

Abstract
We previously established that COX-2 overexpression promotes breast cancer progression and metastasis. As long-term use of COX-2 inhibitors (COX-2i) can promote thrombo-embolic events, we tested an alternative target, prostaglandin E2 receptor EP4 subtype (EP4), downstream of COX-2. Here we used the highly metastatic syngeneic murine C3L5 breast cancer model to test the role of EP4-expressing macrophages in vascular endothelial growth factor (VEGF)-C/D production, angiogenesis, and lymphangiogenesis in situ, the role of EP4 in stem-like cell (SLC) functions of tumor cells, and therapeutic effects of an EP4 antagonist RQ-15986 (EP4A). C3L5 cells expressed all EP receptors, produced VEGF-C/D, and showed high clonogenic tumorsphere forming ability in vitro, functions inhibited with COX-2i or EP4A. Treating murine macrophage RAW 264.7 cell line with COX-2i celecoxib and EP4A significantly reduced VEGF-A/C/D production in vitro, measured with quantitative PCR and Western blots. Orthotopic implants of C3L5 cells in C3H/HeJ mice showed rapid tumor growth, angiogenesis, lymphangiogenesis (CD31/LYVE-1 and CD31/PROX1 immunostaining), and metastasis to lymph nodes and lungs. Tumors revealed high incidence of EP4-expressing, VEGF-C/D producing macrophages identified with dual immunostaining of F4/80 and EP4 or VEGF-C/D. Celecoxib or EP4A therapy at non-toxic doses abrogated tumor growth, lymphangiogenesis, and metastasis to lymph nodes and lungs. Residual tumors in treated mice revealed markedly reduced VEGF-A/C/D and phosphorylated Akt/ERK proteins, VEGF-C/D positive macrophage infiltration, and proliferative/apoptotic cell ratios. Knocking down COX-2 or EP4 in C3L5 cells or treating cells in vitro with celecoxib or EP4A and treating tumor-bearing mice in vivo with the same drug reduced SLC properties of tumor cells including preferential co-expression of COX-2 and SLC markers ALDH1A, CD44, OCT-3/4, β-catenin, and SOX-2. Thus, EP4 is an excellent therapeutic target to block stem-like properties, angiogenesis, and lymphangiogenesis induced by VEGF-A/C/D secreted by cancer cells and tumor infiltrating macrophages.
Keywords: Cancer stem cells, cyclooxygenase 2, macrophages, prostaglandin E2 receptor EP4 subtype, RQ-00015986
Cyclooxygenase-2, an inflammation-inducible enzyme, is associated with progression of epithelial cancers.1–3 Cyclooxygenase-2 overexpression, noted in nearly half of all breast cancer patients, marks poor prognosis.4 High COX-2 activity resulting in elevated prostaglandin (PG) E2 levels in the tumor milieu promotes breast cancer progression and metastasis through multiple events: inactivation of host immune cells;5 stimulation of tumor cell migration/invasiveness;6–8 and tumor-associated angiogenesis6,9 and lymphangiogenesis.10 Expression of vascular endothelial growth factor (VEGF)-C/D in human breast cancer in situ is strongly correlated with lymphangiogenesis, lymphovascular invasion, and lymphatic metastasis.11–14 Cyclooxygenase-2 is a major stimulator of VEGF-C production in human11 and VEGF-C/D production in murine10 breast cancer models. In addition to its lymphangiogenic role, COX-2-upregulated VEGF-C directly promoted breast cancer cell motility, a phenotype for metastasis, by binding to a diverse group of VEGF-C receptors.15
Although the above evidence makes COX-2 a reasonable therapeutic target, increased risks of thrombo-embolic effects of long-term use of high-dose COX-2 inhibitors16,17 suggest the need for identifying alternative target(s) downstream of COX-2 that may spare the risks. The vaso-protective role of COX-2 was attributed to IP receptors interacting with PGI2.18 Thus, targeting one or more of the PGE (EP) receptors should retain IP actions. They are G protein-coupled receptors with differential signaling abilities: EP1 is coupled with Gq, stimulating (Ca++) i; EP2 and EP4 are coupled with Gs, stimulating the adenylate cyclase/PKA pathway; whereas most EP3 isoforms are coupled with Gi, thus inhibiting adenylate cyclase.19
Unlike EP2, EP4 can additionally stimulate phosphatidylinositol 3-kinase (PI3K)/Akt-mediated cell survival pathway as well as the pro-migratory ERK pathway.20 Most of the COX-2 mediated events in breast cancer, such as cancer cell migration/ invasiveness,7,8 VEGF-C or -D upregulation in cancer cells10,11 and inactivation of natural killer cells21 were shown to follow activation of EP4 on these cells, making it an excellent therapeutic target, without crippling the vaso-protective arm of COX-2. This target was validated by preclinical studies in syngeneic murine breast cancer models with a number of EP4 antagonists.10,22
Tumor progression, metastasis, and recurrence after therapy-initiated remission are all believed to result from a tumor cell subpopulation known as “stem-like cells” (SLC).23,24 Interestingly, PGE-2 was shown to stimulate hematopoietic stem cells25 and EP4 activation was reported to be essential for hematopoietic stem cell expansion.26 Recently, EP4 has been implicated in promotion of the SLC phenotype in breast cancer cells.27
Although tumor-associated macrophages (TAMs) can play a complex role in both halting and promoting tumor progression, there is compelling evidence for the latter in established solid tumors.28 Tumor-associated macrophages can facilitate many key processes in breast cancer progression such as immune suppression, production of proteases, and promotion of angiogenesis.29,30 Indeed, macrophage infiltration in the tumor stroma is an independent indicator of poor prognosis in human breast cancer.31 The capacity of macrophages to produce both VEGF-A32 and VEGF-C/D33 explains their stimulatory roles in angiogenesis and lymphangiogenesis. It is presently unclear whether VEGF-A/C/D production by TAMs in breast cancer is COX-2- or EP4-dependent.
