

Abstract
Mutant mouse models are indispensable tools for clarifying gene functions and elucidating the pathogenic mechanisms of human diseases. Here, we describe novel cancer models bearing point mutations in the retinoblastoma gene (Rb1) generated by N-ethyl-N-nitrosourea mutagenesis. Two mutations in splice sites reduced Rb1 expression and led to a tumor spectrum and incidence similar to those observed in the conventional Rb1 knockout mice. The missense mutant, Rb1D326V/+, developed pituitary tumors, but thyroid tumors were completely suppressed. Immunohistochemical analyses of thyroid tissue revealed that E2F1, but not E2F2/3, was selectively inactivated, indicating that the mutant Rb protein (pRb) suppressed thyroid tumors by inactivating E2F1. Interestingly, Rb1D326V/+ mice developed pituitary tumors that originated from the intermediate lobe of the pituitary, despite selective inactivation of E2F1. Furthermore, in the anterior lobe of the pituitary, other E2F were also inactivated. These observations show that pRb mediates the inactivation of E2F function and its contribution to tumorigenesis is highly dependent on the cell type. Last, by using a reconstitution assay of synthesized proteins, we showed that the D326V missense pRb bound to E2F1 but failed to interact with E2F2/3. These results reveal the effect of the pRb N-terminal domain on E2F function and the impact of the protein on tumorigenesis. Thus, this mutant mouse model can be used to investigate human Rb family-bearing mutations at the N-terminal region.
Keywords: E2F transcription factors, endocrine tumors, mice, mutagenesis, retinoblastoma
Humans with a germline mutation of the retinoblastoma gene (Rb1) are predisposed to retinoblastoma and other tumors.1 Rb1 is a tumor suppressor gene and loss of Rb1 heterozygosity, downregulation and mutations in the gene have been associated with various human cancers.2–4 All heterozygous Rb1 knockout (Rb1+/−) mice develop neuroendocrine tumors, which arise in the pituitary and medullary component of thyroid (C-cells).5–8 Retinoblastoma protein (pRb) binds to E2F transcription factors through its C-terminal region and pRb suppresses tumor formation by inhibiting E2F activity.9 Consistent with this model, perturbation of the functions of E2F, including E2F1 and E2F3, reduces the incidence of tumor formation induced by Rb1 mutation. Interestingly, these mice exhibit a distinct tumor spectrum, suggesting distinct contributions of each E2F to tumor formation in a tissue-dependent manner.10,11 However, the molecular basis of this tissue-dependent tumor-suppressive function of pRb has remained elusive thus far.
We previously screened mutant mice generated by N-Ethyl-N-nitrosourea (ENU) mutagenesis, which introduces single base-pair changes.12,13 Here, we report three novel cancer model mice predisposed to neuroendocrine tumors. These three lines harbor novel point mutations in Rb1: two splice-site mutations and one missense mutation, which involve a substitution of aspartic acid by valine on residue 326 (D326V). Heterozygous animals of the splice-site mutations developed pituitary and thyroid tumors, similarly to Rb1+/− mice. However, heterozygous D326V mutants (Rb1D326V/+) developed only pituitary tumors. The unique tumor spectrum observed in Rb1D326V/+ mice renders this mutant a useful tool to investigate the cell- or tissue-specific tumor suppressive mechanism of pRb. Additionally, we report that the D326V-mutated pRb (pRbD326V) is partially deficient in binding E2F in vitro. Thus, Rb1D326V/+ animals will be valuable for understanding missense-mutated, partially affected pRb function in vivo.
Materials and Methods
Animals and ENU mutagenesis
All animal experiments were approved by the Institutional Animal Experiment Committee of RIKEN BioResource Center. The ENU mutagenesis was conducted as previously described.14 We injected male C57BL/6J (B6) mice with ENU (Sigma Aldrich, St Louis, MO, USA); these male mice were mated with DBA/2J (D2) female mice.
Mutation mapping and identification
For mapping, we analyzed genomic DNA from the N2 and N3 progeny backcrossed to D2 by using single nucleotide polymorphism (SNP) and single-strand conformation polymorphism (SSCP) markers. We performed genotype analysis of Rb1 mutations by sequencing the genomic DNA from backcrossed animals that were affected or not affected.
Quantitative real-time RT-PCR (qPCR)
The qPCR analyses were conducted as previously described.14 Reverse transcription was performed with total RNA extracted from the brains of mice using SuperScript II (Life Technologies, Carlsbad, CA, USA) and an oligo-dT primer. The qPCR was performed using the QuantiTect SYBR Green RT-PCR system (Qiagen, Hilden, Germany). Rb1 gene-specific primers covering exons 1–2 were as follows: forward 5′-CGAAGAGCTGCCCCTGG-3′ and reverse 5′-ATGATCGGGTACCTTTAACTTTTGA-3′.
Histological analyses
Tissues were fixed in 4% paraformaldehyde and paraffin embedded. For immunohistochemistry (IHC), specimens were incubated with antibodies against alpha melanocyte-stimulating hormone (α-MSH), calcitonin, E2F1/2/3, prolactin (PRL), luteinizing hormone/follicle-stimulating hormone alpha subunit (LH/α-FSH), thyroid-stimulating hormone (TSH) and growth hormone (GH; antibody sources, Supporting Information Table S1), followed by incubation with Envision reagent (DAKO, Glostrup, Denmark). The HRP substrates and alkaline phosphatase (AP) substrates were used in the chromogenic reactions. Labeled specimens were scored for the number of positive cells per three high-powered fields per sample.
LOH analysis
The LOH analyses were conducted as previously described.14 LOH at the Rb1 locus was determined by PCR using microsatellite markers D14Mit125 and D14Mit262 for thyroid tumors and D14Mit193 and D14Mit225 for pituitary tumors. The PCR products of these markers differ in size between the mutant allele (B6 derived) and wild-type (WT) allele (D2 derived).
