

Abstract
Somatic mutations in splicing factor genes have frequently been reported in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Although aberrant epigenetic changes are frequently implicated in blood cancers, their direct role in suppressing one or both alleles of critical splicing factors has not been previously examined. Here, we examined promoter DNA hypermethylation of nine splicing factors, SF3B1, SRSF2, U2AF1, ZRSR2, SF3A1, HNRNPR, MATR3, ZFR, and YBX3 in 10 leukemic cell lines and 94 MDS or AML patient samples from the Australasian Leukemia and Lymphoma Group Tissue Bank. The only evidence of epigenetic effects was hypermethylation of the YBX3 promoter in U937 cells in conjunction with an enrichment of histone marks associated with gene silencing. In silico analysis of DNA methylation data for 173 AML samples generated by the Cancer Genome Atlas Research Network revealed promoter hypermethylation of the gene encoding Y box binding protein 3, YBX3, in 11/173 (6.4%) AML cases, which was significantly associated with reduced mRNA expression (P < 0.0001). Hypermethylation of the ZRSR2 promoter was also detected in 7/173 (4%) cases but was not associated with decreased mRNA expression (P = 0.1204). Hypermethylation was absent at the promoter of seven other splicing factor genes in all cell lines and patient samples examined. We conclude that DNA hypermethylation does not frequently silence splicing factors in MDS and AML. However, in the case of YBX3, promoter hypermethylation-induced downregulation may contribute to the pathogenesis or maintenance of AML.
Keywords: Acute myeloid leukemia, DNA hypermethylation, epigenetics, myelodysplastic syndromes, splicing factors
Aberrant hypermethylation at the promoter region of genes, leading to their silencing, is a hallmark of human cancers including hematological malignancies. This epigenetic abnormality often affects tumor suppressor genes that can also be inactivated due to sequence mutations. In myeloid neoplasia, hypermethylation of tumor suppressor genes including CDH1, CDKN2B, and HIC1 have been found in myelodysplastic syndromes (MDS) and are associated with poorer clinical outcomes.(1) Hypermethylation of CDH1, CDKN2B, and CEBPA have been described in acute myeloid leukemia (AML) by independent large cohort studies.(2,3) Promoter hypermethylation can be mono or biallelic; for example, the former acting as the “second hit” in silencing a gene when a mutation exists in one allele as the “first hit”.(4)
Recent studies using massively parallel sequencing have identified mutations in genes encoding several components of the mRNA splicing machinery in hematological neoplasia.(5–7) Mutations of splicing factor encoding genes, including SF3B1, SRSF2, U2AF1, and ZRSR2, have been found in over 60% of individuals with MDS.(5,7–12) Splicing factor mutations are strongly associated with specific phenotypic features such as SF3B1 in refractory anemia with ring sideroblasts and SRSF2 with more advanced forms of MDS including secondary AML and refractory anemia with excess blasts.(7) Mutations of SF3B1, U2AF1, HCFC1, SAP130, SRSF6, SON, U2AF26, LUC7L2, PRPF8, and SFPQ have been identified in individuals with AML.(5,7,13,14) However, mutations of splicing factors detected to date are mostly heterozygous missense mutations leading to aberrant RNA splicing. It remains unknown if alterations in splicing factor genes can also be caused by epigenetic defects such as DNA hypermethylation, potentially affecting a second allele when mutation has already occurred in the first. Biallelic promoter hypermethylation may also affect splicing factor genes, as seen in the silencing of other cancer-causing genes.(2,3) Hence, there is a compelling reason to pursue the possibility of epigenetic silencing as a collaborating mechanism in the pathogenesis or maintenance of hematological neoplasms.
Here we have determined the promoter methylation signatures of splicing factor genes in leukemic cell lines and diagnostic bone marrow samples from individuals with MDS and AML. Histone modifications were also investigated in cell lines when promoter hypermethylation was present. We also analyzed publicly available DNA methylation data of 173 AML samples from the Cancer Genome Atlas Research Network (TCGA) for promoter hypermethylation affecting splicing factor genes.
