

Abstract
MicroRNA (miRNA) can function as tumor suppressors or oncogenes, and also as potential specific cancer biomarkers; however, there are few published studies on miRNA in synovial sarcomas, and their function remains unclear. We transfected the OncomiR miRNA Precursor Virus Library into synovial sarcoma Fuji cells followed by a colony formation assay to identify miRNAs to confer an aggressive tumorigenicity, and identified miR-17-5p from the large colonies. MiR-17 was found to be induced by a chimeric oncoprotein SS18-SSX specific for synovial sarcoma, and all examined cases of human synovial sarcoma expressed miR-17, even at high levels in several cases. Overexpression of miR-17 in synovial sarcoma cells, Fuji and HS-SYII, increased colony forming ability in addition to cell growth, but not cell motility and invasion. Tumor volume formed in mice in vivo was significantly increased by miR-17 overexpression with a marked increase of MIB-1 index. According to PicTar and Miranda algorithms, which predicted CDKN1A (p21) as a putative target of miR-17, a luciferase assay was performed and revealed that miR-17 directly targets the 3′-UTR of p21 mRNA. Indeed, p21 protein level was remarkably decreased by miR-17 overexpression in a p53-independent manner. It is noteworthy that miR-17 succeeded in suppressing doxorubicin-evoked higher expression of p21 and conferred the drug resistance. Meanwhile, introduction of anti-miR-17 in Fuji and HS-SYII cells significantly decreased cell growth, consistent with rescued expression of p21. Taken together, miR-17 promotes the tumor growth of synovial sarcomas by post-transcriptional suppression of p21, which may be amenable to innovative therapeutic targeting in synovial sarcoma.
Keywords: Cyclin-dependent kinase inhibitor p21, drug resistance, hsa-mir-17 microRNA, SS18-SSX fusion protein, synovial sarcoma
Synovial sarcoma is a high-grade malignancy and accounts for approximately 5–10% of soft tissue sarcomas. Synovial sarcomas mainly develop in the para-articular regions in adolescents and young adults.1 The combination of surgery and chemotherapy has resulted in an approximate 60% 5-year survival rate,2,3 but the 10-year survival rate is still miserably low; therefore, identification of effective therapeutics for this sarcoma is critical. Synovial sarcoma is characterized by a chromosomal translocation between chromosomes 18 and X, generating oncoproteins such as SS18-SSX1 and SS18-SSX2.4,5 This translocation is present in >95% of cases and is likely to be the driving oncogenic event in the development of this tumor6; however, the precise mechanisms of SS18-SSX transformation remain controversial.
MicroRNA (miRNA) are small, non-coding RNA ranging from 18 to 24 nucleotides in length that negatively regulate gene expression at the post-transcriptional level, primarily through base pairing to the 3′-UTR of target mRNA.7 As miRNA control basic cell functions such as proliferation and apoptosis,7,8 upregulation or downregulation of miRNA expression causes diverse diseases, including cancers,9 as illustrated by their differential expression in carcinomas,10 sarcomas11,12 and hematologic tumors.13 MiRNA can function as tumor suppressor genes or oncogenes, and also as potential specific cancer biomarkers.14,15 However, only a few published studies have reported on miRNA in synovial sarcoma,16,17 which include let-7e, miR-99b, miR-125-3p and miR-183, and their functions remain obscure.
The miR-17-92 cluster is a polycistron encoding six mature micro RNA belonging to four seed families: the miR-17 family (miR-17 and miR-20a), the miR-18 family (miR-18a), the miR-19 family (miR-19a and miR-19b-1) and the miR-92 family (miR-92a-1).9,18 Overexpression of miR-17-92 has been reported in various types of human cancers.19,20 When overexpressed, miR-17-92 promotes cell cycle progression and proliferation,20 inhibits apoptosis21,22 and induces tumor angiogenesis.23 Previous work has confirmed that some target genes of the miR-17-92 cluster, such as E2F, CDKN1A (p21), BIM, Tsp1, CTGF, AIB1 and Cyclin D1, function mainly in cell cycle regulation.23 Among these, p21 (CIP1/WAF1) is a potent cyclin-dependent kinase inhibitor that binds to cyclin-CDK2 or cyclin-CDK4 complexes and inhibits their activities, ultimately inhibiting G1/S transition.
In this study, we identified miR-17 as an oncogene that causes an aggressive growth of synovial sarcoma cells by directly inhibiting p21 expression. In miR-17-overexpressing synovial sarcoma cells, p21 protein levels were dramatically decreased in a p53-independent manner, even in the context of doxorubicin treatment-evoked massive expression of p21, resulting in profound tumor growth in mice both in vitro and in vivo. These findings suggest that miR-17 is a potentially powerful therapeutic agent in synovial sarcoma.