Co-immunoprecipitation (co-IP) assays of E2F1/2/3 and pRb
All proteins tested were synthesized using an in vitro transcription and translation system using T7 RiboMAX Express and Wheat Germ Extract (WGE) Plus (Promega, Madison, WI, USA). Plasmids expressing pRb, E2F1/2/3 and E2F dimerization partner 1 (DP-1) were prepared from FANTOM clones (DNAFORM, Yokohama, Japan). The Rb1D326V mutant was generated using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, La Jolla, CA, USA) and complementary oligonucleotide primers (forward, 5′-CTTAAAAACAAAGATTTAGTTGCAAGACTGTTTTTGG-3′; reverse, 5′-CCAAAAACAGTCTTGCAACTAAATCTTTGTTTTTAAG-3′). A Flag tag and restriction enzyme sites were then added to the WT and mutant Rb1 constructs by PCR.
After the translation reaction, each of the E2F, pRb (WT or mutant) and DP-1 WGE lysate were mixed and incubated. Magnetic beads (Dynabeads; Life Technologies) bound with or without (negative control) anti-Flag antibody were added to the mixture and incubated. Protein immunocomplexes were isolated and analyzed by western blotting using primary antibodies against pRb and E2F1/2/3 (Table S1).
Results
Generation of novel endocrine tumor model mice by ENU mutagenesis
We have previously identified 17 tumor-predisposed mutant lines.14 Among them, three cells lines (M1326, M1032 and M1033) developed endocrine tumors. The pituitary tumor was evident by the moribund necropsies of these three mutant founders (Fig.1a–f). The IHC revealed positive staining for α-MSH, indicating that the tumors were derived from the intermediate lobe of the pituitary (ILP; Fig.1g–i). Additionally, the founders of M1032 and M1033 also developed thyroid tumors at 53 and 56 weeks, respectively (Fig.1j–m). These tumors exhibited positive staining for calcitonin (Fig.1n,o), indicating they originated from C-cells.
Figure 1.
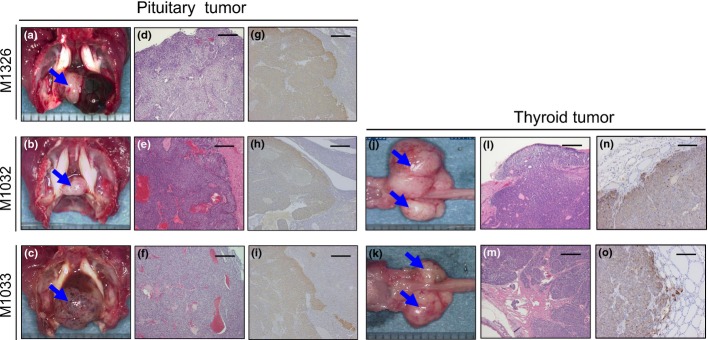
Pathological analysis of tumors from novel cancer model mice generated by N-ethyl-N-nitrosourea mutagenesis. Pituitary and thyroid tumors found in M1326 (a,d,g), M1032 (b,e,h,j,l,n) and M1033 (c,f,i,k,m,o). Alpha melanocyte-stimulating hormone-positive (g,h,i) and calcitonin-positive (n,o) tumors were detected. Bar, 200 μm.
Mutation mapping and identification
To identify the mutation responsible for the cancer phenotype, genome-wide mapping was initially performed with SSCP markers. Pituitary tumor development in all three mutant lines was associated with mouse chromosome 14. We then performed a fine mapping using SNP markers. In the M1326 line, the cancer phenotype was mapped within the region between D14Mit193 and D14Mit225 (Fig.2a). Similarly, tumor development was mapped to the regions between D14Mit235 and rs3080790 and between D14Mit115 and D14Mit35 in M1033 and M1032, respectively. These observations suggest that the same gene was responsible for the development of endocrine tumors observed in the three lines. Among the 17 genes located within the common region (approximately 2.0 Mb), we focused on Rb1, because several Rb1-deficient mice have been reported to develop pituitary and thyroid tumors.5–8 We conducted genomic DNA sequencing of all exons and splice sites of Rb1 and identified distinct base substitutions. The M1326 line contained a base substitution of 977A to T, which resulted in a missense mutation that changed Asp326 (GAT) to Val (GTT). We termed this mutation Rb1D326V (Fig.2b). Two other mutant lines contained base substitutions in the following splice sites: T to A mutation in a donor splice site of intron 21 (M1032 line) and A to G mutation in that of intron 10 (M1033 line; Fig.2b).
Figure 2.
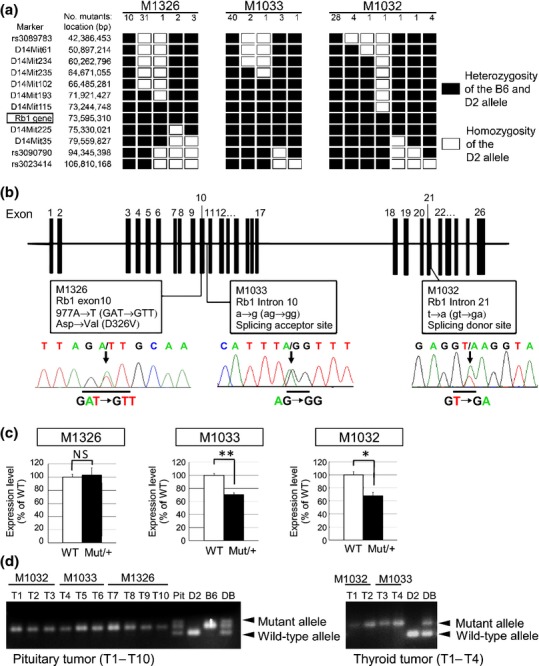
Identification of retinoblastoma gene (Rb1) as causal gene in the three mutant lines generated by N-ethyl-N-nitrosourea mutagenesis. (a) Fine mapping study on chromosome 14 using SSCP and single nucleotide polymorphism markers. Haplotype analysis of D2-backcrossed N2-N3 progeny was performed. (b) The mutations found in exon 10 (M1326), intron 10 (M1033) and intron 21 (M1032) are shown in the exon-intron structure of mouse Rb1. Arrows indicate the mutations in the sequence chromatograph. (c) qPCR of Rb1 mRNA levels in the three Rb1 hetero mutants (Mut/+) or wild-type littermates (WT; n = 3 per genotype). Statistical analysis: unpaired two-tailed Student's t-test. NS, not significant. *P < 0.05. **P < 0.01. Error bar, SD. (d) LOH at the Rb1 locus was confirmed in pituitary tumors (left; T1–T10) and thyroid tumors (right; T1–T4) derived from Rb1 mutants. Pit, pituitary DNA from a WT mouse. D2, B6 and DB: genomic DNA derived from D2, B6 and DBF1 mice, respectively.