Materials and Methods
Cell lines and patient samples
HL-60, KG-1, KG-1A, Kasumi-1, THP-1, CEM, and Jurkat cells were cultured in RPMI-1640 media containing 10% FCS. NB4, U937, and K562 were cultured in DMEM supplemented with 10% FCS. 5U/mL penicillin, 50 μg/mL streptomycin sulphate, and 2 mM L-glutamine were added to all growth media. DNA samples from MDS, AML, and acute promyelocytic leukemia patients (n = 94) were acquired from the Australasian Leukemia and Lymphoma Group (ALLG) Tissue Bank with ethics approval from the Human Research Ethics Committee of the Royal Prince Alfred Hospital (HREC/08/RPAH/222; Camperdown, NSW, Australia).
DNA methylation analysis
Genomic DNA extracted from cell lines and patient samples were bisulfite-converted using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA), according to the manufacturer's instructions. Combined bisulfite restriction analysis (COBRA) and clonal bisulfite sequencing were carried out as previously described.(15,16) For each candidate gene, bisulfite-converted DNA was amplified using primers that coamplify methylated and unmethylated template at an equal efficiency. Primer sequences are provided in Table S1. Following PCR, amplicons were either digested with a restriction enzyme that discriminates between methylated and unmethylated CpG, or cloned into pGEM-T Easy vectors (Promega, Madison, WI, USA) and sequenced using standard Sanger Sequencing. Reversal of DNA hypermethylation was carried out using 500 nM 5-Aza-2′deoxycitidine for 72 h with drug replenished every 24 h.
Chromatin immunoprecipitation
HL-60 or U937 cells (5 × 106) were fixed in 0.1% formaldehyde. Chromatin immunoprecipitation was carried out using the Millipore Magna ChIP A/G Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Anti-H3K4me3, anti-H3K27me3 and IgG control antibodies (Millipore) were used. Following ChIP, quantitative RT-PCRs were carried out using primers designed against the promoter region of YBX3 (Table S1).
Analyses of DNA methylation and mRNA expression data from the TCGA AML cohort
DNA methylation levels determined using Illumina's Infinium 450 K array were downloaded as β-values. We chose a cut-off of 10% to distinguish between methylated and unmethylated loci, as previously published.(17) All promoter methylation probes located 1 kb upstream of the transcriptional start site were considered for each gene. For genes with multiple promoter methylation probes, analyses were carried out for probes least correlated with mRNA expression to minimize false positive results. mRNA expression was downloaded as RNA-Seq by Expectation Maximization values(18) from the cBioPortal freeware (http://www.cbioportal.org/public-portal.(19,20) Only samples with matched DNA methylation and mRNA expression data were included in this analysis (n = 173).
Analyses of clinicopathological and molecular features
Clinical data for the AML cohort were downloaded from the TCGA repository website (http://cancergenome.nih.gov/). Clinicopathological and molecular features including age, sex, white blood cell count at diagnosis, blast percentage, subtypes, cytogenetic risks, cytoplasm-dislocalized leukemic nucleophosmin protein status, and mutations of FLT3, IDH1, RAS and DNMT3A were compared with the methylation status of YBX3 and ZRSR2 in AML samples (n = 173).
Statistical analyses
The significance of fold change in mRNA expression and histone mark enrichment was analyzed using Student's t-test. Categorical variables were compared using the Chi-square test or Fisher's exact test. The Mann–Whitney U-test was used to compare continuous variables that were not normally distributed. Analyses were carried out using GraphPad Prism version 5 (La Jolla, CA, USA). P-values < 0.05 were considered significant.
Results
Epigenetic modifications of splicing factor genes in leukemic cell lines
In order to explore the role of promoter DNA hypermethylation in the regulation of splicing factor expression in leukemia, we used COBRA and Clonal Bisulfite Sequencing to determine the promoter methylation signatures of splicing factor genes in eight myeloid: HL-60, NB4, KG-1, KG-1A, Kasumi-1, U937, THP-1, K562, and two lymphoid leukemia cell lines, CEM and Jurkat. We examined a total of nine splicing factors, five of which were previously reported to be mutated in MDS and AML, namely SF3B1, SRSF2, U2AF1, ZRSR2, and SF3A1.(5,7–14) The other four, HNRNPR, MATR3, ZFR, and YBX3, have been reported to be associated with the spliceosome.(21,22) They have also been found to be aberrantly hypermethylated in AML samples compared to normal reference.(23) Nonetheless, it was not previously reported whether promoter hypermethylation induced their silencing, and how these factors may contribute to the development and/or maintenance of AML.