Materials and Methods
Cell lines, cell culture and synovial sarcoma cases
The human synovial sarcoma cell lines SYO-1, Fuji and HS-SYII were established and maintained as described previously.24 SYO-1 and Fuji cells genetically possess the SS18-SSX2 fusion transcript, whereas HS-SYII cells occur as SS18-SSX1. Human oral cancer cells (HSC-2, HSC-3 and HSC-4), prostate cancer cells (LNCap), fibrosarcoma (HT1080), osteosarcoma (Saos-2), embryonic kidney 293T cells and BJ/t foreskin fibroblast were maintained in DMEM (Sigma, St. Louis, MO, USA) supplemented with 10% FBS (Cansera, Toronto, Ontario, Canada). Human Ewing's sarcoma cells (TC71) were maintained in RPMI1640 with 10% FBS. Mir-17-overexpressig cells were established using the BLOCK-iT HiPerform Lentiviral PolII miR RNAi Expression System with EmGFP (Invitrogen, Carlsbad, CA, USA). Seven surgically resected tumors diagnosed as synovial sarcoma were used to assess the expression levels of miR-17. The present study was approved by the Medical Ethics Committee of the Hokkaido University Graduate School of Medicine.
Reagents, antibodies and immunoblotting
The following antibodies were purchased: antibodies to p21WAF1/CIP1, phospho-Akt S473 and phospho-ERK1/2 were obtained from Cell Signaling Technology (Beverly, MA, USA); those to Cyclin D1 and actin (I-19) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and those to Ki-67 (clone: MIB1) and p53 were obtained from DAKO (Glostrup, Denmark). Immunoblot analyses were performed as described previously.24 Doxorubicin hydrochloride was obtained from Wako Pure Chemical Industries (Osaka, Japan).
Identification of microRNA to promote tumorigenicity in synovial sarcoma
The OncomiR miRNA Precursor Virus Library (System Bioscience, Mountain View, CA, USA) was infected into Fuji and HS-SYII cells, and a colony formation assay in soft agar was performed as described below. RNA was isolated from the two largest colonies formed by Fuji cells, and semi-quantitative RT-PCR using the OncomiR miRNA Precursor Virus Library primer (System Bioscience) and the following sequencing were performed to identify infected oncomiR.
Analysis of cell proliferation and colony formation assay
Analysis of cell proliferation was conducted as described previously.25 To assess the effect of miR-17 on cell growth, cells were transfected with anti-miR-17 reagent (miRCURY LNA microRNA Inhibitors, hsa-miR-17, 410087-00, EXIQON, Vedbaek, Denmark) and counted after 4 days. For the colony formation assay, the numbers of colonies after 4 weeks of <100 μm, 100–250 μm and more than 250 μm in diameter were counted.25
Analysis of cell motility and invasion assay
A wound healing assay assessing cell motility and a Matrigel invasion assay (Corning, Bedford, MA, USA) were performed as described previously.26
RNA isolation and RT-PCR
RNA isolation, cDNA synthesis and RT-PCR for human miR-17, p21, SS18-SSX1, SS18-SSX2, RNU6B and GAPDH (conventional and quantitative real-time PCR) were performed as described previously.26 Primers used were as follows: miR-17 forward, 5′-GCCGGCGTCAGAATAATGTCAAAGTGC-3′, and reverse, 5′-CACCATAATGCTACAAGTGCCTTCACTGC-3′; p21 forward, 5′-GCCCAGTGGACAGCGAGCAG, and reverse, 5′-GCCGGCGTTTGGAGTGGTAGA; SS18-SSX1 forward, 5′-CAACAGCAAGATGCATACCA, and reverse, 5′-AGATCTCTTATTAATCTTCTCAGAAA; SS18-SSX2 forward, 5′-CAACAGCAAGATGCATACCA, and reverse, 5′-TTTTGGGTCCAGATCTCTCGTG; RNU6B forward, 5′-GCTTCGGCAGCACATATACTAA, and reverse, 5′-AAAATATGGAACGCTTCACG; GAPDH, forward 5′-CTCATGACCACAGTCCATGC, and, reverse, 5′-TTACTCCTTGGAGGCCATGT.
siRNA and transfection
siRNA targeting SS18-SSX (si-SS18-SSX, TGACCAGATCATGCCCAAGAA) was purchased from Qiagen (Valencia, CA, USA); 5 × 105 Fuji cells were transfected with 15, 25 and 50 nM of si-SS18-SSX using HiPerfect Transfection Reagent (Qiagen, Valencia, CA, USA).
Luciferase reporter assay for targeting p21-3′UTR
The p21-3′UTR was amplified from BJ/t cells, converted to cDNA and sequenced. The p21-3′UTR was cloned into the region downstream of the Firefly luciferase gene in the pGL3-promoter luciferase reporter vector (Promega, Madison, WI, USA). The luciferase reporter vector was co-transfected with the miR-17-overexpression vector or the control vector in Fuji cells using the Fugene HD transfection reagent (Roche). The Renilla luciferase plasmid pRL-CMV (Promega) was used as a control for transfection efficiency. After 48 h, a dual luciferase assay was performed as described previously.24
Xenograft model
MiR-17-overexpression and its control Fuji cells (5 × 106) were injected s.c. into the left and right abdomen of 6-week-old female nude mice, BALB/cA Jc1 nu/nu (CLEA Japan, Tokyo, Japan), respectively. After injection, the volume of the tumors was measured twice a week using the following formula: volume = 1/2 (length × width2). Fifty days post-cell implantation, the mice were killed. This was followed by standard histopathological and immunohistochemical examination (described below). Mice were maintained under specific pathogen-free conditions, and studies were performed in accordance with the guidelines established by the Hokkaido University Committee on Animal Care and Use.