In view of the above, the present study was designed in our COX-2 expressing syngeneic breast cancer model10 to explore: (i) whether VEGF-C or -D production by TAMs is an additional driver of lymphangiogenesis in situ and, if so, whether it is COX-2- or EP4-dependent; (ii) the role of EP4 in stem-like tumor cell functions; and (iii) the potential therapeutic effects of a COX-2 inhibitor celecoxib and an EP4 antagonist RQ-15986 on these events, including tumor growth and spontaneous metastasis to the lungs and lymph nodes. Effects of these drugs on angiogenesis and lymphangiogenesis were tested with VEGF-A/C/D expression in residual tumors and in situ immunostaining of tumor vasculature for LYVE-1/CD31 and PROX1/CD31. In addition, in vitro effects of the drugs were tested on VEGF-A/C/D production by a murine macrophage cell line. Results revealed that EP4 is an excellent therapeutic target to block stem-like properties in cancer cells and tumor-associated angiogenesis and lymphangiogenesis induced by VEGF-A/C/D production by cancer cells as well as TAMs.
Materials and Methods
Cell line
C3L5 is a highly metastatic derivative of a spontaneous mammary adenocarcinoma in C3H/HeJ mice,34 which expresses high levels of COX-2, the PGE-2 secreting ability primarily attributed to COX-2.6,8 Mouse macrophage cell line RAW 264.7 were purchased from ATCC (Manassas, VA, USA). Cells were maintained in high glucose DMEM (Gibco, Invitrogen, ON, Canada), 10% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin.
Mice
Six-week-old female C3H/HeJ mice from Jackson Laboratories (Bar Harbor, ME, USA) were allowed to acclimatize for 2–3 weeks, maintained on standard mouse chow and tap water (unless otherwise indicated) on a 12:12 h light:dark cycle, and treated in accordance with the guidelines set by the Canadian Council on Animal Care.
Drugs and reagents
NS-398 (COX-2 inhibitor) was from Cayman Chemical, Ann Arbor, MI, USA. Selective COX-2 inhibitor celecoxib and EP4 antagonist CJ-042794 (renamed as RQ-15986) were respective gifts of Pfizer, Groton, CT, USA and RaQualia Pharma, Nagoya, Japan. The structure, binding affinity, high selectivity, and pharmacokinetic properties of RQ-15986 (henceforth called EP4A) has been characterized.35,36 For in vitro treatments and treating tumor-transplanted mice, the details of all drugs and vehicles used to dissolve or administer them are presented in “Materials and Methods” and “Results”. For in vitro or in vivo treatments, respective vehicles served as controls.
Proliferation, migration, and VEGF-C/D production by C3L5 cells
Proliferation and migration at 24 h were measured as reported.14 C3L5 cells had been shown to express all the EP receptors.10 The effects of treating C3L5 and cells with EP4A was measured with real-time quantitative PCR (for VEGF-C and -D mRNA), ELISA (for VEGF-C), and Western blot analysis (for VEGF-D) as reported.10
Vascular endothelial growth factor-A/C/D production by RAW 264.7 cells
Cells were treated for 24 h with DMSO, celecoxib, and EP4A before RNA and protein extraction to carry out TaqMan real-time PCR (VEGF-A, cat # 4453320; VEGF-C, cat # 4453320; VEGF-D, cat # 444889; and Gabpb1, cat # 4448892) and Western blots as described later in Western blot analysis.10
Detection of SLC in vitro by clonogenic spheroid formation assay
Cells were serum-starved for 12 h and serially diluted at 1000, 100, 10, and 1 cell(s)/100 μL in HUMEC medium (Gibco) supplemented with 20 ng/mL basic fibroblast growth factor (Invitrogen, ON, Canada), 20 ng/mL epidermal growth factor (Invitrogen), and B-27 supplement (1:50 dilution; Life Technologies, Burlington, ON, Canada). They were cultured on ultra-low attachment plates (Corning, MA, USA) to observe the temporal kinetics for spheroid (tumorsphere) formation. In some experiments, to explore dependence of spheroid formation on COX-2 or EP4 activity, cells were pretreated with either 0.003% DMSO or 2 μM celecoxib or EP4A for 5 days before plating and the same drug concentrations continued during spheroid formation. Spheroid formation was also evaluated after stable knockdown of COX-2 with shRNA plasmid (sc-29278-SH) and transient knockdown of EP4 with siRNA (sc-40174), both obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The number of spheroids (at least 60 μm in diameter) and their perimeters were measured with ImageJ (http://imagej.nih.gov/ij/) at different time points and spheroids were dissociated and recultured to assess their spheroid-forming ability at successive generations.
Detection of SLC or embryonic stem cell-associated markers with immunofluorescence
Five micrometer-thick frozen sections of C3L5 cell in vitro spheroids and 12-day-old tumors (see later in measurements of angiogenesis and lymphangiogenesis section) were subjected to dual immunostaining for COX-2 (Abcam, Toronto, ON, Canada) and breast cancer stem cell markers aldehyde dehydrogenase (ALDH) and CD44 or embryonic stem (ES) cell markers OCT-3/4 and SOX-2 or hematopoietic stem cell marker β-catenin, with antibodies from BD Biosciences.
Tumor implantation and treatment regimen
Briefly, 5 × 104 C3L5 cells, suspended in diluted growth factor-reduced Matrigel were implanted s.c. in both inguinal mammary regions. Animals were divided into five different groups. Group I (six mice) received implants of Matrigel alone and no other treatment to serve as negative controls for angiogenesis or lymphangiogenesis. The remaining four groups (groups II–V, 24 mice per group) were implanted with tumor cells suspended in Matrigel. Group II received celecoxib (250 mg/kg/day dissolved in 0.5% methylcellulose; Sigma, Oakville, ON, Canada) by oral gavage twice daily. Group III received EP4A (10 mg/kg/day dissolved in 0.003 N NaOH by oral gavage twice daily). Groups IV and V received vehicles alone serving as respective controls for celecoxib and EP4A. The dosage and vehicles for these drugs were guided by earlier reports for celecoxib37 and EP4A.35,36 Mice (two from group I and eight from each of the other groups) were killed on days 8, 12, and 16. After retrieval of the implants (two per mouse), their weights and diameters (maximum and minimum) were recorded and gross morphology photographed. Tumors were then sliced in half, each half fixed or frozen for further histological or immunohistological analysis. Tumor-draining inguinal and distant axillary lymph nodes were removed before fixing them for histopathology.
Evaluation of drug-mediated toxicity in treated mice
All animals were monitored for daily water intakes, and examined at autopsy for any visible bleeding in the gastrointestinal tract.