We then evaluated the effects of Rb1 mutations on mRNA expression in vivo. qPCR analysis revealed that M1326 mutants expressed levels of Rb1 mRNA comparable with the WT (Fig.2c). However, the animals heterozygous for M1032 and M1033 exhibited significant decreases in Rb1 mRNA (67.5% and 70.0%, respectively; Fig.2c), suggesting that these mutations cause nonsense-mediated decay.
Analysis of the genomic DNA from pituitary tumors of N2 and N3 progeny revealed loss of the WT Rb1 allele derived from D2 (Fig.2d). Similarly, the thyroid tumors from M1032 and M1033 exhibited loss of the WT allele. These results indicate that the WT Rb1 gene is lost in the tumors that develop in the mutants. Taken together, we conclude that Rb1 mutations are responsible for endocrine tumor development.
Distinct tumor spectrum and incidence among the mutant lines
To precisely evaluate the tumor spectrum and incidence, we analyzed the phenotypes of N2 and N3 progeny. In M1032 and M1033 more than 90% of the heterozygous animals developed thyroid tumors by 70 weeks (Fig. S1A). These mice also developed pituitary tumors in 81.1% of M1032 and 57.4% of M1033 heterozygotes (Fig. S1B). These tumor profiles recapitulated those reported in Rb1+/− animals,7,10 further supporting our conclusion that Rb1 mutations are responsible for the tumorigenesis.
The progeny of heterozygous M1326 animals also developed pituitary tumors, although the incidence was lower (43.6% at 75 weeks; Fig. S1B) and the onset was delayed compared with that in the splice-site mutants. Strikingly, these animals did not develop thyroid tumors (Fig. S1A). These results suggest that this point mutation altered pRb function, leading to the selective loss of pituitary tumor suppression function.
D326V mutant selectively suppress E2F1 in thyroid C-cells
To understand the mechanism of the D326V mutation on the tumor spectrum, we performed comparative analyses for Rb1D326V/+ and Rb1+/− mice. Because inactivation of the functions of E2F was considered to be the major mechanism by which pRb suppressed tumor development, we focused on the E2F proteins in the mutant mice. For this purpose, we initially tried to determine the protein level in multiple tissues, including thyroid tissues, using immunoblot analysis, but we did not detect any significant differences between the controls and the mutant mice. We hypothesized that this is because each tissue contained multiple types of cells, which respond differently to the inactivation of pRb function. In fact, previous reports indicate that protein levels of E2F are increased by the depletion of pRb; this occurs via the expression of shRNA in prostate cancer cells and by targeted mutation in mouse embryonic fibroblasts.15,16 In contrast, two reports have demonstrated that E2F proteins increased their stabilities by interacting with pRb17,18 (Fig. S2). These observations indicate that pRb deficiency might have a positive or negative influence on the abundance of E2F proteins, depending on the cell context. Hence, there is a need to examine the cell type-specific effect of pRb deficiency. For this purpose, we performed IHC to evaluate the level of E2F and found that E2F1, E2F2 and E2F3 were decreased in the thyroid C-cells of the mutant Rb1+/− mice (Fig.3a). These results indicate that, in the thyroid C-cells, “free” active E2F were unstable, while pRb-bound E2F were stable and accumulated.17,18