Histopathology and immunohistochemistry
Formalin-fixed, paraffin-embedded mouse tumor tissues were stained with H&E using the conventional method. Immunohistochemistry for MIB-1 and p21 was performed and evaluated as described previously.26–28
Statistical analyses
All data represent means and SD of experiments performed in triplicate and were subjected to a one-way analysis of variance, followed by comparison with Student's t-tests. P-values below 0.05 were considered statistically significant, as described in the figure legends.
Results
SS18-SSX-regulated miR-17 functions as oncogene in synovial sarcoma cells
To identify whether microRNA causes marked tumorigenicity on synovial sarcoma, cancer-related miRNA library screening was performed, the OncomiR miRNA Precursor Virus Library was infected into independent human synovial sarcoma cell lines Fuji and HS-SYII, and a colony formation assay in soft agar was performed (Fig.1a). Fuji cells infected with the OncomiR Library, but not HS-SYII, formed numerous large colonies compared to cells without the infection (Fig.1b). Total RNA was isolated from the largest two colonies formed by infected Fuji cells, and semi-quantitative RT-PCR using the OncomiR miRNA Precursor Virus Library primer and the following sequencing identified miR-17-5p (Fig.1c). The miR-17-92 cluster is a polycistron encoding six mature microRNA belonging to four seed families (Fig.1d). MiR-17 was highly expressed in SYO-1, Fuji and HS-SYII cells, and also in all of the human synovial sarcoma cases tested at various amounts (Fig.1e). It is noteworthy that forced expression of SS18-SSX2, but not SS18-SSX1, succeeded in promoting the endogenous miR-17 level in Fuji cells (Fig.1f), whereas the depletion of endogenous SS18-SSX2 by siRNA reduced miR-17 expression levels depending on the concentration of siRNA (Fig.1g), which definitely certified the SS18-SSX2-regulated miR-17 expression in Fuji cells. As miR-17 expression levels increased remarkably in human synovial sarcoma surgical specimens compared with the cell lines, the tumor microenvironment may be involved in miR-17 induction (Fig.1h).
Fig 1.
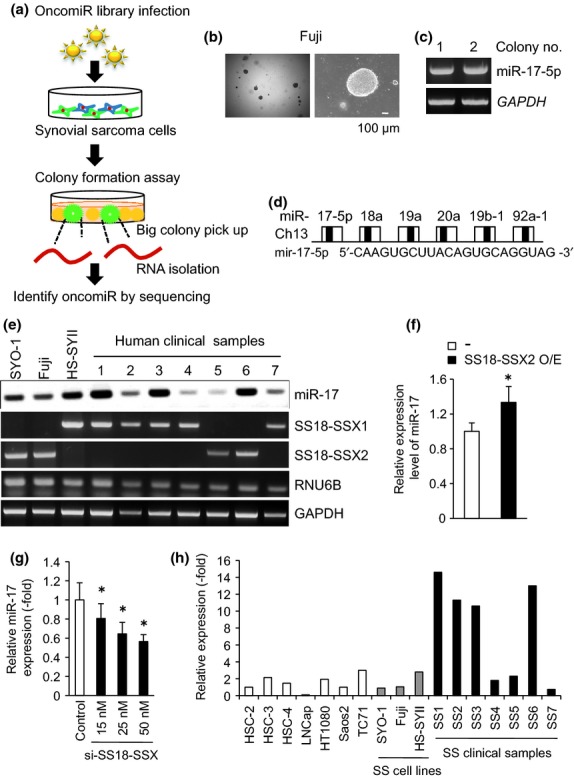
SS18-SSX-regulated miR-17 promotes colony formation ability in synovial sarcoma cells. (a) Schematic diagram identifying microRNA to cause marked tumor growth on synovial sarcoma cells. (b) Fuji cells infected with the OncomiR miRNA Precursor Virus Library formed large colonies in soft agar. (c) Total RNA was isolated from the largest two colonies formed by Fuji cells (b), and subjected to semi-quantitative RT-PCR using the OncomiR miRNA Precursor Virus Library, followed by sequencing. (d) Diagram of a polycistronic microRNA cluster termed miR-17-92. Sequence of miR-17-5p is shown. (e) Endogenous expression levels of miR-17 in indicated samples were examined by semi-quantitative RT-PCR. RNU6B was used as an internal control for evaluating microRNA expression. (f) mir-17 expression levels were examined by quantitative RT-PCR in Fuji cells with enforced expression of SS18-SSX2 (f), with SS18-SSX depletion (g), and in indicated cell lines and clinical samples (h). *P < 0.05 versus control cells.
MiR-17 increases growth of synovial sarcoma cells
To clarify the functions of miR-17 in synovial sarcoma cells, we stably established miR-17-overexpressing Fuji and HS-SYII cells by infecting the cells with miR-17-producing lentivirus (Fig.2a). MiR-17 overexpression significantly promoted cell proliferation in both Fuji and HS-SYII cells, reaching 1.6-fold and 4.0-fold increases compared to the control cells, respectively (Fig.2b). In addition, forced expression of miR-17 in Fuji and HS-SYII cells increased the colony formation ability compared to the corresponding control cells (Fig.2c). Based on these findings, miR-17 might have a similar biological effect on cell proliferation and colony formation both in SS18-SSX1 and SS18-SSX2-harboring synovial sarcoma cells. In contrast, cell motility and invasion capabilities were not altered by miR-17 overexpression (Fig.2d,e). MiR-17 overexpression did not lead to significant alterations on the cell morphologies (data not shown).
