

Abstract
Vascular endothelial growth factor (VEGF)-C is an important lymphangiogenic factor involved in the lymphangiogenesis of gallbladder carcinoma (GBC) and the lymph node metastasis of the tumor. Tumor necrosis factor (TNF)-α, a key inflammatory cytokine responding to chronic inflammation of GBC, has been reported to stimulate the expression of VEGF-C in some nonneoplastic cells. But whether TNF-α promotes the expression of VEGF-C in GBC has yet to be determined. Therefore, in the present study, the concentration of TNF-α and VEGF-C and the lymphatic vessel density (LVD) in the clinical GBC specimens were analyzed, and a linear correlation was found between the concentration of TNF-α and that of VEGF-C, the lymphatic vessel density (LVD); The transcription and protein level of VEGF-C in NOZ cell line were detected by real-time polymerase chain reaction (PCR) and enzyme linked immunosorbent assay (ELISA), and TNF-α enhanced the expression of VEGF-C in NOZ cell lines in a dose and time-dependent manner. Lymphatic tube formation in vitro was observed in a three-dimensional coculture system consisting of HDLECs and NOZ cell lines, and lymphatic vessels of GBC in nude mice model was detected by immunohistochemistry. TNF-α promoted the tube formation of lymphatic endothelial cells in vitro and the lymphangiogenesis of GBC in nude mice; The nuclear factor (NF)-κB binding site on the VEGF-C promoter was identified using Site-directed mutagenesis, electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation assay (ChIP). Taken together, TNF-α can upregulate the expression of VEGF-C and promote the lymphangiogenesis of GBC via NF-κB combining with the promoter of VEGF-C.
Keywords: Gallbladder carcinoma, lymphangiogenesis, nculear factor-κB, tumor necrosis factor-α, vascular endothelial growth factor-C
Gallbladder cancinoma (GBC), which ranks sixth among gastrointestinal cancers, is the most common malignancy of the biliary system, representing 80–95% of biliary tract cancers worldwide.1 There is a widely variable geographic pattern for GBC, and the incidence rates are extraordinarily high in South America and Eastern Asia, where Chile and Korea exhibit the highest rate of GBC: 9.7/100 000 and 6.4/100 000.2 Gallbladder cancinoma is characterized by high malignance and striking invasion and metastasizes mainly through liver and lymph nodes.3,4 Lymph node metastasis often occurs at an early stage of tumorigenesis, which leads to poor prognosis of GBC patients,5–7 the countries with the highest incidence experience the highest mortality, for example, the mortality rate of Chile and Korea were 7.8/100 000 and 4.8/100 000, respectively. However, the incidence and progression mechanisms of this malignance remain blurred, although gallstones and chronic inflammatory changes are considered important risk factors.1
In the chronic inflammatory response, macrophage is the key player and releases a great number of bioactive products, among which tumor necrosis factor (TNF)-α is one of the most important. TNF-α, as a monocyte-derived cytokine, has been documented to mediate widespread systemic effects, such as shock and tissue injury,8 cachexia, anemia and chronic inflammation.9,10 Further research of signaling pathways report the multifaceted actions of TNF-α in cancers, which lead to cell death,11,12 proliferation and migration.13 Its promoting14,15 or inhibiting actions16,17 in the tumor metastasis have also been reported in different tumors. Previous studies have showed that the expression of TNF-α in gallbladder carcinoma is higher than that in calculous cholecystitis, and that TNF-α protein expression increases accordingly with the evolution of gallbladder mucosal epithelium evolves from hyperplasia, dysplasia to carcinoma, and with the progress of tumor stages, which suggests that TNF-α may contribute to the carcinogenesis and progression of gallbladder carcinoma,18 but the mechanism of it is unknown. Whether TNF-α promotes the metastasis of gallbladder carcinoma has yet to be determined.
In some non-tumor cells such as fibroblasts and rheumatoid synoviocytes, TNF-α can stimulate the mRNA and protein expression of VEGF-C,19,20 and vascular endothelial growth factor (VEGF)-C has been identified as a lymphangiogenic factor,21 involving in the lymphangiogenesis of gallbladder carcinoma and the lymph node metastasis of the tumor.22 So we thought of the possibility that TNF-α can promote the expression of VEGF-C and lymphangiogenesis of gallbladder carcinoma. In this study we tried to prove this hypothesis and investigate the potential underlying mechanism.
Material and Methods
Patients and tissue specimens
A total of 20 specimens of gallbladder carcinoma and the bile were randomly obtained from the patients admitted to the Affiliated Union Hospital of Fujian Medical University in China (from 2008 to 2012, background of clinical samples is shown in Table1). Twenty specimens of “control bile” were obtained from the patients with cholesterol gallbladder polyps receiving surgical treatment, as it is practically infeasible to obtain pure normal bile in clinical practice. All patients enrolled in this study had not received any preoperative chemotherapy or radiotherapy. The tissues were collected according to the protocol approved by the Ethics Committee of the Medical Faculty of Fujian Medical University.
Table 1.
Correlation between clinicopathological factors and concentration of vascular endothelial growth factor-C (VEGF-C) and tumor necrosis factor-α (TNF-α) in gallbladder carcinoma (GBC)
| Factor | Concentration of VEGF-C (pg/mL) | P | Concentration of TNF-α(pg/mL) | P | ||
|---|---|---|---|---|---|---|
| >764 | <764 | >609 | <609 | |||
| Age | ||||||
| <60 | 5 | 7 | 0.535 | 4 | 8 | 0.388 |
| ≥60 | 4 | 4 | 4 | 4 | ||
| Gender | ||||||
| Male | 4 | 5 | 0.658 | 4 | 6 | 0.658 |
| Female | 5 | 6 | 4 | 6 | ||
| Clinical stage | ||||||
| I–III | 1 | 7 | 0.025 | 0 | 8 | 0.005 |
| IV–V | 8 | 4 | 8 | 4 | ||
| Lymph node metastasis | ||||||
| Negative | 1 | 8 | 0.009 | 1 | 8 | 0.025 |
| Positive | 8 | 3 | 7 | 4 | ||
| Histological grade | ||||||
| Poorly | 4 | 3 | 0.246 | 4 | 3 | 0.72 |
| Moderately | 4 | 5 | 3 | 6 | ||
| Well | 1 | 3 | 1 | 3 | ||
| Histological type | ||||||
| Adenocarcinoma | 5 | 7 | 0.835 | 5 | 7 | 0.835 |
| Papillary carcinoma | 2 | 3 | 2 | 3 | ||
| Others | 2 | 1 | 1 | 2 | ||
Cell culture
The human gallbladder carcinoma cell line NOZ was originally obtained from Health Science Research Resources Bank (HSRRB) in Japan. Human dermal lymphatic endothelial cells (HDLECs) was from Sciencell (San Diego, California, USA). NOZ cell line was maintained in Dulbecco's Modified Eagle's Medium (DMEM, Gibco, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies Gibco, Carlsbad, California). HDLECs were incubated in endothelial cell medium (ECM, Sciencell). Both of the cell lines were incubated at 37°C in a 5% CO2 humidified incubator.