Scoring metastases
Metastatic lung colonies were scored microscopically in both lungs in H&E stained coronal sections through mid-trachea (providing the maximal surface area of both lungs) in fixed tissues. Mean and median scores were based on eight mice/group, with each score acquired from counts of all lobes. Metastases to tumor-draining inguinal and distant axillary lymph nodes were identified histologically in H&E stain as reported.10 Both pulmonary and lymphatic metastases were scored by two independent observers with less than 8% variation.
Measurements of angiogenesis and lymphangiogenesis
Eight micrometer-thick frozen sections were dual immunostained for LYVE-1/CD31and PROX1/CD31.10,38 Adjacent sections of fixed tissues stained with Masson's trichrome, examined under bright field, provided a map to locate vessels within the peritumoral stroma and tumor parenchyma. With this method, capillaries devoid of red cells could not be adequately discriminated between blood or lymphatic capillaries. Blood microvessel density and lymphatic vessel density were assessed in dual immunostained sections (CD31, red; LYVE-1, PROX1, green) as reported.10,38 Three hotspots per tumor (48/group/day) were examined at ×400 magnification, and mean scores for blood microvessel density and lymphatic vessel density were obtained after background correction, using ImageJ.
Western blot analysis of proteins in tumor tissues
As reported, two vehicle-treated tumor extracts were pooled.10 We used the following primary antibodies: polyclonal rabbit anti-mouse VEGF-A (1:200), polyclonal goat anti-mouse VEGF-C (1:200), monoclonal rabbit anti-mouse VEGF-D (1:200), monoclonal anti-mouse GAPDH (Santa Cruz Biotechnology), polyclonal rabbit anti mouse Akt (cat #9272), mouse monoclonal phospho-Akt (Ser 473) (cat #4051S), and polyclonal rabbit anti-mouse antibodies to detect total ERK1/2 and phospho-ERK1/2 proteins (1:1000 dilution) (Cell Signaling Technology, Danvers, MA, USA). IRDye polyclonal secondary antibodies from LI-COR (Lincoln, NE, USA), either donkey anti-goat or goat anti-rabbit and donkey anti-mouse were used. Dilution for all VEGFs was 1:5000; for GAPDH, it was 1:20 000). Membranes were scanned on an Odyssey infrared imaging system (LI-COR).
Proliferative/apoptotic cell ratios
Proliferative/apoptotic cell ratios were measured in tumors from drug- and vehicle-treated mice by dual immune staining for Ki-67 (proliferation marker) and TUNEL (apoptosis marker).10
Detection of EP4, F4/80, and VEGF-C/D in cells
EP4 was localized in tumor cells in vitro and tumor tissues in vivo by immunostaining with a highly specific antibody (sc-20677; Santa Cruz Biotechnology), replacing the primary antibody with immune IgG in negative controls. EP4 expression in tumor infiltrating macrophages was identified with dual immunostaining of EP4 and F4/80 (sc-71085; Santa Cruz Biotechnology) in frozen sections. Macrophages within tumors expressing VEGF-C/D were identified by dual immunostaining with F4/80 and VEGF-C/D antibodies (sc-9047 and sc-13085 respectively; Santa Cruz Biotechnology).
Statistical analysis
Data were analyzed using GraphPad Prism, GraphPad Software, La Jolla, CA, USA. All parametric data were compared with one-way ANOVA followed by Tukey–Kramer or Dunnett post hoc comparisons; two datasets were compared with Student's t-test. Statistically significant differences were accepted at P < 0.05.
Results
Proliferation, migration, and VEGF-C and -D production inhibited by EP4A in C3L5 cells in vitro
The COX-1/COX-2 inhibitor indomethacin, COX-2 inhibitor NS-398 (both at 20 μM), as well as EP4A (10–5000 nM), all inhibited C3L5 cell proliferation and migration (Fig. S1, respectively). Production of VEGF-C and -D by C3L5 cells (data not shown) after treatment with EP4A (50–5000 nM) was inhibited at a level similar to that reported with another EP4 antagonist (ONO AE3-208, 2 μM).10
Celecoxib and EP4A treatments reduced tumor growth and spontaneous lung metastasis
Implants of Matrigel alone appeared as clear avascular jelly beans (Fig.1a, shown for day 16) at all time points with no evidence of angiogenesis or lymphangiogenesis, as previously reported.10,38 Tumor growth was significantly blocked (at all time points) with both EP4A (as early as day 8) and celecoxib treatments (Fig.1b). Spontaneous lung metastases are shown in Figure1(c), as surface colonies and microscopic colonies (Fig.1d) on day 12. The incidence of microscopic colonies was high in vehicle-treated mice on day 8, with a further small increase on days 12 and 16. There was a significant reduction of lung colonies at all time points with celecoxib and EP4A treatments (Fig.1e).
Fig 1.
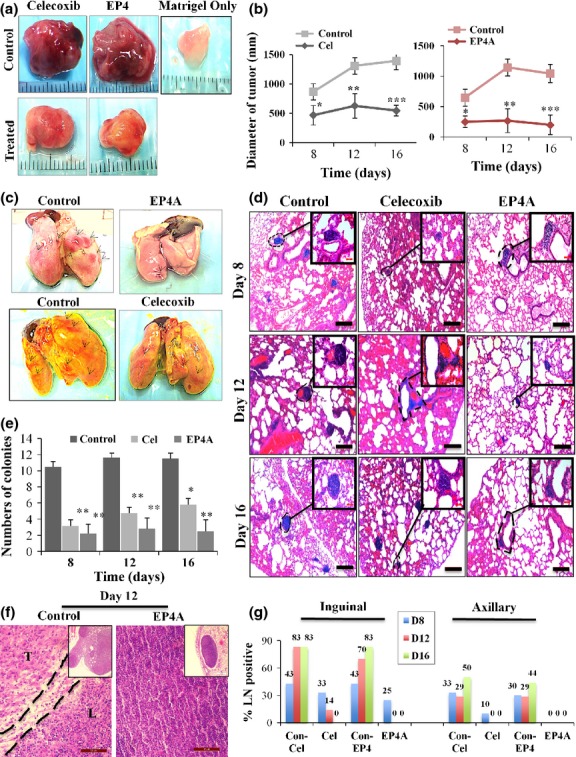
Tumor growth (a, b), lung colonies (c–e), and lymph metastasis (f, g) in a murine C3L5 breast cancer model. (a) Representative images of tumor-inclusive Matrigel or Matrigel only implants retrieved on day 16 (a scale in mm shown in the background). (b) Mean diameters of tumors were reduced significantly in mice treated with celecoxib or RQ-15986 (EP4A), compared to respective vehicle-treated controls. This reduction was significant at all time points for both therapies. Data represent means (n = 16/group/day) ± SE. *P < 0.05; **P < 0.005; ***P < 0.001. (c) Digital camera photographs of surface lung colonies at day 12 (black arrows) on left. (d) Representative images of micrometastases in H&E stained lung sections on days 8, 12, and 16. Scale bar = 200 μm. (e) Charts showing mean numbers of metastatic lung colonies were reduced with celecoxib (Cel) and EP4A therapies at all time points after tumor transplantation. Data represent mean ± SE (n = 8/group/day). *P < 0.05; **P < 0.01. (f) Histological pictures (H&E stained) of representative inguinal lymph nodes in control (vehicle-treated) and EP4A-treated mice on day 12, including low power views in the inset. Control lymph node (L) is nearly completely replaced by tumor cells (T). Lymph node treated with EP4A is tumor-free. Scale bar = 50 μm. (g) Column graphs show percent of lymph nodes (LN) with metastasis in control (Con) and treated mice at days 8, 12, and 16.