Fig 2.
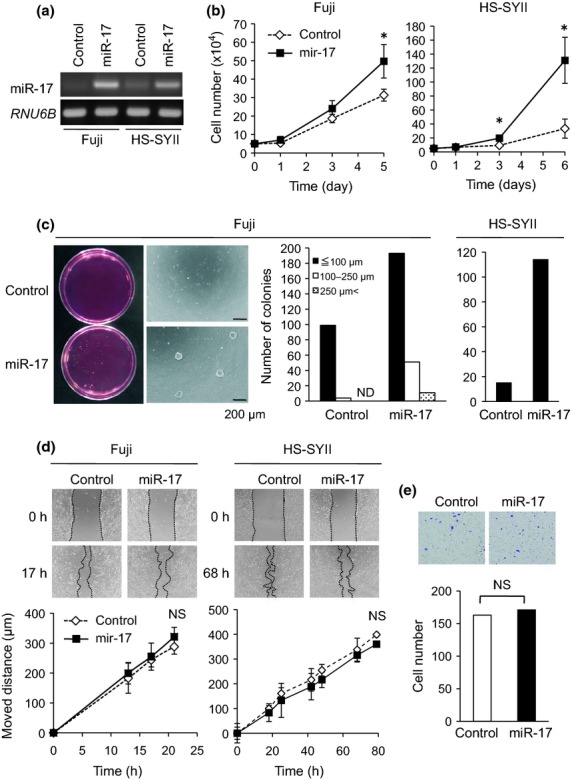
MiR-17 increases growth of synovial sarcoma cells. (a) Fuji and HS-SYII cells were infected with miR-17-producing or its control lentivirus, and the expression levels of miR-17 were examined by semi-quantitative RT-PCR. (b) Proliferation of Fuji and HS-SYII cells with or without MiR-17 overexpression was investigated. *P < 0.05 versus control cells. (c) Colony formation assay was performed in both Fuji and HS-SYII cells with or without miR-17 overexpression. The numbers and size were measured and graphed. ND means not detected. (d) Wound healing assay. Moved distances of Fuji and HS-SYII cells with or without miR-17 overexpression were measured at indicated time points. (e) Matrigel invasion assay. Invaded cells under the filter were counted and graphed.
MiR-17 promotes tumor development of synovial sarcoma in in vivo mice
To evaluate the effect of miR-17 on in vivo tumorigenicity of synovial sarcoma, miR-17-overexpressing Fuji cells were injected s.c. into nude mice, and the tumor volume was measured twice a week. Twenty-nine days post-implantation, miR-17 overexpression produced a significant increase in tumor volume compared with the control cells. Ultimately, massive tumors developed, with a 6.5-fold increase at 50 days (Fig.3a,b). Average weights of the formed tumors with or without miR-17 overexpression were 1.4 and 0.2 g, respectively (Fig.3c), demonstrating the significant contribution of miR-17 in in vivo tumor growth.
Fig 3.
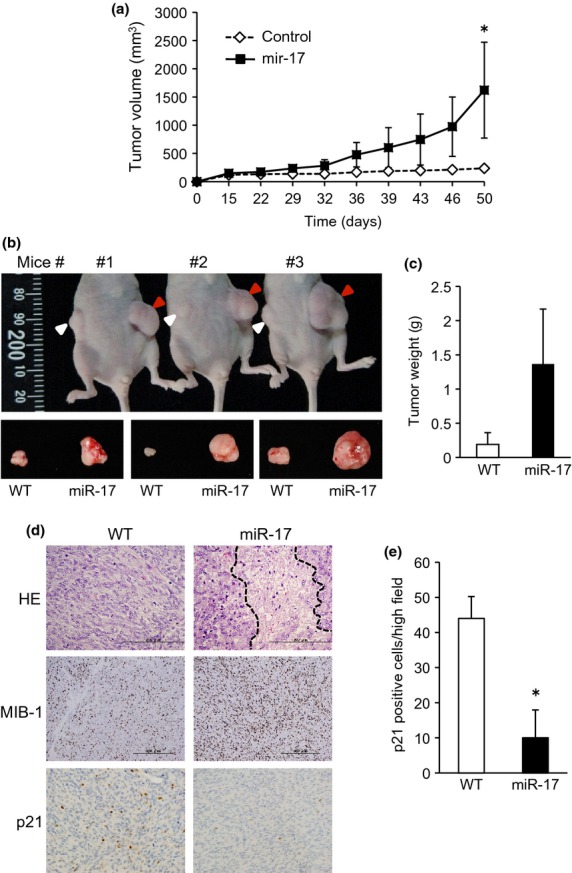
MiR-17 promoted tumor formation of synovial sarcoma in in vivo mice. (a) MiR-17-overexpressing Fuji and its control cells were injected s.c. into nude mice. The tumor volume was measured twice a week, and plotted in the graph. *P < 0.05 versus control tumor. (b, c) 50 days post-implantation, mice were killed and resected tumor weights were measured and graphed (c). Red and white arrowheads indicate miR-17-overexpressing tumors and control, respectively. (d) Tumor tissues were subjected to H&E staining and immunostaining for Ki-67 and p21. Region surrounded by dashed lines shows necrosis in the center of the tumor. (e) p21 positive cells were counted in randomly selected three regions and graphed.