Immunohistochemistry and counting of lymphatic vessel
The lymph vessels of gallbladder carcinoma specimens were detected by immunohistochemistry as previously described.23 The primary antibodies used in clinical specimens and nude mice specimens were mouse anti-human D2-40 rabbit monoclonal antibody (MAIXIN_BIO, Fuzhou, China) and goat anti-mouse LYVE-1 polyclonal antibody (R&D Systems, Minneapolis, MN, USA), respectively. Secondary antibodies used were goat anti-mouse and rabbit anti-goat Ig/HRP (ZSGB-BIO, Beijing, China). The specimens were visualized with stable 3, 3-diaminobenzidine (DAB, ZSGB-BIO, Beijing, China).
Sections were first examined at low magnification (×100) to identify areas with most intense staining and apparent highest density of microvessel (hotspot).24 Three areas of hotspots were selected by three pathologists who independently evaluated the slides for microvessel counting using 400× magnification (0.17 mm2 field), without the knowledge of patient status. In the absence of hotspots, three or more randomly selected areas were counted. Intratumoral and peritumoral lymphatic vessel should be counted separately. Brown vessels without muscle layer or red blood cells (RBCs) in their lumen were considered as lymphatic vessels. Single immunoreactive endothelial cells, or endothelial cell clusters separate from adjacent microvessels, were counted as a vessel.25 The highest number of vessels counted was recorded and used in the statistical analysis.
Real-time RT-PCR
Total RNA was extracted from NOZ cells with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), in accordance with the manufacturer's instructions. It was reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo, Waltham, MA, USA) in accordance with the manufacturer's recommendations. PCR reactions were performed with Fast Start Universal SYBR Green Master (Roche) and fluorescence was measured using the 7500 quantitative real-time thermocycler (Applied Biosystems). β-Actin served as an internal control. The primers for VEGF-C were: 5′-TGTGTGTCCGTCTACAGATGTG-3′ (forward) and 5′-TCGGCAGGAAGTGTGATTGG-3′ (reverse). Each reaction was run in triplicate. All experiments had efficiencies between 95% and 105%, and the gene measure displayed normal melt curves. Fold changes were calculated by 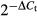 , where ΔCt = Ct (target gene) − Ct (β-Actin) and Δ(ΔCt) = ΔCt (experimental groups) − mean ΔCt.
, where ΔCt = Ct (target gene) − Ct (β-Actin) and Δ(ΔCt) = ΔCt (experimental groups) − mean ΔCt.
Enzyme linked immunosorbent assay
Levels of VEGF-C and TNF-α in bile or cell culture media were assessed by double antibody sandwich Enzyme linked immunosorbent assay using Quantikine ELISA Kits from R&D Systems according to the manufacturer's instructions. Standard curves were prepared before the antibody reaction. Bile samples were diluted twofold with Calibrator Diluent RD6U, and cell culture supernates were not diluted. The wells were read at 450 nm with a Model 550 Microplate Reader (Bio-Rad, Hercules, CA, USA). Each reaction was run in triplicate.
Tube formation assay
To determine whether TNF-α promoted the lymphangiogenesis of gallbladder carcinoma in vitro, we established a three-dimensional coculture system consisting of NOZ cell line and DiI (Beyotime Institute of Biotechnology, Haimen, China)-labeled HDLECs by refering to Yiping Zeng's method31. A mixture of HDLECs (6 × 103 per well) with each NOZ cell line (3 × 103 per well) was seeded in μ-Slide Angiogenesis (ibidi, Martinsried, Germany) polymerized with growth factor–reduced (GFR) Matrigel, and incubated in serum-free DMEM with 50 ng/mL of TNF-α. To assess the role of VEGF-C in the tube formation of HDLECs, we established a VEGF-C-RNAi-LV-transfected NOZ cell line, which was mixed-cultured with HDLECs. All tube formation experiments were observed by inverted fluorescence microscopy (Nikon, Japan), and images were digitally captured at 6 h after plating. The total length, area, and number of tube-like structures formed in each well were measured with Axiovision Rel 4.1 software (Carl Zeiss AG, Jena, Germany).
Establishment of the orthotopic xenograft model
Male athymic BALB/c nude mice in an age range of 4–6 weeks were obtained from Slaccas Laboratory Animal Co. (Shanghai, China). As Jan-Hendrik Egberts' method described,26 after the anesthesia, about 4 × 105 cells were mixed with Matrigel (20 μL of aforementioned cell suspension in DMEM with 20 μL of reduced growth factor Matrigel). The gallbladder was exposed via abdominal midline incision (approximately 0.8–1.0 cm) and the bile in the gallbladder was extruded. Then 40 μL of cell suspension mixed with Matrigel was slowly injected into the gallbladder with a 29G insulin syringe (BD, Research Triangle Park, North Carolina, USA). The syringe was withdrawn from the gallbladder when the cell suspension became white because of solidification. Finally, the gallbladder and liver lobes were replaced and the abdominal wall was sutured. The physical condition of the mice was monitored every day in the first week and then every 3 days in the following 3 weeks. Four weeks later, the mice were euthanized by exposure to CO2 and primary tumors were dissected and excised.
VEGF-C promoter luciferase constructs
Genomic DNA from NOZ cells was extracted with DNeasy Tissue Kit (Bio Teke, Beijing, China) and used as a template for PCR amplification. The plasmid pGL3B- 2000 with the VEGF-C promoter was constructed by ligation of the PCR-generated VEGF-C promoter (nucleotides -2000 – +1, relative to the transcription start site) into the XhoI and HindIII (Thermo) cleaved sites of the luciferase reporter plasmid pGL3-Basic (Promega, Madison, WI, USA). Various VEGF-C promoter deletion constructs, which included pGL3B-1500 (nucleotides -1500 – +1), pGL3B-1000 (nucleotides -1000 – +1), pGL3B-600 (nucleotides -600 – +1), pGL3B-487 (nucleotides -487 – +1), pGL3B-332 (nucleotides -332 – +1) and pGL3B-228 (nucleotides -228 – +1), were also made similarly.