Celecoxib and EP4A therapies abrogated metastasis to lymph nodes
Histopathological analysis of H&E stained sections of all lymph nodes collected from vehicle and drug-treated groups was used to compile the data presented in Figure1(g). Representative H&E images of day 12 lymph node sections of control and EPA-treated mice are shown on Figure1(f). As metastasis at the single cell or small cell cluster level was not identifiable reliably on histological grounds, many putative lymph nodes showing only tumor cells and no lymph node architecture were excluded from the analysis. Thus, the above incidence based on scoring 6–10 nodes in each group can be considered as minimal in all groups.
Celecoxib and EP4A therapies inhibited tumor-associated angiogenesis and lymphangiogenesis
Masson's trichrome staining of tumor sections adjacent to those used for fluorescence microscopy showed blood vessels containing red blood cells, most abundant in the peritumoral stroma (Fig.2a). High levels of angiogenesis (identified with CD31) and lymphangiogenesis (identified with LYVE-1 and PROX1 immunostaining) of “hot spots”, as shown in merged pictures, were evident in all vehicle-treated mice at all time points (days 8, 12, and 16) with very little temporal change (Fig.2a). Treatments with EP4A as well as celecoxib significantly blocked both events as early as day 8 (Fig.2b).
Fig 2.
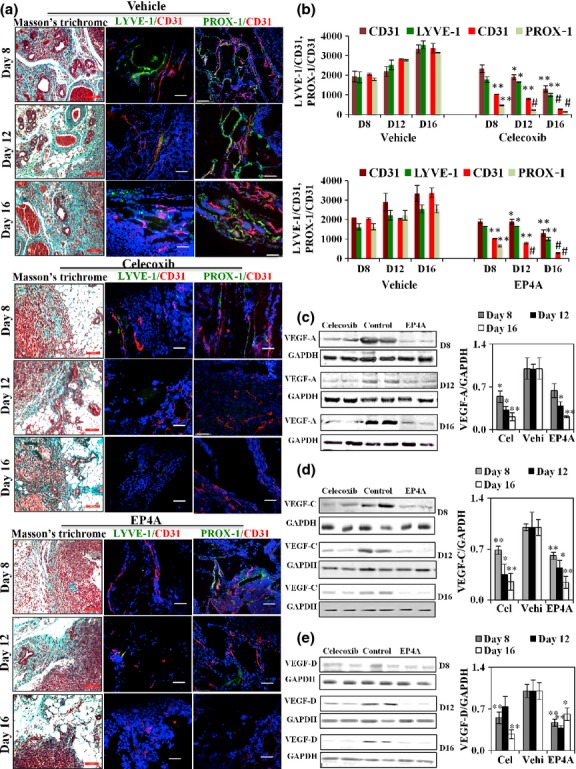
(a, b) Angiogenesis and lymphangiogenesis in a murine C3L5 breast cancer model. (a) Bright field images of Masson's trichrome stained sections of tumor showing peritumoral stroma inclusive of vessels (scale bar = 200 μm), and fluorescence images of corresponding tumors representing “hot spots” within the tumors. Tumor-associated angiogenesis (red, CD31 immuno-staining) and lymphangiogenesis (green, LYVE-1 and PROX1 immunostaining) are shown as representative fluorescent merged images. (b) Quantification of the hot spots of vascularity per unit area (n = 16, using the mean of three hot spots from each of the 16 tumors per group) ± SE. *P < 0.05; **P < 0.005; #P < 0.0005. Therapy with celecoxib and RQ-15986 (EP4A) reduced the levels of both angiogenesis and lymphangiogenesis. (c–e) Immunoblots for vascular endothletial growth factor (VEGF)-A/C/D. Tumor lysate proteins (n = 16, pooled from 16 tumors/group, triplicate measurements, shown in duplicate) were subjected to Western blots for VEGF-A (c), VEGF-C (d), VEGF-D (e), and GAPDH. Both celecoxib (Cel) and EP4A therapies reduced the levels of VEGF-A, -C, and -D proteins in residual tumors as revealed with densitometry, relative to GAPDH (normalized to vehicle (Vehi)-treated controls). Data represent mean ± SE. *P < 0.05; **P < 0.001. D, day.
Therapy with celecoxib and EP4A reduced levels of VEGF-C and -D proteins in residual tumors
As both EP4A and celecoxib significantly reduced angiogenesis and lymphangiogenesis, we examined the expression levels of VEGF-A/C/D proteins in residual tumors. Both therapies significantly reduced the levels of VEGF-A, -C, and -D proteins (Fig.2c–e, respectively) in residual tumors.