Figure 3.
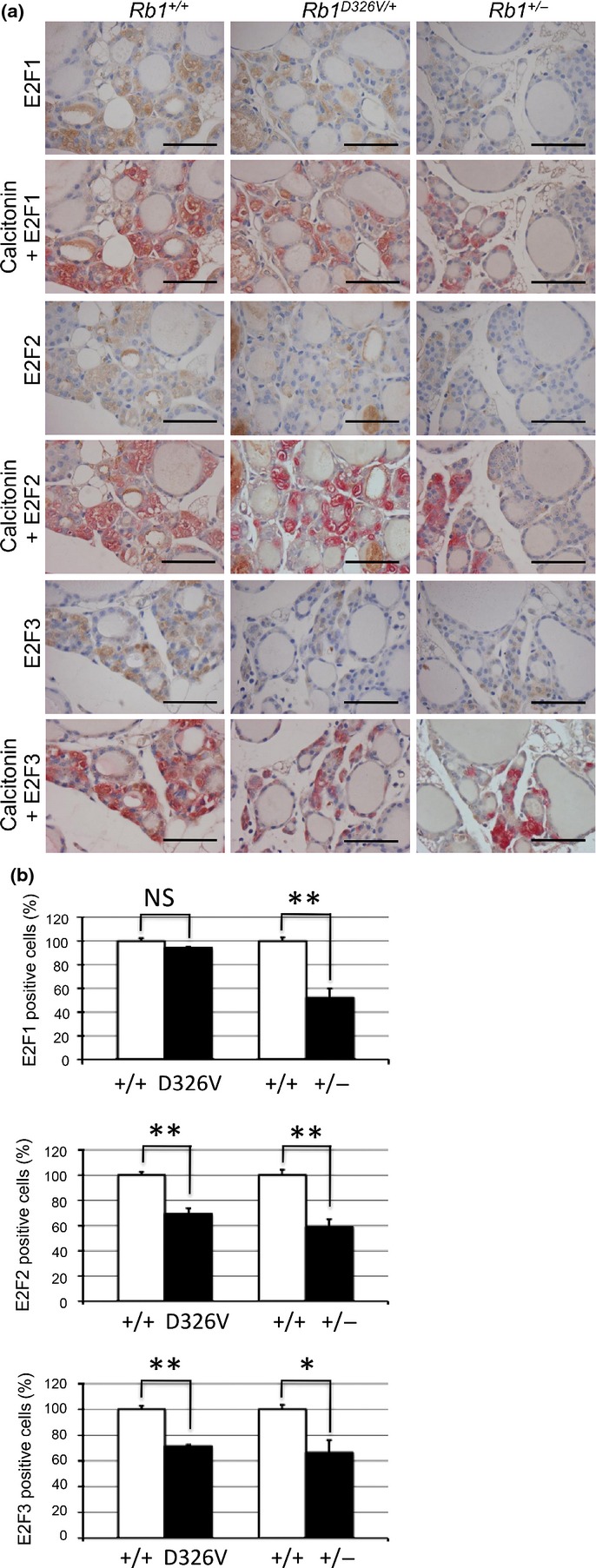
Immunohistochemical analysis of E2F in normal thyroid C-cells. (a) Normal thyroid tissues from retinoblastoma gene (Rb1)+/+ (left column), Rb1D326V/+ (middle column) or Rb1+/− (right column) mice were stained with anti-E2F1 (upper section), anti-E2F2 (middle section) and anti-E2F3 (lower section) antibody using HRP chromogenic substrate, DAB (brown-colored staining). To confirm the C-cell area, each specimen was double stained with anti-calcitonin antibody using alkaline phosphatase chromogenic substrate, Fuchsin (red-colored staining). Bar, 50 μm. (b) The positive-cell ratio was analyzed in C-cells stained with anti-E2F1 (upper graph), anti-E2F2 (middle graph) or anti-E2F3 (lower graph) antibody. Each positive-cell ratio calculated for the C-cells of Rb1D326V/+ (D326V) or Rb1+/− (+/−) mice was normalized with respect to those for the wild-type littermate (+/+), considered 100%. Statistical analysis: unpaired two-tailed Student's t-test. NS, not significant. *P < 0.05. **P < 0.01. Error bar, SD. n = 3 per genotype.
Using this system, we determined the level of E2F in the thyroid C-cells of Rb1D326V/+ mice using IHC (Fig.3a, middle panels) and found a significant decrease in the positive ratios of E2F2 and 3 (Fig.3b, middle and lower graphs). Thyroid C-cells were identified by detecting the expression of calcitonin (Fig.3a). Interestingly, no significant difference in the positive cell ratio of E2F1 between Rb1D326V/+ mice and WT littermates was detected (Fig.3b, upper graph). These results suggest that, in the C-cells of Rb1D326V/+ animals, pRbD326V retains the ability to bind E2F1 and suppress its function, but pRbD326V cannot inhibit E2F2/3 function. Because thyroid tumors were suppressed in this mutant line, these observations indicate that E2F1 is the major oncogenic determinant and its suppression is sufficient to prevent thyroid tumors.
Different regulation of E2F by pRbD326V in ILP cells in Rb1 mutants
In contrast to thyroid tumors, pituitary tumors were observed in Rb1D326V/+ mutant mice. This observation raised the possibility that the suppression of E2F1 was not sufficient to inhibit pituitary tumor formation. Alternatively, perhaps pRbD326V could not inhibit E2F1 function in this type of tumor. To distinguish these possibilities, IHC for E2F was performed in pituitary specimens (Fig.4). It is thought that the primary origin of pituitary tumors induced by Rb1 mutation is the ILP6,8; in fact, we found that most pituitary tumors in Rb1D326V/+ and Rb1+/− mice expressed α-MSH, a marker for ILP cells (Fig.5).
Figure 4.
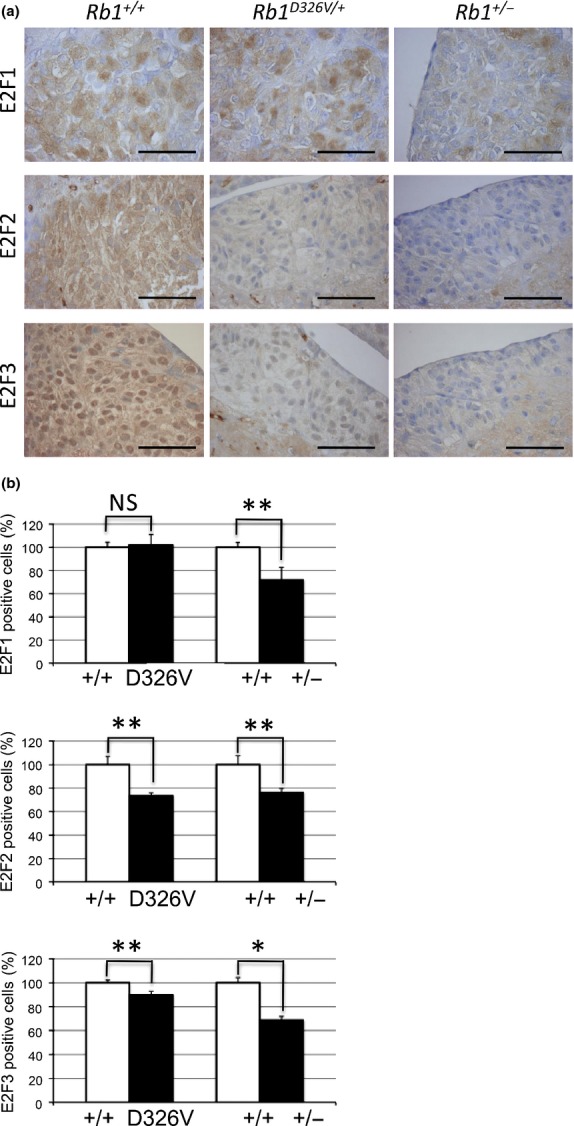
Immunohistochemical analysis of E2F in normal intermediate lobe of the pituitary (ILP) cells. (a) Normal ILP tissues from retinoblastoma gene (Rb1)+/+ (left column), Rb1D326V/+ (middle column) or Rb1+/− (right column) mice were incubated with anti-E2F1 (upper row), anti-E2F2 (middle row) or anti-E2F3 (lower row) antibody. Bar, 50 μm. (b) The positive-cell ratio was analyzed in the ILP specimens treated with anti-E2F1 (upper panel), anti-E2F2 (middle panel) or anti-E2F3 (lower panel) antibody. The positive-cell ratios were calculated as described in Figure3(b). Statistical analysis: unpaired two-tailed Student's t-test. NS, not significant. *P < 0.05. **P < 0.01. Error bar, SD. n = 3 per genotype.