Dual-luciferase reporter assay
A total of 20 mg of cell lysate was used for the detection of intracellular luciferase activity in the Dual-Luciferase Reporter Assay System (Promega), in accordance with the manufacturers' recommendations. Luminescence measurement was carried out on a luminometer (Orion II Microplate Luminometer, Berthold Detection Systems, Prorzheim, Germany).
RNA interference assay
Vascular endothelial growth factor-C gene was knocked down by Lentiviral-mediated small interfering RNA (siRNA) in NOZ cell line, which had been constructed by Genechem (Shanghai, China) and stored in our laboratory.27 The target sequence for NF-κB (p65) siRNA was described by Chuan Bian Lim et al.,28 and the siRNA duplexes, which included a negative control that had no homology with known human genes, were synthesized chemically by Biosune Company (Shanghai, China).
Western blotting
Nucleoprotein was extracted from NOZ cells for measurement of the expression of NF-κB, separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were incubated with monoclonal anti- NF-κB p65 (1:1000; Abcam, Cambridge, UK), with Histone H3 as the internal protein control. Proteins were detected by addition of alkaline phosphatase (AP)-conjugated secondary antibody. Immunoreactive proteins were visualized by addition of CDP STAR reagents (Roche Diagnostics, Germany). Fragment analysis was conducted using Phoretix 1D software.
Identification of putative transcription factor binding sites
Potential transcription factor binding site motifs were searched using TFSEARCH (http://mbs.cbrc.jp/research/db/TFSEARCH.html) and TESS (http://www.cbil.upenn.edu/tess) programs29.
Site-directed mutagenesis
The Site-directed mutagenesis was performed by overlap extension PCR as previously described.30 The NF-κB binding sites, Mut1 (-315 to -306 nt) and Mut2 (-271 to -262 nt), were the target mutation sites. The following three pairs of primers were synthesized chemically by Biosune Company (Shanghai, China): NF-κBmut1F, 5′-AGGCGAGGGAAACGAAGAGCTCCAGGGAGA-3′; NF-κBmut1R: 5′-TCTCCCTGGAGCTCTTCGTTTCCCTCGCCT-3′; NF-κBmut2F: 5′-TGAGAGGGGAGGGCAGCAAGGGCTCGGCACGCT-3′; NF-κBmut2R: 5′-AGCGTGCCGAGCCCTTGCTGCCCTCCCCTCTCA-3′; mutF: 5′-TACGGGAGGTACTTGGAGCGG-3′; mutR: 5′-TTCCAGGAACCAGGGCGTATC-3′ (mutation sites underlined). On step one, the pGL3-332 construct from the VEGF-C promoter was used as a template, and the PCR products (M1F, M1R, M2F and M2R) were respectively obtained with the primers of mutF/NF-κBmut1R, NF-κBmut1F/mutR, mutF/ NF-κBmut2R and NF-κBmut2F/mutR. On step two, mutF/mutR were used as primers, and the templates were the mixture of M1F and M1R or the mixture of M2F and M2R. The PCR products of step two were ligated into the XhoI and HindIII cleaved sites of pGL3-Basic.
Nuclear extraction and electrophoretic mobility shift assay
Nuclear extracts (NE) were prepared from NOZ cells using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime) and nuclear extraction and electrophoretic mobility shift assay (EMSA)was performed with the LightShift Chemiluminescent EMSA kit (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturers' recommendations. A biotin-labeled oligonucleotide probe (5′ biotin-GAGGGAAACGGGGAGCTCCAGGGAG-3′), which contained -315 to -306 nt, was used to confirm the DNA binding of NF-κB. For competition analysis, we used 100-fold excess of unlabeled competitor probe including cold probe (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and mutational cold probe (5′-AGTTGAGGAAACTTGCCCAGGC-3′, mutation sites underlined). Fragment analysis was conducted using Phoretix 1D software.
Chromatin immunoprecipitation assay
The Chromatin immunoprecipitation (ChIP) assay was performed with an EZ-Magna ChIP kit (Merck Millipore, Darmstadt, Germany) according to the manufacturer's instructions. In vivo cross-linking was performed after 1 day with and without TNF-α (50 ng/mL) treatment in the culture of NOZ cells, and then the cell lysates were sonicated to shear genomic DNA. For immunoprecipitation, an antibody against NF-κB p65 and the control normal rabbit immunoglobulin G (IgG) were used. The primers for PCR were: 5′-GACAGGGGCGGGGAGGGAGA-3′ (forward) and 5′-CTCACTCTCCCTCGGAAGCCGTCTC-3′ (reverse), which amplified the area (-389 to -278 nt) including the NF-κB binding site. PCR was performed using personal thermal cycler (Bio-Rad). Digital gel image analysis using Phoretix 1D software.
Statistics
The results were presented as mean ± SEM. Statistical analyses were done with the SPSS software 17.0 (using fisher exact test for enumeration data, employing t-test for means comparison between two groups, applying one-way analysis of variance for means comparison among multi-groups). P < 0.05 was considered statistically significant.
Results
Detection of clinical specimens
Tumor necrosis factor-α and VEGF-C level in the bile specimens from 20 GBC patients and 20 patients with cholesterol gallbladder polyps were assessed by ELISA, and lymphatic vessels density (LVD) of 20 GBC specimens were detected with D2-40 antibody by immunohistochemistry. The concentration of TNF-α in GBC bile specimens was significantly higher than that in control bile (609.0 ± 43.43 vs 193.3 ± 13.74, P < 0.001, Fig.1a). A linear correlation was found between the concentration of TNF-α and that of VEGF-C in the bile of GBC patients (P < 0.01, Fig.1b). A significant correlation was also found between TNF-α level of the GBC bile and LVD of the GBC tissue (P < 0.01, Fig.1c,d).
Figure 1.