Celecoxib and EP4A treatments reduced the incidence of TAMs expressing EP4, VEGF-C/D in situ
Nearly every nucleated cell (DAPI stained) inclusive of tumor cells and macrophages (F4/80 stained) within tumors expressed EP4 (Fig.3a), as did C3L5 cells in culture (Fig.3b). As shown in Figure3(c,d), control tumors in vehicle-treated mice showed an appreciable incidence (25–30%) of F4/80 positive macrophages within the tumor parenchyma. Approximately 75–90% of these macrophages produced VEGF-C/D in situ in control mice, shown by dual immunostaining for F4/80 and VEGF-C or -D (Fig.3c,d). Residual tumors collected from treated mice revealed markedly reduced incidence of total as well as VEGF-C/D positive macrophages amongst nucleated cell population, indicating that the cyto-reductive effects of EP4A were higher on the macrophages than on tumor cells. There was no significant change in the incidence of VEGF-C or VEGF-D-bearing macrophages (75–90%) between control and treated mice. Macrophage cells (RAW 264.7) treated in vitro with COX-2 inhibitor and EP4A, showed significantly reduced VEGF-A/C/D production (Fig.3e,f).
Fig 3.
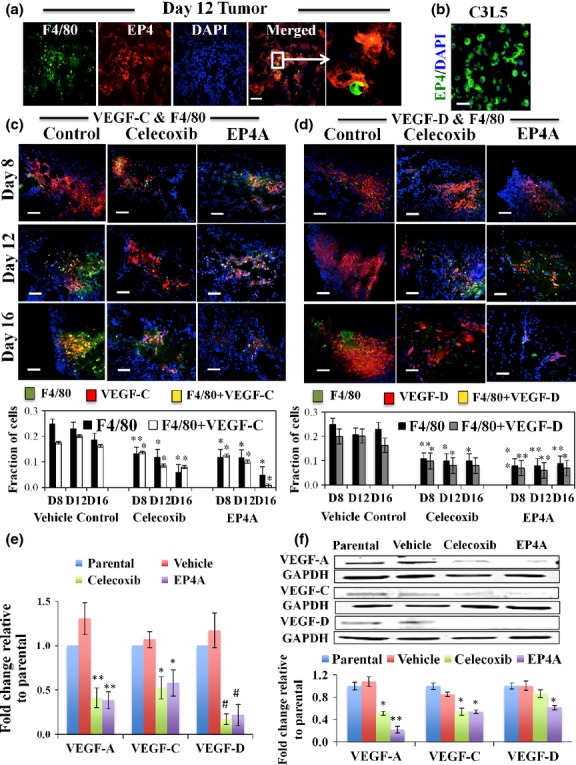
Tumor infiltrating macrophages express EP4 in a murine C3L5 breast cancer model. (a) Day 12 vehicle-treated tumor sections showing F4/80-positive cells (green), expressing EP4 (red) in single channels and yellow in merged channel. The inset is shown enlarged in the next panel. Every nucleated cell (nuclei stained blue with DAPI) inclusive of tumor cells was EP4-positive. (b) Cytoplasmic staining of EP4 in C3L5 cells in culture. (c, d) Treatments with celecoxib and RQ-15986 (EP4A) reduce the incidence of macrophages and vascular endothelial growth factor (VEGF)-C/D-producing macrophages. Merged immunofluorescence images of tumor sections in vehicle-treated control or drug (celecoxib or EP4A)-treated mice showing staining for F4/80 (green), (c) VEGF-C (red, left panel), or (d) VEGF-D (red, right panel) and nuclei (blue for DAPI). Double-stained cells appear yellow. As the vehicle controls for both drugs were similar, a single control is shown. Bottom charts showing incidence of macrophages expressing VEGF-C (bottom left) or VEGF-D (bottom right). Scale bar = 50 μm. (n = 16, using the mean of three hot spots from each of the 16 tumors per group) ± SE. *P < 0.005; **P < 0.0005. (e, f) Treatments with celecoxib and EP4A reduce the VEGF-A/C/D production of macrophages. RAW 264.7 cells treated for 24 h with celecoxib and EP4A significantly reduced VEGF-A/C/D production tested with (e) quantitative PCR and (f) Western blots. Data represents mean fold change (n = 3) ± SE. *P < 0.05; **P < 0.005; #P < 0.0005.
Therapy with celecoxib and EP4A reduced phosphorylated Akt and ERK in residual tumors, concomitant with an increase in apoptotic/proliferative cell ratios
As EP4, unlike EP2, is known to stimulate the PI3K/Akt and Ras/MAPK pathways,20 we examined the activation levels of both Akt and ERK in residual tumors in treated mice. Treatment with EP4A as well as celecoxib effectively reduced the levels of phosphorylation of both ERK and Akt measured with Western blots (Fig.4) and there was a concomitant increase in apoptotic/proliferative cell ratios (Fig. S2).
Fig 4.
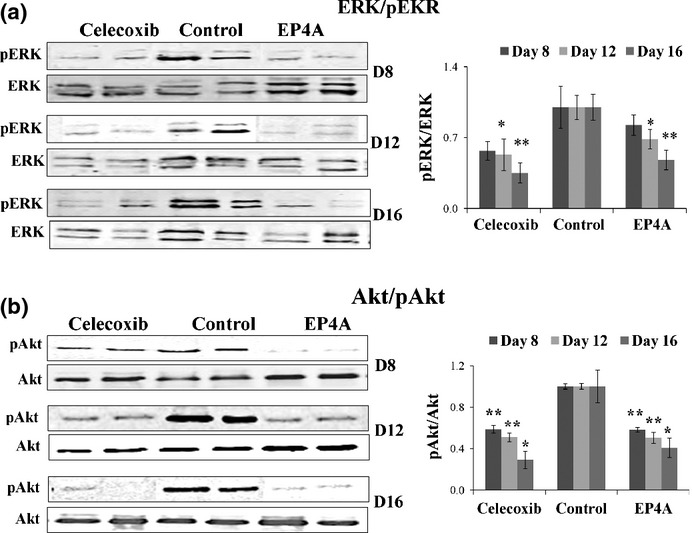
Therapy with celecoxib and RQ-15986 (EP4A) reduced the levels of phosphorylation (p) of both ERK and Akt proteins in residual breast tumors at all time intervals. Total tumor lysate proteins (pooled from 16 tumors per group and experiments run in triplicate, but only duplicate shown) were subjected to Western blot analysis for (a) pERK and total ERK or (b) pAkt and total Akt proteins. Densitometric measurements of phosphorylated versus total ERK (upper right) or Akt (bottom right) proteins relative to vehicle controls (mean of 16 ± SE). *P < 0.005; **P < 0.001.
Assessment of drug-related toxicity
We found no significant differences in daily water intake between vehicle-treated and drug-treated mice (data not presented). Furthermore, during autopsy on day 16, none of the mice revealed visible bleeding in the gastrointestinal tract, indicating that at the current dosage the drugs were non-toxic.