H&E staining of the tumors revealed that the miR-17-overexpressing tumor exhibited brisk mitotic activities, besides necrosis in the center of the tumor. The number of Ki-67-positive proliferating cells within the tumor was significantly increased by miR-17-overexpresion (Fig.3d). Contrary of this, p21 positive cells were significantly reduced in the tumors formed in miR-17 overexpressing Fuji cells (Fig.3d,e).
MiR-17 directly targets p21-3′UTR and induces drug resistance in synovial sarcoma cells
Previous study has demonstrated that miR-17-92 cluster regulates seven target genes, E2F, CDKN1A (p21), BIM, Tsp1, CTGF, AIB1 and Cyclin D1, which are mainly involved in cell-cycle progression.23 We employed target prediction of miR-17 using PicTar and Miranda algorithms, which accurately predicted E2F1, E2F3 and p21 genes. Among them, PicTar and Miranda prediction scores for p21 were 4.19 and −0.8, respectively, suggesting the reliability of this gene as a potential target of miR-17 (Table1). To analyze whether miR-17 directly targets p21-3′UTR in synovial sarcoma cells, we developed the luciferase reporter vector fused to the 3′-UTR of p21 (Fig.4a). In stably miR-17-overexpressing Fuji cells, p21-3′UTR luciferase activity was significantly suppressed compared with the control cells (Fig.4b, left). A similar result was obtained even upon temporary overexpression of miR-17 in Fuji cells (Fig.4b, right), together verifying p21-3′UTR as a direct target of miR-17. Overexpression of miR-17 notably attenuated p21 protein levels in Fuji cells in a p53-independent manner, albeit with an increased p21 mRNA level (Fig.4c,d), indicating miR-17-regulated post-transcriptional degradation of p21. Phosphorylation levels of ERK and Akt remained almost unchanged, irrespective of miR-17 overexpression (Fig.4d). It is noteworthy that marked p21 expression evoked by doxorubicin treatment, a conventional chemotherapy for synovial sarcoma, was also strikingly reduced by miR-17 overexpression (Fig.4e). Consistent with this reduction, forced expression of miR-17 rescued doxorubicin-induced growth suppression in Fuji cells (Fig.4f).
Table 1.
Prediction scores of PicTar and Miranda algorithms in miR-17-targeed genes previously reported
| Gene name | Functions | PicTar score | Miranda score |
|---|---|---|---|
| E2F1 | Positive regulation of cell proliferation, Proapoptotic proteins | 6.57 | −1.06 |
| E2F2 | Positive regulation of cell proliferation, Proapoptotic proteins | ND | −0.14 |
| E2F3 | Positive regulation of cell proliferation, Proapoptotic proteins | 1.24 | −0.46 |
| CDKN1A (p21) | Negative regulator of G1-S checkpoint | 4.19 | −0.8 |
| BIM | Proapoptotic protein | ND | ND |
| Tsp1 | Anti-angiogenic protein | ND | ND |
| CTGF | Anti-angiogenic protein | ND | −0.03 |
| AIB1 | Oncogenic protein | ND | ND |
| CyclinD1 | Oncogenic protein | ND | ND |
ND, not detected.
Fig 4.
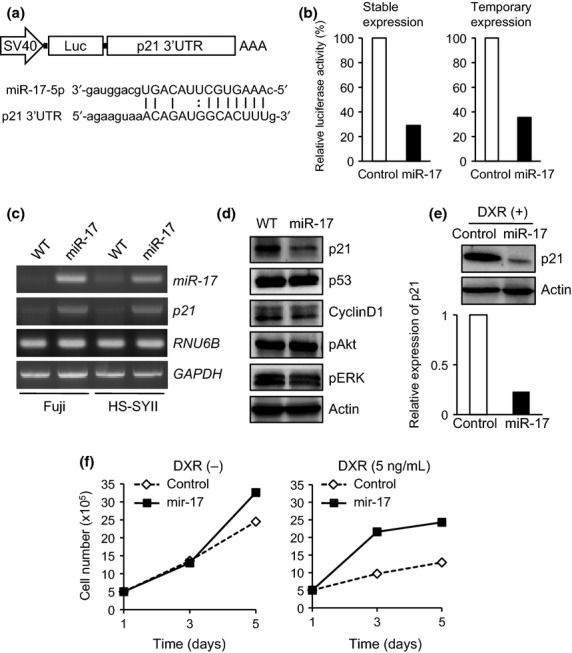
MiR-17 directly targets p21-3′UTR and induces drug resistance in synovial sarcoma cells. (a) Diagram of luciferase reporter vector fused to 3′-UTR of p21 utilized in luciferase assay. Sequences of miR-17-5p and the targeted p21 3′-UTR are shown. (b) Dual luciferase assay. p21-3′UTR luciferase activities were measured in stably (left) and temporarily (right) miR-17-overexpressing Fuji cells. (c) p21 mRNA expression levels were examined in parental and miR-17-overexpressing Fuji and HS-SYII cells by semi-quantitative RT-PCR. (d) In Fuji cells with or without miR-17 overexpression, expression levels of indicated proteins were investigated by immunoblotting. (e) Expression levels of p21 protein in miR-17-overexpressing Fuji and its control cells were examined by immunoblotting in the presence of doxorubicin treatment. (f) Cell proliferation of Fuji cells overexpressing miR-17 was investigated in the presence or absence of doxorubicin treatment.
