–7 mg/kg body weight) of PI polyamide in cyclic form had an acute toxic effect on mice; however, they showed that the hairpin form of PI polyamide did not have any toxic effect at the same dose. In addition, Yang et al.39 report that subcutaneous injection of PI polyamides targeting RNA polymerase II resulted in growth reduction in LNCaP xenografts without detectable DNA damage. Therefore, in the present study, we designed and examined PI polyamide, which binds to break fusion sites, to inhibit double-stranded breaks. It has been reported previously that PI polyamide that recognizes ARE suppresses DHT-dependent gene expression in LNCaP cells,42 and it was revealed that this polyamide inhibits the binding of RNA polymerase II to the transcription start site of AR-driving genes.39
Our present data clearly show that this fusion polyamide suppresses the formation and expression of TMPRSS2-ERG fusion genes. In addition, it suppressed endogenous ERG expression and cellular proliferation in in vitro models and also induced cellular apoptosis in in vivo models. Interestingly, the effect of PI polyamide on the inhibition of cell migration and in vivo tumor growth appeared to be drastic compared to its inhibitory effect on in vitro cell proliferation. Recent study has shown that the TMPRSS2-ERG fusion gene expressed in prostate cancer changes tumor microenvironment to that associated with more aggressive phenotype of cancer, and affects the cellular migration activity.43–45 For example, prostate cancer tissues with higher expression levels of TMPRSS2-ERG fusion transcripts showed increased vascular density, hyaluronan, von Willebrand factor and PDGFRβ, and decreased Caveolin-1. In addition, the in vivo tumor growth of prostate cancer cells is shown to be dependent on the microenvironment.46,47 These factors could explain the difference of the efficacy of the fusion polyamide between in vitro cell proliferation and in vivo tumor growth.
Endogenous ERG expression in prostate tissues was shown to be correlated with biochemical relapse and poor prognosis.48,49 Moreover, ERG overexpression in prostate cancer specimen is a strong predictor of the progression of the disease during active surveillance.50 Recently, it has been reported that the TMPRSS2–ERG fusion gene product binds to the ERG locus and promotes wild-type ERG expression in human prostate cancers.51 This mechanism activated the feed-forward regulation of ERG expression, thereby promoting prostate cancer invasion. This report also showed that the reduction of endogenous ERG expression prevented the invasion of prostate cancer cells. Furthermore, the report showed that overexpression of wild-type ERG was observed in 38% of clinically localized prostate cancers and 27% of metastatic prostate cancers bearing TMPRSS2-ERG fusion genes.51 These reports indicate that TMPRSS2-ERG plays a critical role in prostate cancer progression.
In summary, we developed a PI polyamide that can target the TMPRSS2-ERG fusion site, prevent formation of the fusion gene, and inhibit proliferation and migration of LNCaP cells as well as in vivo tumor growth. The present findings show that break fusion sites, which have a critical role in the formation of AR-dependent fusion genes, could be a novel therapeutic target for prostate cancer.
Acknowledgments
The authors thank Ms Asako Oguni, Ms Noriko Sasaki and Mr Motoaki Kataba for technical support and Ms Emiko Hododa for secretarial assistance.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting Information
Fig. S1. Distribution of FITC-labeled fusion and negative control polyamide in LNCaP cells. LNCaP cells were seeded on 24-well plates and cultured for 24 h, and then 5 μM of FITC-labeled fusion or negative control polyamide were applied to the growth medium. After 2 h incubation, medium was replaced with PBS containing Hoechst 33342, and cells were observed by fluorescence microscopy following 20 min incubation. Scale bar indicates 50 μm.