Figure 5.
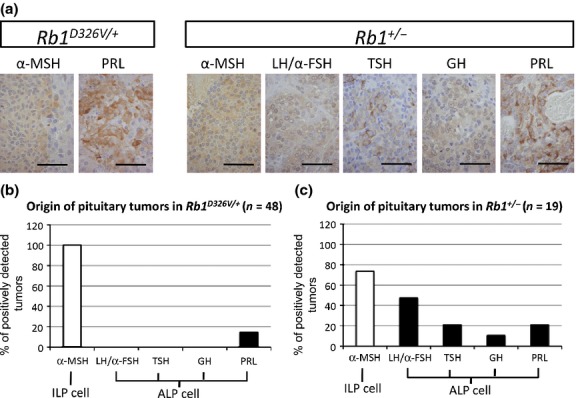
Different tumor distribution in the anterior lobe of the pituitary (ALP) for retinoblastoma gene (Rb1)+/− and Rb1D326V/+ mutant lines. (a) Immunohistochemical analysis of pituitary tumors developed in Rb1D326V/+ and Rb1+/− mutants using antibodies against the pituitary hormones described above each panel. Bar, 50 μm. (b,c) Analysis of cellular origin of the pituitary tumors in Rb1D326V/+ (b) and Rb1+/− (c) mutants. The percentage of the positively stained pituitary tumors analyzed in (a) was calculated. α-MSH, alpha melanocyte-stimulating hormone; GH, growth hormone; LH/α-FSH, luteinizing hormone/follicle-stimulating hormone alpha subunit; PRL, prolactin; TSH, thyroid-stimulating hormone.
Positive cell ratios of E2F were significantly decreased in Rb1+/− mutants, whereas the E2F1 positive cell ratio was selectively maintained in Rb1D326V/+ mutants (Fig.4b, upper graph). Thus, similar to the results for C-cells, pRbD326V retained the ability to inactivate E2F1 but failed to suppress E2F2/3 in ILP cells. Because pituitary tumors developed in this mutant, these results indicate that suppression of E2F1 was not sufficient to inhibit this type of tumor. Although it is unclear which E2F is the key molecule or how E2F contribute to the tumorigenesis of ILP, these observations indicate that E2F have distinct roles in tumorigenesis, depending on cell types.
Different spectrum of anterior lobe of the pituitary (ALP) tumors in Rb1 D326V/+ mutants from those in Rb1+/− animals
During the analysis of pituitary tumors, we found that a certain fraction of the tumors did not express α-MSH, suggesting that those tumors do not originate from the ILP. The ALP tumors are reported in B6-backcrossed Rb1+/− animals19; hence, we performed IHC using antibodies against ALP proteins, including LH/α-FSH, TSH, GH and PRL, to identify their cellular origin (Fig.5a). In Rb1+/− mice, α-MSH-, PRL-, LH/α-FSH-, TSH- and GH-positive cells were observed (73.7%, 21.1%, 47.4%, 21.1% and 10.5%, respectively; Fig.5c). In Rb1D326V/+ mice, all pituitary tumors expressed α-MSH, indicating they originate from the ILP; additionally, 14.6% also expressed PRL. No tumors expressed LH/α-FSH, TSH or GH (Fig.5b); thus, the pRbD326V mutant protein retains its tumor-suppressive activity in cells originating from LH/α-FSH-, TSH- and GH-producing cells. These observations suggest that WT pRb suppresses ALP- and ILP-derived tumors, but pRbD326V only suppresses tumorigenesis originating from LH/α-FSH-, TSH- and GH-producing cells. These results explain the lower incidence of pituitary tumors in Rb1D326V/+ mutants than that of null mutants.
D326V mutant inactivated E2F in a cellular context-dependent manner in ALP cells
To elucidate the mechanism underlying the different ALP cell-derived tumor spectra of Rb1D326V/+ and Rb1+/− mutants, double staining for E2F and pituitary hormones was performed in pituitary specimens (Fig.6).
Figure 6.

Immunohistochemical analysis of E2F in normal anterior lobe of the pituitary (ALP) cells. E2F1- (upper panels), E2F2- (middle panels) and E2F3- (lower panels) positive-cell ratios in normal luteinizing hormone/follicle-stimulating hormone alpha subunit (LH/α-FSH)- (a), thyroid-stimulating hormone (TSH)- (b), growth hormone (GH)- (c) and prolactin (PRL)-producing (d) ALP cells from retinoblastoma gene (Rb1)D326V/+ (D326V) or Rb1+/− (+/−) mutants. The positive-cell ratios were calculated as described in Figure3(b). Statistical analysis: unpaired two-tailed Student's t-test. NS, not significant. *P < 0.05. **P < 0.01. Error bar, SD. n = 3 per genotype.
In Rb1+/− mice, positive cell ratios of E2F significantly decreased in all APL cell types (Fig.6, blue bars). These results are consistent with the observations that Rb1+/− mice developed pituitary tumors that originated from those cell types (Fig.5c). However, in PRL-expressing cells of Rb1D326V/+ mutants, the E2F2-positive cell ratio significantly decreased, while E2F1/3-positive cell ratios were maintained (Fig.6d, red bars). This result indicates that activation of E2F2 is important for the tumorigenic change of PRL-positive cells. However, the positive cell ratios of all E2F in other cell types in Rb1D326V/+ heterozygous mutants were comparable with those observed in WT animals (Fig.6a–c; red bars), which is consistent with the observation that the pituitary tumors originating from these cell types were suppressed in Rb1D326V/+ mutants. These observations demonstrate that pRbD326V retains the ability to inactivate E2F in these cell types. Because pRbD326V failed to inactivate E2F2 in PRL-expressing cells, these results also indicate that suppression of E2F function by pRb depends on the cell type.