Stem-like cell-associated functions and phenotype of C3L5 cells in vitro and in vivo are dependent on COX-2 and EP4 activity
Tumorsphere forming ability was used as an in vitro surrogate of SLC function.39 C3L5 cells quickly formed spheroids for successive generations with the lowest dilution of 1 cell/well (Fig.5a). Tumorsphere formation (both size and number) in vitro was significantly reduced by treatment with celecoxib or EP4A at non-toxic concentrations for successive generations (Fig.5a) or by knockdown of COX-2 or EP4 in C3L5 cells (Fig.5b). Figure S3 shows COX-2 and EP4 mRNA levels after knockdown.
Fig 5.

(a) Treatment with celecoxib or RQ-15986 (EP4A) significantly blocked tumorsphere-forming ability of C3L5 breast cancer cells. With limiting cell dilutions (1000, 100, 10, or 1 cell/100 μL), C3L5 cells rapidly formed spheroids for successive generations (without overcrowding at 1 cell/well). Spheroid formation was inhibited with celecoxib (2 μM) and EP4A (2 μM) at successive passages. (b) Both spheroid forming efficiency (SFE) and sizes (representing spheroid perimeters), shown for serial passages, were significantly reduced with treatments. (c) Knockdown of COX-2 (COX-2-KD) (with shRNA plasmid) and EP4 (EP4-KD) (with siRNA) in C3L5 cells significantly reduced spheroid formation. (d) Quantitative measurement presented as numbers and sizes of spheroids showing that COX-2 knockdown can reduce spheroid formation for successive generations and with EP4 transient knockdown for one generation. Data represents mean of three experiments ± SE. *P < 0.005; **P < 0.001.
We tested co-expression of COX-2 and breast cancer stem cell markers CD44 and ALDH1A, ES cell markers SOX-2 and OCT-3/4, and hematopoietic stem cell marker β-catenin in frozen sections of both tumorspheres formed in vitro (Fig.6a) and day 12 tumors in vivo (Fig.6b). Knockdown of COX-2 resulted in a significant reduction of marker-bearing cells within the spheroids (image in Fig.6a, upper; quantitated in Fig.6a, bottom). Similarly, treatments of tumor-bearing mice with celecoxib and EP4A significantly reduced the incidence of marker-bearing cells in residual tumor sections, as compared to those in mock or vehicle-treated control mice (image in Fig.6b, upper; quantitated in Fig.6b, bottom). A dramatic reduction (to <20%) in spheroid forming efficiency as well as spheroid sizes after EP4 knockdown precluded immunostaining of spheroids for SLC or ES cell markers.
Fig 6.

Co-expression (Co-exp) of COX-2 and several stem cell markers in frozen sections of C3L5 breast carcinoma tumorspheres (parental, mock, COX-2 knockdown [KD]) in vitro and in day 12 tumors in control and treated (with celecoxib and RQ-15986 [EP4A]) mice. (a) COX-2 knockdown in C3L5 cells reduced the incidence of stem cell marker-bearing cells and cells co-expressing COX-2 and any of the stem cell markers in tumorspheres (image, top left; quantification, bottom left). Data represent mean (n = 3) ± SE. *P < 0.0001; #P < 0.004; †P < 0.05. (b) In vivo treatments with celecoxib (Cel) or EP4A also reduced the incidence of marker bearing cells in residual tumors, day 12 tumor sections (image, top right; quantification, bottom right). Data presented as (n = 16, using the mean of three hot spots from each of the 16 tumors per group) ± SE. ALDH, aldehyde dehydrogenase; Con, control.
Discussion
In earlier studies, using high COX-2-expressing C3L5 mammary adenocarcinoma cells we showed that primarily EP4, and to a minor extent EP1, activity promoted cellular migration.7 Furthermore, a COX-2 inhibitor celecoxib and an EP4 antagonist ONO AE3-208, but not an EP1 antagonist ONO-8713, reduced VEGF-C and -D production by the tumor cells in vitro, and inhibited tumor growth, tumor-associated angiogenesis, and lymphangiogenesis, as well as metastasis to the lungs and lymph nodes in vivo.10 In that study, residual tumors in treated mice showed reduced VEGF-C and -D proteins, Akt phosphorylation, and increased apoptotic/proliferative cell ratios. The present study confirmed all these results in the same tumor model with another EP4 antagonist, RQ-15986. In addition, we show here that: (i) tumorsphere formation and growth were reduced with a COX-2 inhibitor, COX-2 or EP4 knockdown, and also with EP4A, indicating their dependence on COX-2 and EP4 expression or activity; (ii) macrophages produced VEGF-A/C/D in vitro; (iii) tumor-infiltrating macrophages expressed EP4 and further contributed to VEGF-C and -D production in situ in promoting lymphangiogenesis; (iv) VEGF-C/D production in situ was curtailed with EP4A; and (iv) there was reduced ERK phosphorylation in the treated residual tumors. Taken together, these findings show the key role of EP4 receptor on tumor cells as well as TAMs in multiple cellular events required for tumor progression and metastasis, which can be effectively blocked with currently available EP4 antagonists.
Among four EP receptors, EP2 and EP4 are both Gs linked and thus their activation leads to an increase in intracellular cAMP followed by activation of PKA.40 In addition, EP4 activation can also stimulate ERK41 and PI3K/Akt42 pathways, promoting PGE-2 dependent migration43 and cell survival, respectively.42 In the present study, residual tumors in mice treated with celecoxib or EP4A showed a parallel reduction of Akt and ERK phosphorylation, concomitant with an increase in apoptotic/proliferative cell ratios. Our in vitro studies with C3L5 cells (Xin X and Lala PK, 2014, unpublsihed data) have validated the need for PI3K/Akt for cell survival and ERK1/2 for migratory functions. Taken together, the present results indicate that a blockade in the above EP4 signaling pathways contributed to antitumor and antimetastatic effects of the drug. However, a contribution of PKA, shared by EP2/EP4 cannot be excluded.