References
- Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Mehra R, Rhodes DR, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Varambally S, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra R, Tomlins SA, Shen R, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–44. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- Mehra R, Tomlins SA, Yu J, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–90. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347–51. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- Perner S, Demichelis F, Beroukhim R, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–41. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–83. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastus NC, Boyd LK, Mao X, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70:9544–8. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, Aryee MJ, Toubaji A, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–75. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani RS, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dervan PB, Edelson BS. Recognition of the DNA minor groove by pyrrole–imidazole polyamides. Curr Opin Struct Biol. 2003;13:284–99. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- Dervan PB. Molecular recognition of DNA by small molecules. Bioorg Med Chem. 2001;9:2215–35. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- Trauger JW, Baird EE, Dervan PB. Recognition of DNA by designed ligands at subnanomolar concentrations. Nature. 1996;382:559–61. doi: 10.1038/382559a0. [DOI] [PubMed] [Google Scholar]
- Wang X, Nagase H, Watanabe T, et al. Inhibition of MMP-9 transcription and suppression of tumor metastasis by pyrrole–imidazole polyamide. Cancer Sci. 2010;101:759–66. doi: 10.1111/j.1349-7006.2009.01435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, Fukuda N, Ueno T, et al. Transcriptional inhibition of progressive renal disease by gene silencing pyrrole–imidazole polyamide targeting of the transforming growth factor-beta1 promoter. Kidney Int. 2011;79:46–56. doi: 10.1038/ki.2010.330. [DOI] [PubMed] [Google Scholar]
- Nickols NG, Szablowski JO, Hargrove AE, Li BC, Raskatov JA, Dervan PB. Activity of a py-im polyamide targeted to the estrogen response element. Mol Cancer Ther. 2013;12:675–84. doi: 10.1158/1535-7163.MCT-12-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sicot G, Cui X, et al. Targeting a DNA binding motif of the EVI1 protein by a pyrrole–imidazole polyamide. Biochemistry. 2011;50:10431–41. doi: 10.1021/bi200962u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno T, Fukuda N, Tsunemi A, et al. A novel gene silencer, pyrrole-imidazole polyamide targeting human lectin-like oxidized low-density lipoprotein receptor-1 gene improves endothelial cell function. J Hypertens. 2009;27:508–16. doi: 10.1097/hjh.0b013e3283207fe1. [DOI] [PubMed] [Google Scholar]
- Takayama K, Kaneshiro K, Tsutsumi S, et al. Identification of novel androgen response genes in prostate cancer cells by coupling chromatin immunoprecipitation and genomic microarray analysis. Oncogene. 2007;26:4453–63. doi: 10.1038/sj.onc.1210229. [DOI] [PubMed] [Google Scholar]
- Hu Q, Kwon YS, Nunez E, et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci USA. 2008;105:19199–204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obinata D, Takayama K, Urano T, et al. Oct1 regulates cell growth of LNCaP cells and is a prognostic factor for prostate cancer. Int J Cancer. 2012;130:1021–8. doi: 10.1002/ijc.26043. [DOI] [PubMed] [Google Scholar]
- Obinata D, Takayama K, Urano T, et al. ARFGAP3, an androgen target gene, promotes prostate cancer cell proliferation and migration. Int J Cancer. 2012;130:2240–8. doi: 10.1002/ijc.26224. [DOI] [PubMed] [Google Scholar]
- Lu S, Tsai SY, Tsai MJ. Regulation of androgen-dependent prostatic cancer cell growth: androgen regulation of CDK2, CDK4, and CKI p16 genes. Cancer Res. 1997;57:4511–6. [PubMed] [Google Scholar]
- Li R, Wheeler T, Dai H, Frolov A, Thompson T, Ayala G. High level of androgen receptor is associated with aggressive clinicopathologic features and decreased biochemical recurrence-free survival in prostate: cancer patients treated with radical prostatectomy. Am J Surg Pathol. 2004;28:928–34. doi: 10.1097/00000478-200407000-00013. [DOI] [PubMed] [Google Scholar]
- Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6:429–39. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- Han B, Mehra R, Dhanasekaran SM, et al. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res. 2008;68:7629–37. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra R, Han B, Tomlins SA, et al. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer Res. 2007;67:7991–5. doi: 10.1158/0008-5472.CAN-07-2043. [DOI] [PubMed] [Google Scholar]