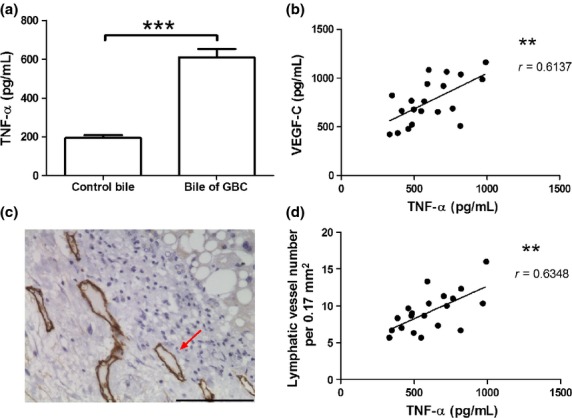
Detection of tumor necrosis factor-α (TNF-α), vascular endothelial growth factor-C (VEGF-C) and lymphatic vessel density (LVD) in clinical specimens of gallbladder carcinoma (GBC). (a). The concentration of TNF-α in the GBC bile specimens and in control bile. (c). Lymphatic vessels (marked by D2-40 antibody) of the GBC were detected by immunohistochemistry. The brown tubular structures (indicated by red arrows) were positive lymphatic vessels. (b, d). Correlation between TNF-α concentration in the GBC bile specimens and VEGF-C level or LVD. r: Spearman's correlation coefficient. (**P < 0.01; ***P < 0.001).
The correlation between clinicopathological factors and concentration of VEGF-C and TNF-α in GBC were also analyzed (Table1). The data showed that clinical stage and lymph node metastasis of GBC were related with the concentration of VEGF-C and TNF-α.
TNF-α promotes the expression of VEGF-C in vitro
NOZ cells were incubated in cell culture plates and treated respectively with different doses of TNF-α (10, 20, 50 and 100 ng/mL) for 12, 24 and 48 h, compared with control group without TNF-α. Relative mRNA of VEGF-C was detected by real-time PCR, and protein expression of VEGF-C was assayed by ELISA. As shown in Figure2, TNF-α promoted the transcription and protein expression of VEGF-C in NOZ cells in a dose and time-dependent manner, and the peak effect appeared when the dose was 50 ng/mL and the time was 24 h or 48 h.
Figure 2.
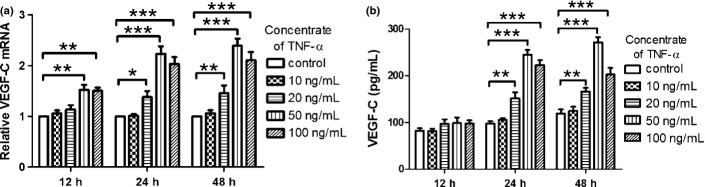
(a) Tumor necrosis factor-a (TNF-α) promoted the transcription of vascular endothelial growth factor-C (VEGF-C). (b) TNF-a promoted protein level of VEGF-C. After TNF-α (10, 20, 50 and 100 ng/mL) acting on NOZ cells for 12, 24 and 48 h, the mRNA and protein level of VEGF-C, which were respectively detected by real-time polymerase chain reaction (PCR) and enzyme linked immunosorbent assay (ELISA), were dose and time-dependently improved. (*P < 0.05; **P < 0.01; ***P < 0.001).
TNF-α promotes the tube formation of HDLECs in vitro
To observe the role of VEGF-C gene in the the lymphangiogenesis of GBC, we structured NOZ cell line, of which VEGF-C was knocked down by Lentiviral-mediated siRNA. As shown in Figure3, the mRNA and protein expression of VEGF-C in NOZ/VEGF-C siRNA gorup (NOZ cells transfected with lentiviral-mediated VEGF-C siRNA) sharply decreased (P < 0.01), compared with NOZ and NOZ/Ctrl group (NOZ cells transfected with empty vector). Before exploring whether TNF-α could enhance tube formation of HDLECs, we firstly observed the effects of TNF-α on the cell number of HDLECs by MTT assay. As shown in Figure4(a), 50 ng/mL of TNF-α had no effect on the growth of HDLECs in 6–48 h (P > 0.05), and the number of cells reduced after the treatment with TNF-α for 72 h (P < 0.05). In the experiment of tube formation, the HDLECs cultured alone had formed a few tubes 2–6 h after seeding, but the tube number did not increase with the formation of small tight clusters 10 h after seeding (Fig.4b). So we decided to observe the tube formation for 6 h in the following mixed-culture experiment. HDLECs were separately cocultured with three cell lines: NOZ, NOZ/Ctrl and NOZ/VEGF-C siRNA on GFR Matrigel. In the treatment with TNF-α, the tube number (10.89 ± 1.10 vs 6.82 ± 0.62, P < 0.05), tube length (10.26 ± 1.034 mm vs 6.14 ± 0.74 mm, P < 0.05) and tube area (0.86 ± 0.04 mm2 vs 0.65 ± 0.06 mm2, P < 0.05) of HDLECs increased obviously, and with the knockdown of VEGF-C, the tube number of treated group (4.11 ± 0.35) was still higher than that of non-treated group (3.44 ± 0.29) (P < 0.05), but the increase rate of VEGF-C siRNA group (39.27% ± 5.60%) was lower than that of control (71.14% ± 5.15%) P < 0.05). As for the tube length and tube area, there was no significant differences between the treated group and non-treated group (P > 0.05). (Fig.4c,d).
Figure 3.
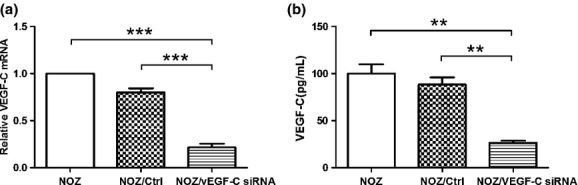
Downregulating vascular endothelial growth factor-C (VEGF-C) mRNA and protein expression by lentiviral-mediated VEGF-C siRNA in NOZ cell lines. (a) VEGF-C mRNA expression of NOZ cells was analyzed by realtime reverse transcription-polymerase chain reaction (RT-PCR), β-Actin served as a interal control; (b) VEGF-C protein expression of NOZ cells was detected by enzyme linked immunosorbent assay (ELISA). (**P < 0.01); ***P < 0.001).
Figure 4.

Tumor necrosis factor-α (TNF-α) promoted the tube formation of human dermal lymphatic endothelial cells (HDLECs). (a). MMT assay were used to observe the growth of HDLECs from 6 to 72 h. (b). When cultured alone, HDLECs formed a few tubes with 2–6 h after seeding. Compared with the 6th hour, the tube number of the 10th hour did not increase (P > 0.05) (c). The total length, area, and number of tubes formed by HDLECs in the assays of cocultured tube formation without or with the treatment with TNF-α.); (d). DiI-labeled HDLECs (emit red fluorescence) were cocultured with three NOZ cell lines and treated with TNF-α (50 ng/mL) for 6 h, the tube formation of HDLECs was observed under an fluorescence microscopy. (*P < 0.05).