Prostaglandin E2 produced by tumor cells has been shown to contribute to the immunosuppressive function of TAMs. For example, tumor-derived PGE-2 was shown to inhibit the production of T-cell stimulating chemokine CCL5 by LPS-activated macrophages, which could be restored with COX-2 inhibitor or EP2/EP4 antagonists.44 Peritumoral lymphangiogenesis is a well-known phenomenon in human breast cancer that promotes lymphatic invasion and metastasis.13,34
In the present study, we show that TAMs expressed EP4 and produced VEGF-C/D. Furthermore, therapy with EP4A reduced the incidence of VEGF-C/D-producing macrophages in situ. This could be an indirect effect of the blockade of PGE-2 production by EP4-bearing tumor cells, leading to reduced migration of VEGF-C/D producing macrophages into the tumor site, or a direct effect on macrophages to block VEGF-C/D production by these cells, or both. As there were no major differences in VEGF-C or -D expression (75–90%) of macrophages between vehicle-treated versus drug-treated tumors, we could not clearly distinguish between these possibilities. However, as previously reported,33,34 our in vitro results revealed that macrophages produced VEGF-A/C/D, and their production was inhibited with both celecoxib and EP4A. Thus, reduced macrophage migration as well as VEGF-C/D production by macrophages in situ in EP4A-treated mice would contribute to reduced lymphangiogenesis. We have further evidence for EP4 activity on lymphatic endothelial cells contributing to tumor-induced lymphangiogenesis.45 These findings, taken together, may explain the role of host EP4 in metastasis promotion.46
Stem-like cells constitute a minor subpopulation of tumor cells, which can perpetuate tumors and escape traditional therapies.24,25 The tumorsphere assay is an in vitro surrogate of SLC function in vivo.39 We show here that C3L5 cells formed tumorspheres at the single cell level for successive generations, and both COX-2 inhibitor and EP4A inhibited this ability. Similarly, COX-2 and EP4 knockdown produced profound inhibitory effects. Numerous SLC-associated markers such as CD4447 and ALDH148 have been reported in breast cancer cells. OCT-4 was implicated as an SLC marker within MCF-7 breast cancer cell-derived mammospheres.49 Embryonic stem cell markers OCT-3/4 and SOX-250 and hematopoietic stem cell marker β-catenin have also been reported to be associated with SLCs in cancer cells.51 Although COX-2 is not considered as an SLC marker, we found co-expression of CD44, ALDH, β- catenin, OCT 3/4, and SOX-2 in a subset of COX-2-positive cells within the tumorspheres as well in tumors in vivo, reinforcing their SLC phenotype. Although very little is known about the molecular pathways responsible for SLC induction, it is likely that multiple pathways can converge on the phenotypes that represent SLCs. Tumor-associated macrophages can contribute to an SLC-stimulating tumor microenvironment52 and conversely, SLCs can recruit myeloid-derived tumor-promoting TAMs.53 We suggest that COX-2 and the PGE-2/EP4 signaling axis are partners in SLC induction in breast cancer. This suggestion is supported by: (i) a reduction in tumorsphere formation with COX-2 inhibition or EP4 antagonism or gene knockdown of COX2/EP4 in vitro; and (ii) a reduction of the marker-bearing cells in residual tumorspheres after COX-2 knockdown in vitro or residual tumors in situ in EP4A-treated mice. A recent report on the role of EP4 in promoting the SLC phenotype in breast cancer cells concurs with our results.27
Long-term use of COX-2 inhibitors at higher doses has raised concerns about increased thrombo-embolic incidents.16,17 By IP gene disruption, combined with the use of IP agonists, IP receptors interacting with PGI-2 were shown to be obligatory for the vaso-protective roles of COX-2.18 Since EP4 antagonists do not inhibit PGI production, they should be safer than COX-2 inhibitors, a view that can only be tested with human trials. As shown earlier for ONO-AE3-208,10 presently used EP4A was also found to be non-toxic in mice at the current dosage regimen. Currently, unpublished results of a closely-related, well-characterized EP4 antagonist AAT-007 (CJ-023423),54 used in several phase1/II human trials on osteoarthritis patients (n > 800) have validated its safety and tolerance at therapeutic doses (Dr Yukinori Take, AskAt Inc./RaQualia Pharma Inc., Japan, personal communication presented with permission). Based on the evidence of multiple roles of EP4 on tumor and host cell-mediated mechanisms in breast cancer progression, we suggest that testing the use of EP4 antagonists as an adjuvant in human breast cancer is timely.
Acknowledgments
We thank Dr. Yukinori Take (AskAt Inc./RaQualia Pharma Inc., Japan) for donating EP4A RQ-15986 and Claire Kwon, Jose Torres-Garcia, Aya Abdou, and Qian Xing for technical help. This study was supported by grants from the Ontario Institute of Cancer Research and Canadian Breast Cancer Foundation, Ontario Chapter (to PKL) and Translational Breast Cancer Research Unit and CIHR Strategic Training Program in Cancer Research and Technology Transfer (CaRTT) postdoctoral fellowships (to MM).
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. We tested the effect of different COX-2 inhibitors (indomethacin, NS-398) and EP4 antagonists (CJ-0427994, GW627368X) on proliferation and migration of C3L5 cells.
Fig. S2. Therapy with RQ-15986 (EP4A) caused a reduction in proliferative cells and an increase in apoptotic cells.
Fig S3. Cyclooxygenase-2 and EP4 knockdown.