Our COBRA analysis indicated that methylation was absent at the promoter regions of SF3B1, SRSF2, U2AF1, ZRSR2, SF3A1, HNRNPR, MATR3, and ZFR in cell lines (Fig. 1). The YBX3 promoter was hypermethylated only in the monocytic leukemia cell line U937 (Fig. 2a–c). Hypermethylation of YBX3 in U937 cells was associated with its reduced mRNA expression. Inhibition of DNA methylation using the DNA methyltransferase inhibitor 5-Aza-2′deoxycytidine increased the expression of YBX3 following reversal of CpG methylation (Fig. 2d,e), indicating that promoter DNA hypermethylation was associated with the regulation of its expression. DNA methylation also occurred in conjunction with altered histone modifications at the promoter of this gene. We found >100-fold enrichment of the histone mark associated with active transcription, H3K4me3, at the YBX3 promoter in YBX3-expressing HL-60 cells (Fig. 2f). Enrichment of H3K4me3 was not present at the YBX3 promoter in U937 cells, which lacks YBX3 expression (Fig. 2f). The silencing mark, H3K27me3 was enriched twofold at the promoter of YBX3 in U937 but not HL-60 cells (Fig. 2g). Thus, appropriate histone modifications occur concomitantly with promoter DNA methylation to induce silencing of YBX3 in U937 cells.
Fig 1.

Absence of promoter DNA hypermethylation of splicing factor genes in leukemic cell lines. Gene promoter maps show CpG sites (vertical black lines) and their distance from the transcriptional start site (+1) for each splicing factor gene. Recognition sites for respective restriction enzymes used in combined bisulfite restriction analysis (COBRA) assays are shown on the maps. For each splicing factor, the gel electrophoretogram of COBRA is shown below its respective gene promoter map. +ve Con, positive control, completely methylated human genomic DNA; N1, N2, N3, negative controls, peripheral blood DNA from three healthy individuals.
Fig 2.
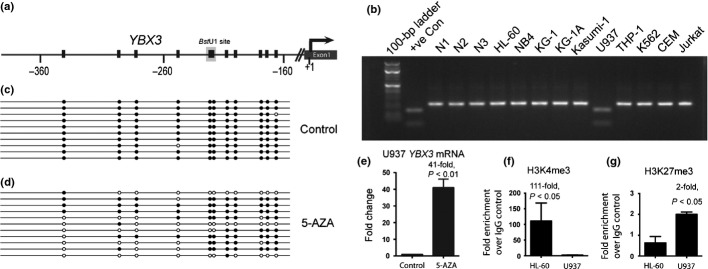
Silencing of YBX3 in U937 cells is associated with promoter DNA hypermethylation and altered histone modifications. (a) YBX3 promoter showing individual CpGs (vertical black line) and their distance from the transcriptional start site (+1). Recognition site for BstU1 used in combined bisulfite restriction analysis (COBRA) assays for YBX3 is shown on the map. (b) Gel electrophoretogram of COBRA showing PCR amplicons of the YBX3 promoter following bisulfite conversion and digestion with BstU1 of genomic DNA from leukemic cell lines and controls. (c) Clonal bisulfite sequencing confirming hypermethylation at the YBX3 promoter in U937 cells. Each row represents a single cloned PCR amplicon aligned to the map shown in (a). Black and white circles denote methylated and unmethylated CpGs, respectively. (d) Reversal of DNA hypermethylation in 5-Aza-2′deoxycytidine (5-AZA)-treated U937 cells as detected by clonal bisulfite sequencing. (e) Quantitative RT-PCR showing increased expression of YBX3 following treatment of U937 cells with 5-AZA. (f) Fold enrichment of YBX3 promoter sequence bound to H3K4me3 (activation mark) and (g) H3K27me3 (silencing mark) in HL-60 and U937 cells normalized to IgG control. +ve Con, positive control, completely methylated human genomic DNA; N1, N2, N3, negative controls, peripheral blood DNA from three healthy individuals.