TNF-α promotes the lymphangiogenesis of GBC in vivo
To study the effect of TNF-α on lymphangiogenesis in vivo, we established three orhtotopic xenograft models of GBC in nude mice, which were respectively inoculated the abovementioned three NOZ cell lines: NOZ, NOZ/Ctrl and NOZ/VEGF-C siRNA in nude mice. Two weeks after inoculation, a daily dose of TNF-α (2 μg/kg) was injected into the abdominal cavity for 2 weeks. The lymphatic vessels of tumors were observed by immunohistochemistry. Since the D2-40 antibody against lymphatic vessel marker podoplanin does not recognize the murine antigen, mouse lymphatic vessels were detected using LYVE-1.31 The intratumoral lymphatic vessels of the tumors in the nude mice models were hardly to be observed, so all of the lymphatic vessels are from peritumor. As shown in Figure5, TNF-α increased the LVD (14.33 ± 1.35 vs 9.89 ± 0.80, P < 0.05) of orhtotopic xenograft tumors. In the VEGF-C siRNA group, TNF-α also increased the LVD (7.44 ± 0.44 vs 5.89 ± 0.29, P < 0.05) of the tumors, but the increase rate (26.37% ± 4.85%) was lower than that of control (44.19% ± 3.85%) (P < 0.05). Meanwhile, the lymph node metastasis rates of orhtotopic xenograft tumors were increased by TNF-α (Table2).
Figure 5.

Tumor necrosis factor-α (TNF-α) promoted lymphangiogenesis in the orthotopic xenograft model of gallbladder carcinoma (GBC). (a) Three groups of orthotopic xenograft model of gallbladder carcinoma were separately established using three NOZ cell lines: NOZ, NOZ/Ctrl and NOZ/Ctrl VEGF-C siRNA (left image). After the treatment with a daily dose of TNF-α (2 μg/kg) for 2 weeks, the tumors were excised. As shown in the middle image, the tumor of the orthotopic xenograft model demonstrated invasive growth with liver and LN metastasis (yellow arrow), In the right image of H-E staining of LN, lymphoid follicle (white arrow) and invasive tumor cells (green arrow) could be observed. (b). Lymphatic vessels (marked by LYVE-1) of the orthotopic xenograft tumors were detected by immunohistochemistry. The brown tubular structures (indicated by red arrows) were lymphatic vessels. (c). Lymphatic vessel number of the orthotopic xenograft tumors. TNF-α increased the lymphatic vessels number of NOZ and NOZ/Ctrl group, while the knockdown of VEGF-C decreased this effect. (*P < 0.05).
Table 2.
Lymphatic vessel density (LVD)and LN metastasis of the orhtotopic xenograft tumors in nude mice
| Control | TNF-α (50 ng/mL) | |||
|---|---|---|---|---|
| LVD | LN metastasis rate | LVD | LN metastasis rate | |
| NOZ | 9.89 ± 0.8 | 3/5 | 14.33 ± 1.35* | 5/5 |
| NOZ/Ctrl | 9.33 ± 0.69 | 3/5 | 13.45 ± 0.97* | 5/5 |
| NOZ/VEGF-C siRNA | 5.89 ± 0.29 | 1/5 | 7.44 ± 0.44* | 2/5 |
P < 0.05.
Activity analysis of VEGF-C promoter
To further investigate the mechanism by which TNF-α increased the expression of VEGF-C, we analyzed the promoter of VEGF-C. A series of 5′-deletion constructs of the VEGF-C gene promoter (products of PCR amplification shown in Fig.6a) were transiently transfected into the NOZ cell line. Cells transfected with pGL3B-2000, pGL3B-1500, pGL3B-1000, pGL3B-600, pGL3B-487 and pGL3B-332 plasmids displayed higher relative luciferase activities than pGL3B-228 plasmid constructs P < 0.01, Fig.6b), which suggests that the short region containing nucleotides -332 is crucial for the full activity of the VEGF-C promoter. The plasmid pGL3B-332 was therefore selected for further studies.
Figure 6.
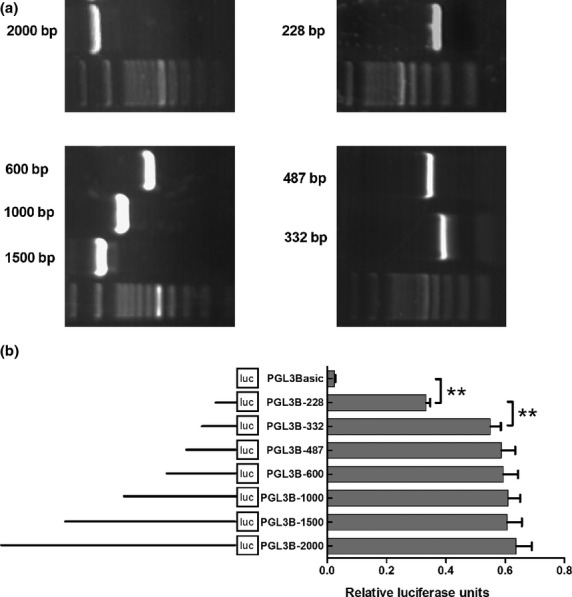
Activity analysis of vascular endothelial growth factor-C (VEGF-C) promoter. (a) A series of polymerase chain reaction (PCR) amplification products of VEGF-C promoter. (b) Activity analysis of VEGF-C promoter. NOZ cells were transfected with each of the VEGF-C promoter constructs (0.8 μg); 100 ng of the renilla luciferase expression vector pRL-TK was used for normalization, and pGL3-Basic served as the negative control. The promoter activity decreased when it was truncated to 228 nt. Each transfection was performed in duplicate and the data were expressed as mean ± SEM of three separate experiments. (**P < 0.01).