- King JC, Xu J, Wongvipat J, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Tran J, Gopalan A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxman B, Tomlins SA, Mehra R, et al. Noninvasive detection of TMPRSS2:ERG fusion transcripts in the urine of men with prostate cancer. Neoplasia. 2006;8:885–8. doi: 10.1593/neo.06625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyten GH, Hessels D, Jannink SA, et al. Prospective multicentre evaluation of PCA3 and TMPRSS2-ERG gene fusions as diagnostic and prognostic urinary biomarkers for prostate cancer. Eur Urol. 2014;65:534–42. doi: 10.1016/j.eururo.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Cerveira N, Ribeiro FR, Peixoto A, et al. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia. 2006;8:826–32. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichelis F, Fall K, Perner S, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–9. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- Shao L, Tekedereli I, Wang J, et al. Highly specific targeting of the TMPRSS2/ERG fusion gene using liposomal nanovectors. Clin Cancer Res. 2012;18:6648–57. doi: 10.1158/1078-0432.CCR-12-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Nickols NG, Li BC, Marinov GK, Said JW, Dervan PB. Antitumor activity of a pyrrole–imidazole polyamide. Proc Natl Acad Sci USA. 2013;110:1863–8. doi: 10.1073/pnas.1222035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskatov JA, Nickols NG, Hargrove AE, Marinov GK, Wold B, Dervan PB. Gene expression changes in a tumor xenograft by a pyrrole-imidazole polyamide. Proc Natl Acad Sci USA. 2012;109:16041–5. doi: 10.1073/pnas.1214267109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskatov JA, Hargrove AE, So AY, Dervan PB. Pharmacokinetics of Py-Im polyamides depend on architecture: cyclic versus linear. J Am Chem Soc. 2012;134:7995–9. doi: 10.1021/ja302588v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci USA. 2007;104:10418–23. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagglof C, Hammarsten P, Stromvall K, et al. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS ONE. 2014;9:e86824. doi: 10.1371/journal.pone.0086824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj N, Budka JA, Ferris MW, Jerde TJ, Hollenhorst PC. Prostate cancer ETS rearrangements switch a cell migration gene expression program from RAS/ERK to PI3K/AKT regulation. Mol Cancer. 2014;13:61. doi: 10.1186/1476-4598-13-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian TV, Tomavo N, Huot L, et al. Identification of novel TMPRSS2:ERG mechanisms in prostate cancer metastasis: involvement of MMP9 and PLXNA2. Oncogene. 2014;33:2204–14. doi: 10.1038/onc.2013.176. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hagglof C, Bergh A. The stroma-a key regulator in prostate function and malignancy. Cancers. 2012;4:531–48. doi: 10.3390/cancers4020531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JI, Tomlins SA, Carskadon SL, et al. Evaluation of tissue PCA3 expression in prostate cancer by RNA in situ hybridization–a correlative study with urine PCA3 and TMPRSS2-ERG. Mod Pathol. 2014;27:609–20. doi: 10.1038/modpathol.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KC, Dolph M, Donnelly B, Bismar TA. ERG expression is associated with increased risk of biochemical relapse following radical prostatectomy in early onset prostate cancer. Clin Transl Oncol. 2014 doi: 10.1007/s12094-014-1182-x. ; in press. [DOI] [PubMed] [Google Scholar]
- Berg KD, Vainer B, Thomsen FB, et al. ERG protein expression in diagnostic specimens is associated with increased risk of progression during active surveillance for prostate cancer. Eur Urol. 2014 doi: 10.1016/j.eururo.2014.02.058. ; in press. [DOI] [PubMed] [Google Scholar]
- Mani RS, Iyer MK, Cao Q, et al. TMPRSS2-ERG-mediated feed-forward regulation of wild-type ERG in human prostate cancers. Cancer Res. 2011;71:5387–92. doi: 10.1158/0008-5472.CAN-11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Distribution of FITC-labeled fusion and negative control polyamide in LNCaP cells. LNCaP cells were seeded on 24-well plates and cultured for 24 h, and then 5 μM of FITC-labeled fusion or negative control polyamide were applied to the growth medium. After 2 h incubation, medium was replaced with PBS containing Hoechst 33342, and cells were observed by fluorescence microscopy following 20 min incubation. Scale bar indicates 50 μm.