Promoter hypermethylation of splicing factor genes in primary MDS and AML samples
We next determined the frequency of promoter DNA hypermethylation affecting splicing factor genes in the diagnostic bone marrow of 32 individuals with MDS, 50 with AML, and 12 with acute promyelocytic leukemia (n = 94), from the ALLG Tissue Bank. The clinical characteristics of patients are summarized in Table S2. For MDS samples, the male to female ratio and mean age were comparable to published large cohort studies.(24,25) The AML samples were representative of a broad range of major and minor cytogenetic AML subgroups.
Consistent with our findings in cell lines, we did not observe hypermethylation at the promoter of any of the eight splicing factor genes, SF3B1, SRSF2, U2AF1, ZRSR2, SF3A1, HNRNPR, MATR3, and ZFR in MDS and AML diagnostic samples (Table S3). The YBX3 promoter was also not hypermethylated in these samples (Table S3). One caveat in our study is the absence of certain subtypes of MDS and AML, such as refractory anemia with ring sideroblasts associated with the highest levels of SF3B1 mutations. Promoter hypermethylation of other splicing factors may be present in other MDS and AML subtypes. However, promoter hypermethylation of splicing factors would have been readily detected in our samples if they occur at a similar frequency as known sequence mutations (over 60% for MDS and 14% in AML).(5,7,8,14) We further analyzed publicly available data from TCGA to determine the presence of promoter methylation of these nine splicing factors in 173 AML samples.(14) With the exception of YBX3 and ZRSR2, promoter methylation was not detected (Fig. 3a). The YBX3 promoter was methylated in 11 samples (6.4%) (Fig. 3a). Importantly, there was a significant association between the presence of promoter methylation and mRNA expression of this gene (P < 0.0001) in AML (n = 173) (Fig. 3b). The ZRSR2 promoter was methylated in seven samples (4%). However, the presence of methylation was not associated with ZRSR2 mRNA expression (P = 0.1204) (Fig. 3c).
Fig 3.
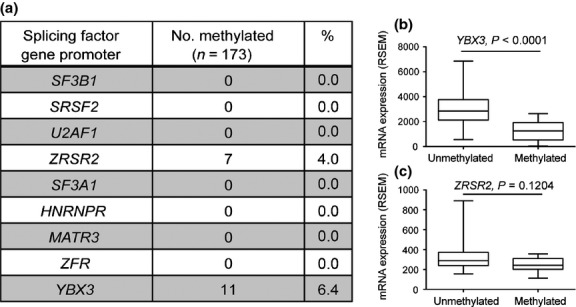
Hypermethylation-induced downregulation of YBX3 in the Cancer Genome Atlas Research Network acute myeloid leukemia cohort. (a) ZRSR2 and YBX3 promoters were hypermethylated in 7/173 (4.0%) and 11/173 (6.4%) acute myeloid leukemia cases, respectively. Promoter hypermethylation was not detected in other splicing factors. (b) YBX3 promoter hypermethylation was significantly associated with the loss of YBX3 mRNA expression (P < 0.0001). (c) Promoter hypermethylation of ZRSR2 was not associated with expression levels of ZRSR2 mRNA (P = 0.1204). RSEM, RNA-Seq by Expectation Maximization.
We determined the clinicopathological features associated with YBX3 or ZRSR2 promoter methylation. The presence of methylation at the YBX3 and ZRSR2 promoters was significantly associated with age (P = 0.0399) and sex (P = 0.0079), respectively, but no other features analyzed (Table 1).
Table 1.