Identification of NF-κB binding sites in -332 nt to 228 nt region of VEGF-C promoter
As shown in Figure6, luciferase activity in cells transfected with pGL3B-228 plasmid was much lower than that in cells transfected with pGL3B-332, which suggests the nucleotides -332 to -228 plays an important role in the regulation of VEGF-C promoter. TFSEARCH and TESS programs were used to search for putative transcription factor binding sites in this region, which reports two putative NF-κB binding sites to be respectively situated in the region -315 to -306 nt and -271 to -262 nt. Site-directed mutagenesis of the putative NF-κB binding sites were then generated, and the promoter activities of the corresponding constructs were measured. As shown in Figure7(a), mutations in the mut1 binding site (-315 nt to -306 nt) led to a significant reduction in the promoter activity when compared with the control non-mutated construct (pGL3B-332). Meanwhile, the mut2 binding site (-271 nt to -262 nt) had no effect on the promoter activity. So mut1 binding site was further confirmed by EMSA. The results demonstrate that the nuclear extract can combine with biotin-labeled probe (Fig.7b1, lane 3). In addition, a competition assay showed that preincubation with a 100-fold molar excess of cold probe (Fig.7b1, lane 1) diminished the intensity of the bands, but not with the cold mutated probe (Fig.7b1, lane 2), Meanwhile, TNF-α enhanced the combined effect of the probe and the binding site by 89.7% ± 8.74% (P < 0.01, Fig.7b1, lane 4 and b2). To determine whether the NF-κB transcription factor was associated with VEGF-C promoter in vivo, we performed ChIP assay with specific antibody and PCR using the primers against the regulatory elements of the VEGF-C promoter. As RelA/p65 is a key active subunit in NF-κB transcription in several cell types,32,33 it was used for the antibody reactions. As shown in Figure7c1, a 112-bp DNA fragment covering the NF-κB binding site was amplified by chromatin immunoprecipitation with an anti-NF-κB antibody. The same band was obtained with the input DNA, whereas the normal IgG control and no antibody control did not result in the immunoprecipitation of DNA fragments detectable by PCR amplification. In line with the results by EMSA, TNF-α enhanced the intensity of the anti-NF-κB band. Taken together, these results demonstrate that NF-κB transcription factor can bind directly to their corresponding consensus binding site in the VEGF-C promoter region, and TNF-α can improve the combined effect by 32.6% ± 6.5% (P < 0.05, Fig.7c2).
Figure 7.

Nucklear factor (NF-κB) binding sites in vascular endothelial growth factor-C (VEGF-C) promoter. (a) Effect of mutation of the NF-κB binding sites on the activity of the VEGF-C promoter. Mutants, indicated as black rectangle, were depicted schematically on the left. A total of 0.2 mg of mutants together with 10 ng of pRL-TK was co-transfected into NOZ cells. pGL3-Basic served as the negative control. The luciferase units were obtained (right) by comparison with pGL3B-332. Each transfection was performed in duplicate and the data were expressed as mean ± SEM of three separate experiments. (**P < 0.01). (b1) Electrophoretic mobility shift assay (EMSA) of NF-κB. The 5′-biotin end-labeled probe was incubated in the absence (lane 0) or presence (lane 1-4) of nuclear extracts from transfected NOZ cells. Unlabeled cold probe (lane 1) and cold mutated probe (lane 2) were used as competitors. The 5′-biotin end-labeled probe and nuclear extracts were incubated without (lane 3) or with (lane 4) tumor necrosis factor-α (TNF-α). (b2). The densitometric value for lane 0–4. TNF-α improved the gray value of lane 4(**, P < 0.01), which indicate TNF-α enhanced the combined effect of the probe and the binding site. (c1) ChIP assay. Chromatin from NOZ cells was immunoprecipitated with the anti-NF-κB. The total extracted DNA (Input) and the immunoprecipitated samples were PCR-amplified using primers specific to a region that spanned -389 to -278 nt (containing the NF-κB binding sites) of the VEGF-C promoter. The normal rabbit IgG or pre-blocked protein A/G (no antibody control) was also performed for control purpose. Another experiment group was treated with 50 ng/mL of TNF-α (bottom row). (c2). The densitometric value for treated and non-treated groups. Relative expression was represented by value of anti-NF-κB/value of input (*P < 0.05; **P < 0.01).
Upregulation of VEGF-C promoter by TNF-α/NF-κB
To determine the effects of TNF-α/NF-κB signaling pathway on the VEGF-C promoter, we used pGL3B-332 as a luciferase reporter plasmid, which was treated with TNF-α or co-transfected with NF-κB (p65) siRNAs into NOZ cells. TNF-α was demonstrated to enhance the expression of NF-κB (P < 0.01, Fig.8a1,a2) and increase the luciferase activity of VEGF-C promoter (P < 0.01, Fig.8c), consequently leading to an increase in VEGF-C protein level (P < 0.001, Fig.8d). In contrast, when the expression of NF-κB in NOZ cells was knocked down by the addition of siRNA (P < 0.01, Fig.8b1,b2), the upregulation of TNF-α on the luciferase activities (75.4% ± 5.7% vs 47.6% ± 5.2%, P < 0.05) and the protein expression (101.7% ± 7.2% vs 24.8% ± 3.2%, P < 0.001) of VEGF-C were reduced (Fig.8c,d).
Figure 8.
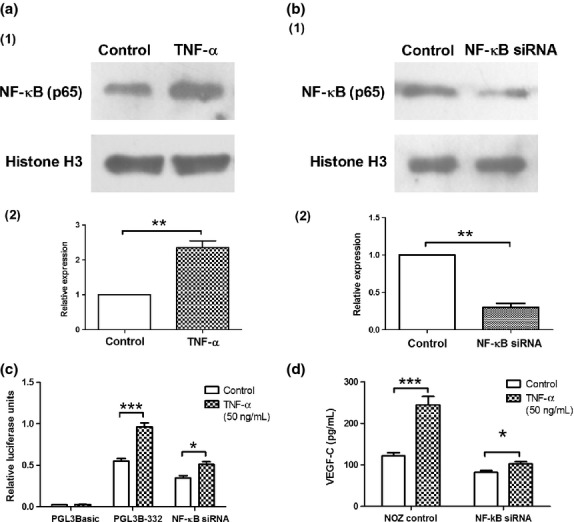
Effects of tumor necrosis factor-α (TNF-α) /nuclear factor (NF)-κB on promoter activity and protein expression of vascular endothelial growth factor-C (VEGF-C). (a1) Western blot analysis of NF-κB (p65) in NOZ cells treated with TNF-α (50 ng/mL). Histone H3 served as a loading control. (a2). The densitometric value for TNF-α treated and non-treated groups. TNF-α improved the relative expression of NF-κB in nucleus. (b1) Western blot analysis of NF-κB in NOZ cells treated with siRNAs. Histone H3 served as a loading control. (b2). The densitometric value for control and NF-κB siRNA groups. NF-κB siRNA reduced the relative expression of NF-κB in nucleus (c) TNF-α enhanced VEGF-C promoter activities in NOZ cells, which was sharply weakened with the knockdown of NF-κB by transfection with siRNA. (d) Enzyme linked immunosorbent assay (ELISA) showed that TNF-α increased VEGF-C protein expression in NOZ cells, which was decreased after the knockdown of NF-κB. Each transfection was performed in duplicate and the data were expressed as mean ± SEM of three separate experiments. (*P < 0.05; ***P < 0.001).