Anti-miR-17-reagent suppresses cell proliferation through restored p21 expression
To assess the effectiveness of miR-17 in molecular targeted therapy of synovial sarcomas, we established Fuji and HS-SYII cells transduced by anti-miR-17 reagent. The depletion of miR-17 by anti-miR-17 treatment reliably rescued p21 protein levels in Fuji cells (Fig.5a), and this was followed by a substantial decline in the cell growth (Fig.5b). Similar suppression of the cell growth was also observed in HS-SYII cells treated with anti-miR-17 reagent (Fig.5b).
Fig 5.
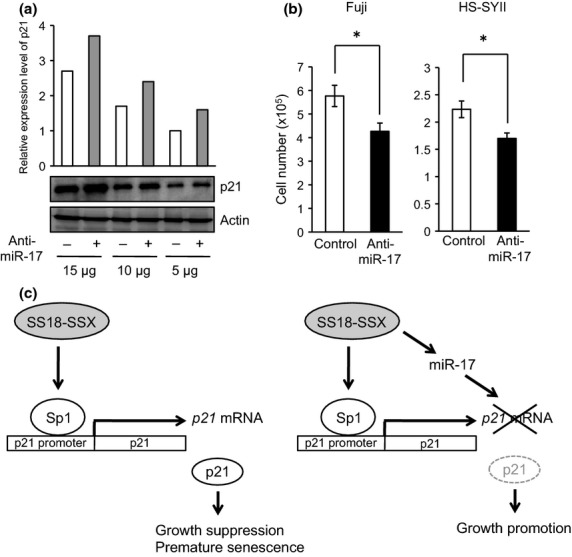
Anti-miR-17 reagent increased p21 protein level followed by suppressed cell proliferation. (a) In Fuji cells treated with or without anti-miR-17 reagent, expression levels of p21 protein were examined by immunoblotting using 15, 10, and 5 μg of total protein. (b) Cell proliferation of Fuji and HS-SYII cells treated with or without anti-miR-17 reagent was investigated. *P < 0.05 versus control cells. (c) Mechanism of SS18-SSX/miR-17-regulated growth promotion by targeting p21. (Left) SS18-SSX has an ability to induce p21 protein in a Sp1/Sp3-dependent manner, leading to growth suppression and premature senescence in synovial sarcoma cells. Involvement of Sp1 has previously demonstrated.24 (Right) SS18-SSX, particularly SS18-SSX2, is also capable of upregulating miR-17 that disrupts 3′UTR of p21 mRNA, leading to an increased cell proliferation.
Discussion
Synovial sarcoma is a relatively common soft tissue sarcoma with poor prognosis. Synovial sarcomas occur in adolescents and young adults mainly in para-articular regions,1 and an effective therapy bringing an improved prognosis is urgently desired. Functional analyses of miRNA have demonstrated the potential for a novel molecular targeted therapy; however, little is known concerning the engagement of miRNA in synovial sarcoma.12,17 In the present study, we found that forced expression of miR-17 in synovial sarcoma cells strikingly promoted tumorigenicity in in vivo mice (Fig.3) by directly targeting p21CIP1/WAF1 (Fig.4b,d). It is noteworthy that SS18-SSX fusion oncoprotein possesses an ability to induce miR-17 expression (Fig.1f). MiR-17 overexpression reduced p21 protein level (Fig.4e). These lines of evidence indicate that miR-17 elevates the growth of synovial sarcoma cells by promoting cell cycle progression due to elimination of the inhibitory machinery of G1-S transition, targeting p21.
We previously demonstrated that SS18-SSX1 per se has an ability to induce p21 protein in an Sp1/Sp3-dependent manner but not hBRM and p53, leading to premature senescence in synovial sarcoma cells.24 Therefore, for full transforming activity of SS18-SSX to overcome such senescence, it has been assumed that other factor(s) should contribute to escape p21-induced growth suppression in this tumor, probably by downregulating p21 expression. In this study, we unraveled this long-standing issue using the OncomiR Library transfection system, and found that SS18-SSX is also capable of upregulating miR-17 that disrupts 3′UTR of p21 mRNA, leading to increased cell proliferation (Fig.5c). Hence, our findings propose miR-17 as a candidate for p53-independent p21 downregulation in synovial sarcoma. We further revealed the SS18-SSX2-dependent, SS18-SSX1-independent induction of miR-17 in synovial sarcoma cells (Fig.1f, data not shown). SS18-SSX was recently shown to induce the assembly of aberrant SWI/SNF (BAF) chromatin remodeling complexes by excluding BAF47, and reversing H3K27me3-mediated repression, leading to proliferation of this tumor. The ability of SS18-SSX to disrupt BAF complexes depends on two regions of the SSX protein: the C-terminal 8 aa (SDPEEDDE) and a polar region of 2 aa.29 The fact that the latter 2 aa is different between SSX1 (KR) and SSX2 (ER) might affect the ability of inducing miR-17. Alternatively, SS18-SSX1 and SS18-SSX2 have been shown to distinctly regulate the expression of Snail and Slug, respectively.30,31 Because these transcription factors were demonstrated to regulate expression of several miRNA such as miR-128 and miR-221 in breast cancer,32,33 they might provide SS18-SSX2/Slug/miR-17 axis in synovial sarcoma.