In vitro interaction of pRbD326V with E2F
To test the physical interaction of pRbD326V with E2F, a co-IP analysis was performed. The Flag-tagged, WT pRb or pRbD326V were synthesized in vitro and then mixed with DP-1 and each of E2F1/2/3. In each mixture, pRb or pRbD326V was immunoprecipitated by an anti-Flag antibody and the bound E2F were analyzed by specific antibodies. Only E2F1 was detected in the precipitate from pRbD326V, but not E2F2/3 (Fig.7). These results indicate that pRbD326V retains intrinsic binding activity to E2F1, but cannot interact with E2F2/3. These observations reveal the D326V mutation selectively abrogates the direct interaction with E2F2/3, but not E2F1, leading to the selective suppression of E2F1 function.
Figure 7.
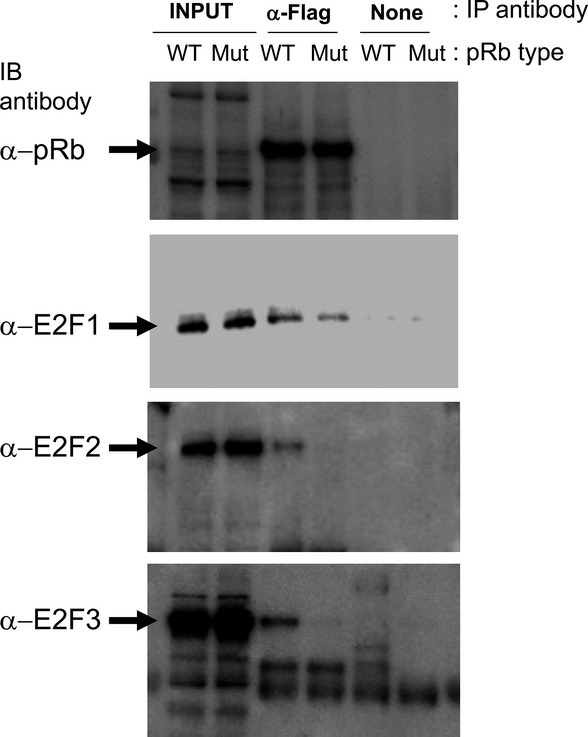
Retained E2F1 binding activity of retinoblastoma protein (pRb)D326V. Flag-tagged WT pRb (WT) or pRbD326V (Mut) were mixed with DP-1 and each of E2F1/2/3. The pRb were then immunoprecipitated (IP) with (α-Flag) or without (none) anti-Flag antibody. The immunoprecipitates were immunoblotted (IB) with anti-pRb, anti-E2F1, anti-E2F2 or anti-E2F3 antibody.
Discussion
In the present study we generated three mutant alleles of Rb1. Two of them contained base substitutions at splice sites, leading to significantly decreased mRNA levels. These mutant mice recapitulated the phenotypes observed in conventional Rb1 knockout mice, confirming the results of previous reports (Fig.8b).7,8 In contrast to these mutants, mice with the third mutant allele, containing the missense at codon 326, exhibited a unique tumor spectrum. (Fig. S1).
Figure 8.
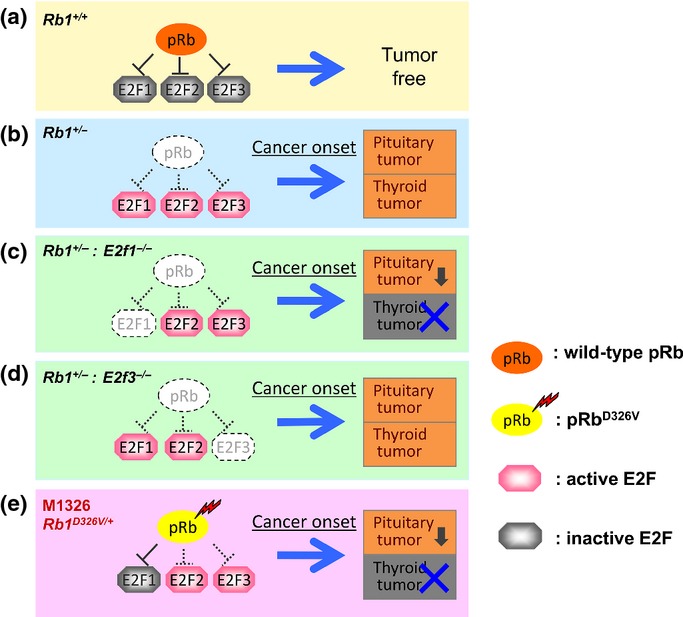
Tissue-specific contribution of E2F to tumorigenesis. Wild-type retinoblastoma protein (pRb) inactivates E2F and suppresses tumor development (a). In retinoblastoma gene (Rb1)+/− mice, loss of Rb1 function leads to activation of E2F1/2/3, which consequently results in development of both pituitary (incidence, 95–100%) and thyroid (incidence, 56–70%) tumors (b).6,7,10,11 In Rb1+/−:E2f1−/− double mutants, the loss of Rb1 function is accompanied by E2f1 deletion, which results in 62% incidence of pituitary tumors and complete suppression of thyroid tumors, indicating that E2F1 is mainly responsible for thyroid tumor development (c).10 In Rb1+/−:E2f3−/− mutants, the loss of Rb1 function is accompanied by E2f3 deletion, which results in suppressed growth of pituitary tumors, indicating that E2F3 is responsible for pituitary tumor growth. These Rb1+/−:E2f3−/− mutants show the promotion of malignancy in thyroid tumor (d).11 In Rb1D326V/+ mutants, pRbD326V binds and inactivates E2F1 only, which results in 44% incidence of pituitary tumors and complete suppression of thyroid tumors (e).