Discussion
In this study we firstly found that TNF-α in GBC specimens was significantly correlated with the expression of VEGF-C and lymphatic vessel density, which is consistent with CHA's finding in the study of rheumatoid synoviocytes.20 It is noteworthy that we obtained the results in gallbladder carcinoma, a kind of malignancy that mostly metastasizes through lymph channels. This result suggests that TNF-α may promote the lymphangiogenesis of gallbladder carcinoma via enhancing the expression of VEGF-C. In the subsequent in vitro and in vivo experiments, we demonstrated that TNF-α strongly enhanced the transcription and protein expression of VEGF-C, and elevated the lymphangiogenesis of gallbladder carcinoma in nude mice. Further study on the molecular mechanism showed that TNF-α-stimulated upregulation of VEGF-C is dependent on NF-κB.
Gallstones and subsequent inflammatory changes are believed to be important risk factors for gallbladder carcinoma, and the macrophage is the key player of the chronic inflammatory response. This is because it releases a great number of bioactive products, such as TNFα,IL-1 and IL-8.34 In gallbladder, TNF-α and IL-1α were found to directly affect epithelial cell absorptive function of the gallbladder wall. This alteration in absorptive function may play a role in gallstone pathogenesis,35 and gallstone leads to chronic inflammation and the producing of cytokines, which sets up a vicious circle. Nevertheless, TNF-α play a pivotal role in orchestrating the production of a proinflammatory cytokine cascade and is thus considered to be a “master regulator” of proinflammatory cytokine production.36,37 So we can speculate that TNF-α plays an important role in the evolution from gallstones and chronic inflammation to GBC development. In the present research, we detected 20 clinical bile specimens, and observed that the concentration of TNF-α in the GBC bile specimens was higher than that of controls. These results suggest that the promoter polymorphisms may not fully reflect the physiological environment of the gallbladder carcinomas, or gene and protein levels may not be consistent in the gallbladder carcinoma.
Although TNF-α has been documented to induce a variety of biological effects, its role in tumor progression remains controversial. TNF-α can trigger pro-cancer or anti-cancer signaling under different circumstances.38 Pei-Wen Tsai reported that TNF-α was not capable of upregulating VEGF-C in human breast cancer cells,39 but the present study showed that TNF-α promoted the transcription and protein level of VEGF-C within the dose range of 10–50 ng/μL in a dose-dependent manner. However, when the concentration of TNF-α was increased to 100 ng/μL, this pro-expression action dropped, the cell number of NOZ reduced, and the cell activity seemed to decline. These data suggest that the conflicting pro-cancer and anti-cancer effects may also coexist in this particular carcinoma.
Previous studies have demonstrated that the dual action of TNF-α in cancers is due to the different signaling pathways. TNF-α trimer binds to TNFR1. The latter can activate the transcription factor NF-κB and evokes the transcription of pro-inflammatory and survival genes through a complex signaling pathway.37,40,41 TNFR1 can also initiate pro-inflammatory, survival and cell death signals by activating apoptosis signaling kinase 1 (ASK1), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase. These kinases, in turn, activate various forms of the heterodimeric transcription factor AP-1. Alternatively, TNFR1 can initiate the formation of a death-inducing signaling complex (DISC) that can trigger cell death through apoptosis or through necroptosis.11,12,38 In addition, TNF-α binding to TNFR2 can regulate cell–cell interactions and signal cell migration.13,38 On the basis of these research findings, we further investigated whether TNF-α could upregulate VEGF-C through the NF-κB signaling pathway and validated this hypothesis experimentally. It should be noted that the knockdown of NF-κB did not completely block the promotion effect of TNF-α on VEGF-C, which may be due to the involvement of AP-1 or other transcription factors binding with the VEGF-C promoter.
Although some growth factors have been found to regulate the expression of VEGF-C,39,42 we demonstrated for the first time that TNF-α can upregulate VEGF-C in the gallbladder carcinoma cell line, which is dependent on NF-κB. In addition, we identified the core activity area of VEGF-C promoter and the specific binding site of NF-κB on the VEGF-C promoter. The interconnected signaling networks of biological factors are very complicated, and the regulation of VEGF-C cannot be completely explained by our experiment results, but the role of NF-κB was defined in this regulation.
Above all, our research documents that TNF-α can upregulate the expression of VEGF-C via NF-κB combining with the promoter of VEGF-C and upregulating the activity of it, which reveals, at least in part, the molecular mechanism by which TNF-α promotes lymphangiogenesis of gallbladder carcinoma. Moreover, the study suggests that TNF-α, a key cytokine in chronic inflammation, is an important accelerator in the progression and metastasis of gallbaldder carcinoma.
Acknowledgments
This study was supported by the grants from The National Natural Science Foundation of China (No. 81272373), Key Project of Science and Technology Research Program in Fujian Province (No. 2009Y0024) and Key Project of Science Research in Fujian Medical University (No. 09ZD017).
Disclosure Statement
No conflicts of interest exist for either of the authors of this manuscript.