Previous papers revealed the overexpression of miR-17 in various types of human cancers19,20 and its involvement in cell cycle regulation.20 p21 is an inhibitory regulator in cell cycle progression of G1-S transition. Regarding synovial sarcoma cells, previous works have revealed that p53 protein acting upstream of p21 is effectively induced in response to DNA damage.34 When DNA damage occurs after radiation therapy and conventional chemotherapy in synovial sarcoma, a depletion of miR-17 might therefore reinforce the effect of p21-induced growth suppression. Indeed, we revealed that miR-17 overexpression conversely promotes the growth of Fuji and HS-SYII cells even in the presence of doxorubicin at an effective dose (Fig.4f).
It has been reported that several molecules, such as cyclin D135 and insulin-like growth factor-1 receptor,36 are essential for cell proliferation and oncogenesis of synovial sarcoma. Cai et al.37 suggest that the SS18-SSX protein plays an important role in synovial sarcoma cell growth through the ERK pathway. The MAPK tyrosine kinase inhibitor sorafenib inhibits cell growth in synovial sarcoma,38 and PI 3-kinase/AKT signaling is also important for cell proliferation of this sarcoma.36 We have previously shown that the Crk adaptor protein enhances proliferation of synovial sarcoma cells through the phosphorylation of Gab1 by Src and focal adhesion kinase and the consequent activation of a DOCK180-p38 MAPK signaling.25 Furthermore, our data demonstrated that dual inhibition of Src and Aurora kinases is a powerful approach to suppress the growth of this sarcoma.26 Although kinase-targeted therapy is, therefore, a promising approach in the treatment of this sarcoma, a kinase-irrelevant strategy using anti-miR-17 reagent might be innovative and effective in avoiding the emergence of serious drug resistance in the clinical application.
Among the synovial sarcoma cell lines we tested, miR-17 overexpression produced a significant increase in proliferation of Fuji and HS-SYII cells, but not SYO-1 distinctly (data not shown). Given that the PicTar algorithm predicted 711 genes as direct targets of miR-17 with prediction scores ranging from 8.63 to 0.83, of which CDKN1A (p21) is ranked as 140/711 genes with the score of 4.19, miR-17 may become exhausted by many of the targets in SYO-1 cells. Supporting this possibility, the effect of miR-17 on cell motility and invasion has been controversial in a cellular context.39–41 Thus, special attention should be paid to assess the balance of miR-17-targeted genes such as oncogenes or tumor suppressors, which definitely determines the physiological property of synovial sarcoma cells.
In the present study, our findings provide the first evidence that miR-17 plays an important role in regulating the growth of human synovial sarcoma. Forced expression of miR-17 drives colony formation capacity, cell proliferation and tumorigenecity both in vitro and in vivo by directly targeting the p21-3′UTR. Synovial sarcoma is largely resistant to conventional chemotherapy. Upregulation of p21 protein by miR-17 depletion might exert potent effects in the therapy of human synovial sarcoma.
Acknowledgments
We thank Dr Akira Kawai and Dr Hiroshi Sonobe for the use of SYO-1 and HS-SYII cells, Dr Hiroaki Hiraga for the use of human samples, and members of our laboratory for helpful discussions. This work was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology, from the Japan Society for the Promotion of Science, and from the Ministry of Health, Labor, and Welfare of Japan, as well as a grant from the Japan Science and Technology Agency.
Disclosure Statement
The authors have no conflict of interest to declare.
References
- Fisher C. Synovial sarcoma. Ann Diagn Pathol. 1998;2:401–21. doi: 10.1016/s1092-9134(98)80042-7. [DOI] [PubMed] [Google Scholar]
- Lewis JJ, Antonescu CR, Leung DH, et al. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000;18:2087–94. doi: 10.1200/JCO.2000.18.10.2087. [DOI] [PubMed] [Google Scholar]
- Krieg AH, Hefti F, Speth BM, et al. Synovial sarcomas usually metastasize after >5 years: a multicenter retrospective analysis with minimum follow-up of 10 years for survivors. Ann Oncol. 2011;22:458–67. doi: 10.1093/annonc/mdq394. [DOI] [PubMed] [Google Scholar]
- Clark J, Rocques PJ, Crew AJ, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. 1994;7:502–8. doi: 10.1038/ng0894-502. [DOI] [PubMed] [Google Scholar]
- Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors. Synovial sarcoma. Cancer Genet Cytogenet. 2002;133:1–23. doi: 10.1016/s0165-4608(01)00626-4. [DOI] [PubMed] [Google Scholar]
- Nagai M, Tanaka S, Tsuda M, et al. Analysis of transforming activity of human synovial sarcoma-associated chimeric protein SYT-SSX1 bound to chromatin remodeling factor hBRM/hSNF2 alpha. Proc Natl Acad Sci U S A. 2001;98:3843–8. doi: 10.1073/pnas.061036798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carè A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Lui WO, Lee CH, et al. MicroRNA expression signature of human sarcomas. Oncogene. 2008;27:2015–26. doi: 10.1038/sj.onc.1210836. [DOI] [PubMed] [Google Scholar]