Assuming that pRb-bound, inactivated E2F are stable and detectable by IHC, pRbD326V suppressed the activity of E2F1 but could not inactivate E2F2/3 in C-cell or ILP cells (Figs4). Consistent with these observations, our biochemical analysis demonstrated the specific binding of pRbD326V to E2F1 (Fig.7). Importantly, the tumor spectrum found in the Rb1D326V/+ mutants resembled those of E2F1−/−:Rb1+/− double-knockout mice reported previously, which only develop pituitary tumors but not thyroid tumors. Furthermore, the incidence of pituitary tumors in the compound mutant mice was reported to be approximately 30% lower than that in the Rb1+/− mice10 and this reduced incidence was recapitulated in the Rb1D326V/+ mutant mice (Fig.8c,e). These observations strongly support our conclusion that pRbD326V selectively inactivated the function of E2F1, but not of E2F2 or 3, which led to these unique tumor spectra. Although the molecular basis for the thyroid tumors being more dependent on E2F1 than pituitary tumors remains unclear, one possible explanation is the expression level of E2F1. We found that the E2F1 level in the thyroid was much higher than that in other organs (Fig. S3). Another possibility is the functional difference among the E2F family proteins in different organs. In fact, several individual E2F-specific targets were reported in cultured cells and mouse tissues, suggesting that each E2F protein has a cell type-specific biologically distinct role.20–22 In future, clarifying the functional redundancy and specificity of the E2F proteins and their contributions to tumorigenesis would be therapeutically important.
In addition to the G1-S transition mediated by transcriptional regulation via E2F proteins, accumulating evidence indicates the role of pRb in the regulation of chromatin structure. The pRb regulates heterochromatin domains around the centromere and the loss of function of pRb reduces chromosome cohesion.23 Although the exact molecular basis for this phenomena remains to be fully elucidated, several molecular mechanisms, including trimethylation of Lys20 on histone H4 (H4K20me3) by Suv4-20 and chromosome cohesion mediated by condensin II, have been proposed.24 Because the aberration in chromosome structure and segregation leads to chromosome instability, which is a common feature of solid tumors, it is important to explore the effects of Rb1D326V mutation on chromosome structure.
A previous report shows that pituitary tumors developing in Rb1+/− mice arise in the ILP and ALP.19 These results were confirmed in the present study using conventional Rb1+/− mice and splice-site mutants. However, IHC of pituitary tumors from Rb1D326V/+ mutants revealed a striking reduction in the incidence of ALP tumors. Detailed analysis of ALP tumors revealed that LH/α-FSH-, TSH- and GH-expressing tumors were not present in Rb1D326V/+ mutants (Fig.5b). Surprisingly, unlike in thyroid C-cells and ILP cells, E2F2 and 3 were also suppressed in these cells, which is in a stark contrast to the observations made in experiments using Rb1+/− mice (Fig.6). These observations suggest the regulation of E2F is cellular context dependent, leading to a distinct tumor spectrum in pituitary tumors arising in the ALP. Similar observations were reported by Leung et al.,19 who showed that the Rb1+/− tumor profile changes dramatically with the genetic background. It would be of interest to clarify the cellular context-dependent mechanisms that determine susceptibility to ALP tumors.
Retinoblastoma protein associates with E2F through the C-terminal region, that is, A/B pocket domain and C-domain.25–27 Several oncoproteins such as E1A and SV40 large T activate E2F by binding directly to the A/B pocket, thus releasing E2F from pRb. Consequently, a point mutation of the B pocket region, R654W, fails to interact with E2F1/2/3, and RbR654W/+ mutants develop both thyroid and pituitary tumors, recapitulating phenotypes observed in Rb1+/− mice.28,29 Several studies indicate that the N-terminal region of pRb modulates the binding capacity of pRb to E2F by acting through structural conformational change.30,31 Furthermore, it has been suggested that the N-terminal region of pRb interacts with proteins and regulates the interaction of pRb and E2F. For example, Inoue et al.32 report Hsc70 interacts with the N-terminal region of pRb (residues 301–372). Hsc70 also interacts with oncoproteins and helps to release E2F from pRb as a molecular chaperone.33 Notably, we showed in vitro that the D326V mutation selectively perturbed the direct interaction with E2F2/3 (Fig.7). These observations raise the possibility that Hsc70 or other unidentified proteins modify these interactions and that the D326V mutation might mimic or disrupt these modifications. This model might also explain the cellular context-dependent inactivation of E2F by this Rb1 mutant.
The contribution of the pRb N-terminal region to tumorigenesis is under debate. Using transgenic mice expressing mutant pRb, Riley et al.34 reported that pRb mutants with deletions of N-terminal regions fail to prevent pituitary tumors, indicating that the N-terminal region plays critical roles in tumor suppression. However, Yang et al.35 found pRb lacking residues 1–378 suppressed pituitary tumors in Rb+/+:Rb−/− chimeric mice, indicating that the N-terminal residues are dispensable for pRb tumor-suppression activity. Because these mutant proteins lack different portions of the N-terminal region, this discrepancy may be explained by the distinct functions of each mutant protein. For example, the former mutant protein, which failed to suppress pituitary tumor development, may lose the binding capacity to E2F, while it is retained in the latter mutant. It is also possible that different sequence deletions lead to differences in interacting proteins. It should be noted that, in contrast to partial or complete deletion of the N-terminal region, we established mutant mice with a point mutation, which compromised tumor suppression by pRb. Furthermore, we modulated endogenous Rb1, rather than exogenously expressing transgenes. In humans, some families with low-penetrance retinoblastoma carry mutations in the N-terminal region of pRb.36 The functional analyses of exon 4 (127–166 amino acid) deletion in Rb1, an N-terminal mutation found in the patients reported by Otterson et.al.37, shows intact E2F1-binding capacity and colony suppression is retained in vitro.37 Thus, Rb1D326V/+ mice can be used as models to elucidate the in vivo functions of mutations in the pRb N-terminal region.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Fig. S1. Incidence of tumors in the three Rb1 mutant lines generated by ENU mutagenesis.
Fig. S2. Effect of pRb on E2F1, 2 and 3 protein abundance.