References
- Lazcano-Ponce EC, Miquel JF, Munoz N, et al. Epidemiology and molecular pathology of gallbladder cancer. CA Cancer J Clin. 2001;51:349–64. doi: 10.3322/canjclin.51.6.349. [DOI] [PubMed] [Google Scholar]
- Cancer IAfRo. Globocan 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Lyon, France: IARC; 2013. [Google Scholar]
- Kokudo N, Makuuchi M, Natori T, et al. Strategies for surgical treatment of gallbladder carcinoma based on information available before resection. Arch Surg. 2003;138:741–50. doi: 10.1001/archsurg.138.7.741. ; discussion 50. [DOI] [PubMed] [Google Scholar]
- Ohtani T, Shirai Y, Tsukada K, Muto T, Hatakeyama K. Spread of gallbladder carcinoma: CT evaluation with pathologic correlation. Abdom Imaging. 1996;21:195–201. doi: 10.1007/s002619900045. [DOI] [PubMed] [Google Scholar]
- Tsukada K, Kurosaki I, Uchida K, et al. Lymph node spread from carcinoma of the gallbladder. Cancer. 1997;80:661–7. [PubMed] [Google Scholar]
- Kondo S, Nimura Y, Kamiya J, et al. Mode of tumor spread and surgical strategy in gallbladder carcinoma. Langenbecks Arch Surg. 2002;387:222–8. doi: 10.1007/s00423-002-0318-6. [DOI] [PubMed] [Google Scholar]
- Izumi T, Shimada H, Maehara M, et al. Modes of spread and surgical strategy for gallbladder carcinoma with subserosal invasion. Nihon Geka Gakkai Zasshi. 1993;94:722–9. [PubMed] [Google Scholar]
- Tracey KJ, Beutler B, Lowry SF, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–4. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- Cerami A, Ikeda Y, Le Trang N, Hotez PJ, Beutler B. Weight loss associated with an endotoxin-induced mediator from peritoneal macrophages: the role of cachectin (tumor necrosis factor) Immunol Lett. 1985;11:173–7. doi: 10.1016/0165-2478(85)90165-8. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Wei H, Manogue KR, et al. Cachectin/tumor necrosis factor induces cachexia, anemia, and inflammation. J Exp Med. 1988;167:1211–27. doi: 10.1084/jem.167.3.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: role in endothelial cell migration and angiogenesis. Mol Cell Biol. 2002;22:7512–23. doi: 10.1128/MCB.22.21.7512-7523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Yamaura T, Murakami K, et al. Involvement of TNF-alpha in enhancement of invasion and metastasis of colon 26-L5 carcinoma cells in mice by social isolation stress. Oncol Res. 1999;11:461–9. [PubMed] [Google Scholar]
- Mochizuki Y, Nakanishi H, Kodera Y, et al. TNF-alpha promotes progression of peritoneal metastasis as demonstrated using a green fluorescence protein (GFP)-tagged human gastric cancer cell line. Clin Exp Metastasis. 2004;21:39–47. doi: 10.1023/b:clin.0000017181.01474.35. [DOI] [PubMed] [Google Scholar]
- Tada Y, O-Wang J, Takenaga K, et al. Expression of the TNF-alpha gene on mouse lung carcinoma cells suppresses spontaneous lung metastasis without affecting tumorigenicity. Oncol Rep. 2002;9:585–8. [PubMed] [Google Scholar]
- Hamaguchi T, Wakabayashi H, Matsumine A, Sudo A, Uchida A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem Biophys Res Commun. 2011;407:525–30. doi: 10.1016/j.bbrc.2011.03.051. [DOI] [PubMed] [Google Scholar]
- Shi JS, Zhou LS, Han Y, Zhu AJ, Sun XJ, Yang YJ. Expression of tumor necrosis factor and its receptor in gallstone and gallbladder carcinoma tissue. Hepatobiliary Pancreat Dis Int. 2004;3:448–52. [PubMed] [Google Scholar]
- Ristimaki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- Cha HS, Bae EK, Koh JH, et al. Tumor necrosis factor-alpha induces vascular endothelial growth factor-C expression in rheumatoid synoviocytes. J Rheumatol. 2007;34:16–9. [PubMed] [Google Scholar]
- Joukov V, Pajusola K, Kaipainen A, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–8. [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Chen YL, She FF, Tang NH, Li XJ, Wang XX. Expressions of VEGF-C and VEGF-D and their correlation with lymphangiogenesis and angiogenesis in gallbladder carcinoma. Zhonghua Zhong Liu Za Zhi. 2010;32:190–5. [PubMed] [Google Scholar]
- Lin W, Jiang L, Chen Y, et al. Vascular endothelial growth factor-D promotes growth, lymphangiogenesis and lymphatic metastasis in gallbladder cancer. Cancer Lett. 2012;314:127–36. doi: 10.1016/j.canlet.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- Vermeulen PB, Gasparini G, Fox SB, et al. Quantification of angiogenesis in solid human tumours: an international consensus on the methodology and criteria of evaluation. Eur J Cancer. 1996;32A:2474–84. doi: 10.1016/s0959-8049(96)00379-6. [DOI] [PubMed] [Google Scholar]
- Egberts JH, Schniewind B, Schafmayer C, et al. Establishment of a novel orthotopic xenograft model of human gallbladder carcinoma. Clin Exp Metastasis. 2007;24:141–8. doi: 10.1007/s10585-007-9058-x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Jiang L, She F, et al. Vascular endothelial growth factor-C promotes the growth and invasion of gallbladder cancer via an autocrine mechanism. Mol Cell Biochem. 2010;345:77–89. doi: 10.1007/s11010-010-0562-y. [DOI] [PubMed] [Google Scholar]
- Lim CB, Fu PY, Ky N, et al. NF-kappaB p65 repression by the sesquiterpene lactone, Helenalin, contributes to the induction of autophagy cell death. BMC Complement Altern Med. 2012;12:93. doi: 10.1186/1472-6882-12-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YL, Peng XE, Wang D, Chen WN, Lin X. Human liver fatty acid binding protein (hFABP1) gene is regulated by liver-enriched transcription factors HNF3beta and C/EBPalpha. Biochimie. 2012;94:384–92. doi: 10.1016/j.biochi.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2:924–32. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Opeskin K, Goad J, Williams ED. Tumor-induced activation of lymphatic endothelial cells via vascular endothelial growth factor receptor-2 is critical for prostate cancer lymphatic metastasis. Cancer Res. 2006;66:9566–75. doi: 10.1158/0008-5472.CAN-06-1488. [DOI] [PubMed] [Google Scholar]
- De Martin R, Hoeth M, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000;20:E83–8. doi: 10.1161/01.atv.20.11.e83. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Macarthur M, Hold GL, El-Omar EM. Inflammation and Cancer II. Role of chronic inflammation and cytokine gene polymorphisms in the pathogenesis of gastrointestinal malignancy. Am J Physiol Gastrointest Liver Physiol. 2004;286:G515–20. doi: 10.1152/ajpgi.00475.2003. [DOI] [PubMed] [Google Scholar]
- Rege RV. Inflammatory cytokines alter human gallbladder epithelial cell absorption/secretion. J Gastrointest Surg. 2000;4:185–92. doi: 10.1016/s1091-255x(00)80055-4. [DOI] [PubMed] [Google Scholar]
- Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18:335–43. doi: 10.1016/j.cytogfr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JP, Pober JS, Bradley JR. Tumour necrosis factor and cancer. J Pathol. 2013;230:241–8. doi: 10.1002/path.4188. [DOI] [PubMed] [Google Scholar]
- Tsai PW, Shiah SG, Lin MT, Wu CW, Kuo ML. Upregulation of vascular endothelial growth factor C in breast cancer cells by heregulin-beta 1. A critical role of p38/nuclear factor-kappa B signaling pathway. J Biol Chem. 2003;278:5750–9. doi: 10.1074/jbc.M204863200. [DOI] [PubMed] [Google Scholar]
- Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Wang CA, Jedlicka P, Patrick AN, et al. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122:1895–906. doi: 10.1172/JCI59858. [DOI] [PMC free article] [PubMed] [Google Scholar]