Clinicopathological and molecular associations of YBX3 and ZRSR2 promoter hypermethylation in the Cancer Genome Atlas Research Network acute myeloid leukemia (AML) cohort
| YBX3 hypermethylation | ZRSR2 hypermethylation | |||||
|---|---|---|---|---|---|---|
| Present (n = 11) | Absent (n = 162) | P-value | Present (n = 7) | Absent (n = 166) | P-value | |
| Age, years | ||||||
| Median(range) | 39 (18–68) | 59 (18–88) | 0.0399 | 53 (23–66) | 59 (18–88) | 0.1316 |
| Sex, n (%) | ||||||
| Male | 7 (64) | 86 (53) | 0.4971 | 7 (100) | 86 (52) | 0.0079 |
| Female | 4 (36) | 76 (47) | 0 (0) | 80 (48) | ||
| WBC, k/μL | ||||||
| Median (range) | 27 (3–203) | 16 (1–297) | 0.5248 | 22 (1–88) | 18 (1–297) | 0.8154 |
| Blast, % | ||||||
| Median (range) | 62 (34–86) | 72 (0–100) | 0.2908 | 72 (35–86) | 71 (0–100) | 0.9025 |
| Subtypes, n (%) | ||||||
| M0 | 1 (10) | 15 (9) | 0.6555 | 0 (0) | 16 (10) | 0.8374 |
| M1 | 2 (18) | 40 (25) | 1 (14) | 41 (25) | ||
| M2 | 5 (45) | 34 (21) | 3 (43) | 36 (22) | ||
| M3 | 0 (0) | 16 (10) | 1 (14) | 15 (9) | ||
| M4 | 3 (27) | 32 (20) | 2 (29) | 33 (20) | ||
| M5 | 0 (0) | 18 (11) | 0 (0) | 18 (11) | ||
| M6 | 0 (0) | 2 (1) | 0 (0) | 2 (1) | ||
| M7 | 0 (0) | 3 (2) | 0 (0) | 3 (2) | ||
| Not classified | 0 (0) | 2 (1) | 0 (0) | 2 (1) | ||
| Cytogenetic risks, n (%) | ||||||
| Low | 0 (0) | 32 (20) | 0.2065 | 3 (43) | 29 (18) | 0.2468 |
| Intermediate | 9 (82) | 94 (58) | 3 (43) | 100 (60) | ||
| High | 2 (18) | 34 (21) | 1 (14) | 35 (21) | ||
| Not available | 0 (0) | 2 (1) | 0 (0) | 2 (1) | ||
| FLT3 mutation, n (%) | ||||||
| Mutant | 1 (9) | 49 (30) | 0.1763 | 2 (29) | 48 (29) | 0.3163 |
| Wild-type | 9 (82) | 107 (66) | 5 (71) | 111 (67) | ||
| Not determined | 1 (9) | 6 (4) | 0 (9) | 7 (4) | ||
| RAS mutation, n (%) | ||||||
| Mutant | 0 (0) | 9 (6) | 1.0000 | 0 (0) | 9 (5) | 1.0000 |
| Wild-type | 11 (100) | 150 (92) | 7 (100) | 154 (93) | ||
| Not determined | 0 (0) | 3 (2) | 0 (0) | 3 (2) | ||
| DNMT3A mutation, n (%) | ||||||
| Mutant | 2 (18) | 40 (25) | 1.0000 | 0 (0) | 42 (25) | 0.2027 |
| Wild-type | 9 (82) | 122 (75) | 7 (100) | 124 (75) | ||
| NPMc status, n (%) | ||||||
| Positive | 0 (0) | 42 (26) | 0.0708 | 1 (14) | 41 (25) | 1.0000 |
| Negative | 11 (100) | 117 (72) | 6 (86) | 122 (73) | ||
| Not determined | 0 (0) | 3 (2) | 0 (0) | 3 (2) | ||
Significant P-values are in bold. M0, AML with minimal differentiation; M1, AML without differentiation; M2, AML with maturation; M3, acute promyelocytic leukemia; M4, acute myelomonocytic leukemia; M5, acute monoblastic and monocytic leukemia; M6/M7, acute erythroid leukemia/acute megakaryoblastic leukemia; NPMc, cytoplasm-dislocalized leukemic nucleophosmin protein.
Discussion
Many studies have reported frequent mutations of splicing factors in MDS and AML. Importantly, mutations of specific splicing factors were strongly associated with particular phenotypic features, such as SF3B1 in MDS with ring sideroblasts(5) and SRSF2 with more advanced forms of MDS including secondary AML and refractory anemia with excess blasts.(7) Mutations of splicing factors may have prognostic significance, although the impact remains controversial. Several independent studies indicated improved overall survival for MDS patients with SF3B1 mutations,(8,26,27) but others did not.(28,29) One study reported that mutations of SRSF2 but not SF3B1, U2AF1, or ZRSR2 were associated with poorer overall survival in MDS.(30) Regardless, there is a strong association between altered expression or mutations of splicing genes and splicing changes including exon skipping and intron retention.(31,32) Mutations of individual splicing factors also show particular patterns of splicing in specific gene subsets. For example, U2AF1 mutations in myeloid neoplasms resulted in specific changes in splicing patterns affecting mainly cell cycle and RNA processing genes.(31) These gene ontology subsets were distinct from those associated with mutations of SF3B1 and SRSF2.(31) These results indicate the potential role of individual splicing factor mutations in the pathogenesis and/or maintenance of MDS and AML through selective impairment of tumor-associated genes within different molecular pathways.