Fig. S3. Different expression levels of E2F1 in multiple organs.
Table S1. Antibodies used for immunohistochemistry and co-IP analysis.
References
- Matsunaga E. Retinoblastoma: host resistance and 13q-chromosomal deletion. Hum Genet. 1980;56:53–8. doi: 10.1007/BF00281568. [DOI] [PubMed] [Google Scholar]
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–24. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–7. [PubMed] [Google Scholar]
- Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994;7:480–4. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- Harrison DJ, Hooper ML, Armstrong JF, Clarke AR. Effects of heterozygosity for the Rb-1t19neo allele in the mouse. Oncogene. 1995;10:1615–20. [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−) mice. Nat Genet. 1998;18:360–4. doi: 10.1038/ng0498-360. [DOI] [PubMed] [Google Scholar]
- Ziebold U, Lee EY, Bronson RT, Lees JA. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol. 2003;23:6542–52. doi: 10.1128/MCB.23.18.6542-6552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitotsumachi S, Carpenter DA, Russell WL. Dose-repetition increases the mutagenic effectiveness of N-ethyl-N-nitrosourea in mouse spermatogonia. Proc Natl Acad Sci U S A. 1985;82:6619–21. doi: 10.1073/pnas.82.19.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet. 1999;8:1955–63. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- Toki H, Inoue M, Motegi H, et al. Novel mouse model for Gardner syndrome generated by a large-scale N-ethyl-N-nitrosourea mutagenesis program. Cancer Sci. 2013;104:937–44. doi: 10.1111/cas.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Yeow WS, Ertel A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–92. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Lane AN, Robertson B, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2014;33:556–66. doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Martelli F, Livingston DM, Wang Z. The retinoblastoma gene product protects E2F-1 from degradation by the ubiquitin-proteasome pathway. Genes Dev. 1996;10:2949–59. doi: 10.1101/gad.10.23.2949. [DOI] [PubMed] [Google Scholar]
- Hateboer G, Kerkhoven RM, Shvarts A, Bernards R, Beijersbergen RL. Degradation of E2F by the ubiquitin-proteasome pathway: regulation by retinoblastoma family proteins and adenovirus transforming proteins. Genes Dev. 1996;10:2960–70. doi: 10.1101/gad.10.23.2960. [DOI] [PubMed] [Google Scholar]
- Leung SW, Wloga EH, Castro AF, Nguyen T, Bronson RT, Yamasaki L. A dynamic switch in Rb+/− mediated neuroendocrine tumorigenesis. Oncogene. 2004;23:3296–307. doi: 10.1038/sj.onc.1207457. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–16. [PMC free article] [PubMed] [Google Scholar]
- Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20:5797–807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J, Graveel CR, Bartley SM, Madore SJ, Farnham PJ. The identification of E2F1-specific target genes. Proc Natl Acad Sci U S A. 2002;99:3890–5. doi: 10.1073/pnas.062047499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Yazinski SA, Nicolay B, Bryll A, Zou L, Dyson NJ. Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol Cell. 2014;53:993–1004. doi: 10.1016/j.molcel.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemington EK, Speck SH, Kaelin WG., Jr E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci U S A. 1993;90:6914–8. doi: 10.1073/pnas.90.15.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–8. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin SM, Gall AL, Zheng N, Pavletich NP. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell. 2005;123:1093–106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Sun H, Chang Y, Schweers B. An E2F binding-deficient Rb1 protein partially rescues developmental defects associated with Rb1 nullizygosity. Mol Cell Biol. 2006;26:1527–37. doi: 10.1128/MCB.26.4.1527-1537.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Wang Y, Chinnam M, et al. E2f binding-deficient Rb1 protein suppresses prostate tumor progression in vivo. Proc Natl Acad Sci U S A. 2011;108:704–9. doi: 10.1073/pnas.1015027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J Biol Chem. 2010;285:16286–93. doi: 10.1074/jbc.M110.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamber EP, Beuron F, Morris EP, Svergun DI, Mittnacht S. Structural insights into the mechanism of phosphoregulation of the retinoblastoma protein. PLoS One. 2013;8:e58463. doi: 10.1371/journal.pone.0058463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Torigoe T, Sogahata K, et al. 70-kDa heat shock cognate protein interacts directly with the N-terminal region of the retinoblastoma gene product pRb. Identification of a novel region of pRb-mediating protein interaction. J Biol Chem. 1995;270:22571–6. doi: 10.1074/jbc.270.38.22571. [DOI] [PubMed] [Google Scholar]
- Sheng Q, Denis D, Ratnofsky M, Roberts TM, DeCaprio JA, Schaffhausen B. The DnaJ domain of polyomavirus large T antigen is required to regulate Rb family tumor suppressor function. J Virol. 1997;71:9410–6. doi: 10.1128/jvi.71.12.9410-9416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley DJ, Liu CY, Lee WH. Mutations of N-terminal regions render the retinoblastoma protein insufficient for functions in development and tumor suppression. Mol Cell Biol. 1997;17:7342–52. doi: 10.1128/mcb.17.12.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Williams BO, Hinds PW, et al. Tumor suppression by a severely truncated species of retinoblastoma protein. Mol Cell Biol. 2002;22:3103–10. doi: 10.1128/MCB.22.9.3103-3110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouzeid H, Schorderet DF, Balmer A, Munier FL. Germline mutations in retinoma patients: relevance to low-penetrance and low-expressivity molecular basis. Mol Vis. 2009;15:771–7. [PMC free article] [PubMed] [Google Scholar]
- Otterson GA. Chen Wd, Coxon AB, Khleif SN, Kaye FJ. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc Natl Acad Sci U S A. 1997;94:12036–40. doi: 10.1073/pnas.94.22.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Incidence of tumors in the three Rb1 mutant lines generated by ENU mutagenesis.
Fig. S2. Effect of pRb on E2F1, 2 and 3 protein abundance.
Fig. S3. Different expression levels of E2F1 in multiple organs.
Table S1. Antibodies used for immunohistochemistry and co-IP analysis.