- Sarver AL, Phalak R, Thayanithy V, Subramanian S. S-MED: sarcoma microRNA expression database. Lab Invest. 2010;90:753–61. doi: 10.1038/labinvest.2010.53. [DOI] [PubMed] [Google Scholar]
- Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs – MicroRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarver AL, Li L, Subramanian S. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res. 2010;70:9570–80. doi: 10.1158/0008-5472.CAN-10-2074. [DOI] [PubMed] [Google Scholar]
- Hisaoka M, Matsuyama A, Nagao Y, et al. Identification of altered MicroRNA expression patterns in synovial sarcoma. Genes Chromosom Cancer. 2011;50:137–45. doi: 10.1002/gcc.20837. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Ota A, Tagawa H, Karnan S, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–95. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Wu J. Feud or friend? The role of the miR-17-92 cluster in tumorigenesis. Curr Genomics. 2010;11:129–35. doi: 10.2174/138920210790886853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Watanabe T, Seki T, et al. Induction of p21WAF1/CIP1 by human synovial sarcoma-associated chimeric oncoprotein SYT-SSX1. Oncogene. 2005;24:7984–90. doi: 10.1038/sj.onc.1208942. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Tsuda M, Tanaka S, et al. Adaptor protein Crk induces Src-dependent activation of p38 MAPK in regulation of synovial sarcoma cell proliferation. Mol Cancer Res. 2009;7:1582–92. doi: 10.1158/1541-7786.MCR-09-0064. [DOI] [PubMed] [Google Scholar]
- Arai R, Tsuda M, Watanabe T, et al. Simultaneous inhibition of Src and Aurora kinases by SU6656 induces therapeutic synergy in human synovial sarcoma growth, invasion and angiogenesis in vivo. Eur J Cancer. 2012;48:2417–30. doi: 10.1016/j.ejca.2011.12.028. [DOI] [PubMed] [Google Scholar]
- Trojani M, Contesso G, Coindre JM, et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer. 1984;33:37–42. doi: 10.1002/ijc.2910330108. [DOI] [PubMed] [Google Scholar]
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol. 1997;15:350–62. doi: 10.1200/JCO.1997.15.1.350. [DOI] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nagai M, Ladanyi M. SYT-SSX1 and SYT-SSX2 interfere with repression of E-cadherin by snail and slug: a potential mechanism for aberrant mesenchymal to epithelial transition in human synovial sarcoma. Cancer Res. 2006;66:6919–27. doi: 10.1158/0008-5472.CAN-05-3697. [DOI] [PubMed] [Google Scholar]
- Saito T. The SYT-SSX fusion protein and histological epithelial differentiation in synovial sarcoma: relationship with extracellular matrix remodeling. Int J Clin Exp Pathol. 2013;6:2272–9. [PMC free article] [PubMed] [Google Scholar]
- Lambertini E, Lolli A, Vezzali F, Penolazzi L, Gambari R, Piva R. Correlation between Slug transcription factor and miR-221 in MDA-MB-231 breast cancer cells. BMC Cancer. 2012;12:445. doi: 10.1186/1471-2407-12-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P, Banerjee A, Wu ZS, et al. Loss of SNAIL regulated miR-128-2 on chromosome 3p22.3 targets multiple stem cell factors to promote transformation of mammary epithelial cells. Cancer Res. 2012;72:6036–50. doi: 10.1158/0008-5472.CAN-12-1507. [DOI] [PubMed] [Google Scholar]
- D'Arcy P, Ryan BA, Brodin B. Reactivation of p53 function in synovial sarcoma cells by inhibition of p53-HDM2 interaction. Cancer Lett. 2009;275:285–92. doi: 10.1016/j.canlet.2008.10.030. [DOI] [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- Friedrichs N, Küchler J, Endl E, et al. Insulin-like growth factor-1 receptor acts as a growth regulator in synovial sarcoma. J Pathol. 2008;216:428–39. doi: 10.1002/path.2438. [DOI] [PubMed] [Google Scholar]
- Cai W, Sun Y, Wang W, et al. The effect of SYT-SSX and extracellular signal-regulated kinase (ERK) on cell proliferation in synovial sarcoma. Pathol Oncol Res. 2011;17:357–67. doi: 10.1007/s12253-010-9334-y. [DOI] [PubMed] [Google Scholar]
- Peng CL, Guo W, Ji T, et al. Sorafenib induces growth inhibition and apoptosis in human synovial sarcoma cells via inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol Ther. 2009;8:1729–36. doi: 10.4161/cbt.8.18.9208. [DOI] [PubMed] [Google Scholar]
- Yang F, Yin Y, Wang F, et al. miR-17-5p Promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology. 2010;51:1614–23. doi: 10.1002/hep.23566. [DOI] [PubMed] [Google Scholar]
- Li H, Bian C, Liao L, Li J, Zhao RC. miR-17-5p promotes human breast cancer cell migration and invasion through suppression of HBP1. Breast Cancer Res Treat. 2011;126:565–75. doi: 10.1007/s10549-010-0954-4. [DOI] [PubMed] [Google Scholar]
- Yu Z, Willmarth NE, Zhou J, et al. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci U S A. 2010;107:8231–6. doi: 10.1073/pnas.1002080107. [DOI] [PMC free article] [PubMed] [Google Scholar]