It is important to recognise that most mutations affecting splicing factors such as SF3B1, U2AF1, and SRSF2 are more likely to cause gain of functions,(9,33) indicating that they are unlikely to function as tumor suppressors. Therefore, it is not surprising that haploinsufficiency of Sf3b1 alone was insufficient to cause MDS in mice.(34,35) Rather, mutations of these splicing factors may promote further splicing-related aberrancies affecting a series of genes, potentially in a multistep process. The exact sequence of such defects in the pathogenesis of myeloid neoplasm remains elusive and is an important subject of future studies.
In contrast to SF3B1, U2AF1, and SRSF2, mutations of ZRSR2 have been associated with loss of functions as the majority of these lead to nonsense or frameshift changes.(9,33) Whether loss of ZRSR2 expression is sufficient to cause MDS or AML awaits the results of in vivo experiments.
Irrespective of the consequences of splicing factor mutations in MDS and AML, it is unknown if splicing factors can be rendered defective by other mechanisms. Understanding whether expression of splicing factors can be affected by epigenetic changes is important to obtain a clearer insight into their role in MDS and AML. If promoter hypermethylation of splicing factors had been consistently identified, it would have suggested their role as tumor suppressor genes.
Our data indicate that promoter methylation leads to the silencing of the YBX3 gene in the myelomonocytic cell line U937 and ∼6% of AML samples in the TCGA cohort. It was surprising that YBX3 methylation was significantly associated with age but no other clinicopathological or molecular features. Independent analysis in a larger cohort would be necessary to confirm this observation.
YBX3 was found to be associated with spliceosomes in large-scale spliceosome capture and mass spectrometry analyses and is therefore considered a splicing factor.(21,22) Nevertheless, its exact role in splicing is as yet undetermined. A previous study reported upregulation of YBX3 mRNA levels during myeloid differentiation.(36) Therefore, downregulation of YBX3 may lead to perturbation of myeloid differentiation, which is a characteristic of AML. The exact role of promoter hypermethylation-induced downregulation of YBX3 in AML pathogenesis is currently unclear and is worthy of future studies.
Although promoter methylation of ZRSR2 was found in 4% of AML samples in the TCGA cohort, it is unlikely to exert an effect on ZRSR2 expression. Promoter methylation levels for ZRSR2 were also much lower (average β-value = 13.1%) than YBX3 (average β-value = 33.1%), indicating that subtle promoter methylation levels affecting ZRSR2 may not play a substantial role in its regulation.
Overall, our study showed that promoter hypermethylation-induced silencing of splicing factors occurs uncommonly in the subtypes of MDS and AML examined. Epigenetic modification is unlikely either to act as a second hit in the silencing of many splicing factors or to cause biallelic silencing of splicing factors in these types of myeloid neoplasia.
Acknowledgments
We thank Paula Marlton and Megan Ellis from the ALLG Tissue Bank for the provision of patients’ data and samples, and Jane Gordon for karyotype risk analysis. We thank the TCGA Research Network for the availability of data from AML patients. This work was supported in part by a Young Centenary Foundation Grant (JJ-LW), National Health and Medical Research Council Project Grants (#1061906 and #1030830, to JEJR), Cure the Future (JEJR), Tour de Cure (JEJR), NSW Cancer Council Grant (#RG1409 to JEJR) and an anonymous foundation (JEJR). JJ-LW is a Cancer Institute of NSW Fellow.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Table S1. Primer sequences used in this study.
Table S2. Characteristics of individuals with myelodysplastic syndromes or acute myeloid leukemia from the Australasian Leukemia and Lymphoma Group Tissue Bank.
Table S3. Absence of hypermethylation at the promoter of splicing factor genes in myelodysplastic syndrome and acute myeloid leukemia samples from the Australasian Leukemia and Lymphoma Group Tissue Bank.
References
- 1.Grövdal M, Khan R, Aggerholm A, et al. Negative efect of DNA hypermethylation on the outcome of intensive chemotherapy in older patients with high-risk myelodysplastic syndromes and acute myeloid leukemia following myelodysplastic syndrome. Clin Cancer Res. 2007;13:7107–12. doi: 10.1158/1078-0432.CCR-07-1193. [DOI] [PubMed] [Google Scholar]
- 2.Schoofs T, Berdel WE, Müller-Tidow C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia. 2013;28:1–14. doi: 10.1038/leu.2013.242. [DOI] [PubMed] [Google Scholar]
- 3.Lin T-C, Hou H-A, Chou W-C, et al. CEBPA methylation as a prognostic biomarker in patients with de novo acute myeloid leukemia. Leukemia. 2011;25:32–40. doi: 10.1038/leu.2010.222. [DOI] [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 5.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–10. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malcovati L, Papaemmanuil E, Bowen DT, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118:6239–46. doi: 10.1182/blood-2011-09-377275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 10.Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–7. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120:e83–92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- 14.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hitchins M, Suter C, Wong J, et al. Germline epimutations of APC are not associated with inherited colorectal polyposis. Gut. 2006;55:586–7. doi: 10.1136/gut.2005.087486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong JJ-L, Hawkins NJ, Ward RL, Hitchins MP. Methylation of the 3p22 region encompassing MLH1 is representative of the CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2011;24:396–411. doi: 10.1038/modpathol.2010.212. [DOI] [PubMed] [Google Scholar]
- 17.Etcheverry A, Aubry M, de Tayrac M, et al. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genomics. 2010;11:701. doi: 10.1186/1471-2164-11-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Dewey C. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–45. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–5. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
- 23.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strom SS, Gu Y, Gruschkus SK, Pierce SA, Estey EH. Risk factors of myelodysplastic syndromes: a case–control study. Leukemia. 2005;19:1912–8. doi: 10.1038/sj.leu.2403945. [DOI] [PubMed] [Google Scholar]
- 25.Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820–9. doi: 10.1200/JCO.2011.35.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patnaik MM, Lasho TL, Hodnefield JM, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119:569–72. doi: 10.1182/blood-2011-09-377994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mian SA, Smith AE, Kulasekararaj AG, et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica. 2013;98:1058–66. doi: 10.3324/haematol.2012.075325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376–82. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Damm F, Kosmider O, Gelsi-Boyer V, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–8. doi: 10.1182/blood-2011-12-400994. [DOI] [PubMed] [Google Scholar]
- 30.Thol F, Kade S, Schlarmann C, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–84. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- 31.Przychodzen B, Andres J, Kathryn G, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong JJ-L, Ritchie W, Ebner OA, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154:583–95. doi: 10.1016/j.cell.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa S. Splicing factor mutations in myelodysplasia. Int J Hematol. 2012;96:438–42. doi: 10.1007/s12185-012-1182-y. [DOI] [PubMed] [Google Scholar]
- 34.Matsunawa M, Yamamoto R, Sanada M, et al. Haploinsufficiency of Sf3b1 leads to compromised stem cell function but not to myelodysplasia. Leukemia. 2014;28:1844–1850. doi: 10.1038/leu.2014.73. [DOI] [PubMed] [Google Scholar]
- 35.Wang C, Sashida G, Saraya A, et al. Depletion of Sf3b1 impairs proliferative capacity of hematopoietic stem cells but is not sufficient to induce myelodysplasia. Blood. 2014;123:3336–43. doi: 10.1182/blood-2013-12-544544. [DOI] [PubMed] [Google Scholar]
- 36.Lian Z, Wang L, Yamaga S, et al. Genomic and proteomic analysis of the myeloid differentiation program. Blood. 2001;98:513–24. doi: 10.1182/blood.v98.3.513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences used in this study.
Table S2. Characteristics of individuals with myelodysplastic syndromes or acute myeloid leukemia from the Australasian Leukemia and Lymphoma Group Tissue Bank.
Table S3. Absence of hypermethylation at the promoter of splicing factor genes in myelodysplastic syndrome and acute myeloid leukemia samples from the Australasian Leukemia and Lymphoma Group Tissue Bank.