References
- Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA. 1997;94:3336–40. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soslow RA, Dannenberg AJ, Rush D, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000;89:2637–45. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Parrett ML, Harris RE, Jorder FA, Ross MS, Clausen KP, Robertson FK. Cyclooxygenase-2 gene expression in human breast cancer. Int J Oncol. 1997;10:503–7. doi: 10.3892/ijo.10.3.503. [DOI] [PubMed] [Google Scholar]
- Ristimäki A, Sivula A, Lundin J, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62:632–5. [PubMed] [Google Scholar]
- Lala PK, Parhar RS, Singh P. Indomethacin therapy abrogates the prostaglandin-mediated suppression of natural killer activity in tumor-bearing mice and prevents tumor metastasis. Cell Immunol. 1986;99:108–18. doi: 10.1016/0008-8749(86)90220-0. [DOI] [PubMed] [Google Scholar]
- Rozic JG, Chakraborty C, Lala PK. Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int J Cancer. 2001;93:497–506. doi: 10.1002/ijc.1376. [DOI] [PubMed] [Google Scholar]
- Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res. 2003;289:265–74. doi: 10.1016/s0014-4827(03)00269-6. [DOI] [PubMed] [Google Scholar]
- Timoshenko AV, Lala PK, Chakraborty C. PGE2-mediated upregulation of iNOS in murine breast cancer cells through the activation of EP4 receptors. Int J Cancer. 2004;108:384–9. doi: 10.1002/ijc.11575. [DOI] [PubMed] [Google Scholar]
- Chang SH, Liu CH, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA. 2004;101:591–6. doi: 10.1073/pnas.2535911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin X, Majumder M, Girish GV, Lala PK. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab Invest. 2012;92:1115–28. doi: 10.1038/labinvest.2012.90. [DOI] [PubMed] [Google Scholar]
- Timoshenko AV, Chakraborty C, Wagner GF, Lala PK. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br J Cancer. 2006;94:1154–63. doi: 10.1038/sj.bjc.6603067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iterson V, Leidenius M, von Smitten K, Bono P, Heikkilä P. VEGF-D in association with VEGFR-3 promotes nodal metastasis in human invasive lobular breast cancer. Am J Clin Pathol. 2007;128:759–66. doi: 10.1309/7FXVRMXF58PVRJUH. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee RN, Timoshenko AV, Cai J, Lala PK. Relationship between cyclooxygenase-2 and human epidermal growth factor receptor 2 in vascular endothelial growth factor C up-regulation and lymphangiogenesis in human breast cancer. Cancer Sci. 2010;101:2026–32. doi: 10.1111/j.1349-7006.2010.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M, Tutunea-Fatan E, Xin X, et al. Co-expression of α9β1 integrin and VEGF-D confers lymphatic metastatic ability to a human breast cancer cell line MDA-MB-468LN. PLoS One. 2012;7:e35094. doi: 10.1371/journal.pone.0035094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoshenko AV, Rastogi S, Lala PK. Migration-promoting role of VEGF-C and VEGF- C binding receptors in human breast cancer cells. Br J Cancer. 2007;97:1090–8. doi: 10.1038/sj.bjc.6603993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;35:1709–11. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- Graham DJ. COX-2 inhibitors, other NSAIDs, and cardiovascular risk: the seduction of common sense. JAMA. 2006;296:1653–6. doi: 10.1001/jama.296.13.jed60058. [DOI] [PubMed] [Google Scholar]
- Guo Y, Tukaye DN, Wu WJ, et al. The COX-2/PGI2 receptor axis plays an obligatory role in mediating the cardio protection conferred by the late phase of ischemic preconditioning. PLoS One. 2012;7(7):e41178. doi: 10.1371/journal.pone.0041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–90. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by ep4, but not ep2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem. 2003;278:12151–6. doi: 10.1074/jbc.M212665200. [DOI] [PubMed] [Google Scholar]
- Ma X, Holt D, Kundu N, et al. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology. 2013;2:e22647. doi: 10.4161/onci.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Kundu N, Collin PD, Goloubeva O, Fulton AM. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res Treat. 2012;132:1001–8. doi: 10.1007/s10549-011-1675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- Tysnes BB. Tumor-initiating and -propagating cells: cells that we would like to identify and control. Neoplasia. 2010;12:506–15. doi: 10.1593/neo.10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North TE, Goessling W, Walkley CR, et al. Prostaglandin E2 regulates vertebrate hematopoietic stem cell homeostasis. Nature. 2007;447:1007–11. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima YM, Arai F, Hosokawa K, et al. Prostaglandin E2 regulates murine hematopoietic stem/progenitor cells directly via EP4 receptor and indirectly through mesenchymal progenitor cells. Blood. 2013;121:1995–2007. doi: 10.1182/blood-2012-06-437889. [DOI] [PubMed] [Google Scholar]
- Kundu N, Ma X, Kochel T, et al. Prostaglandin E receptor EP4 is a therapeutic target in breast cancer cells with stem-like properties. Breast Cancer Res Treat. 2014;143:19–31. doi: 10.1007/s10549-013-2779-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Tumor-educated macrophages promote tumor progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- Medrek C, Pontén F, Jirström K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012;12:306. doi: 10.1186/1471-2407-12-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmey JH, Dimitriadis E, Kay E, Redmond HP, Bouchier-Hayes D. Regulation of macrophage production of vascular endothelial growth factor (VEGF) by hypoxia and transforming growth factor beta-1. Ann Surg Oncol. 1998;5:271–8. doi: 10.1007/BF02303785. [DOI] [PubMed] [Google Scholar]
- Schoppmann SF, Birner P, Stöckl J, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002;16:947–56. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lala PK, Parhar RS. Eradication of spontaneous and experimental adenocarcinoma metastases with chronic indomethacin and intermittent IL-2 therapy. Int J Cancer. 1993;54:677–84. doi: 10.1002/ijc.2910540425. [DOI] [PubMed] [Google Scholar]
- Murase A, Taniguchi Y, Tonai-Kachi H, Nakao K, Takada J. In vitro pharmacological characterization of CJ-042794, a novel, potent, and selective prostaglandin EP (4) receptor antagonist. Life Sci. 2008;82:226–32. doi: 10.1016/j.lfs.2007.11.002. (Jan 16); [DOI] [PubMed] [Google Scholar]
- Murase A, Okumura T, Sakakibara A, Tonai-Kachi H, Nakao K, Takada J. Effect of prostanoid EP4 receptor antagonist, CJ-042, 794, in rat models of pain and inflammation. Eur J Pharmacol. 2008;580:116–21. doi: 10.1016/j.ejphar.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Trifan OC, Durham WF, Salazar VS, et al. Cyclooxygenase-2 inhibition with celecoxib enhances antitumor efficacy and reduces diarrhea side effect of CPT-11. Cancer Res. 2002;62:5778–84. [PubMed] [Google Scholar]
- Majumder M, Xin X, Lala PK. A practical and sensitive method of quantitating lymphangiogenesis in vivo. Lab Invest. 2013;93:779–91. doi: 10.1038/labinvest.2013.72. [DOI] [PubMed] [Google Scholar]
- Morrison BJ, Steel JC, Morris JC. Sphere culture of murine lung cancer cell lines are enriched with cancer initiating cells. PLoS One. 2012;7(11):e49752. doi: 10.1371/journal.pone.0049752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Pozzi A, Yan X, Macias-Perez I, Wei S, Hata AN, Breyer RM. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem. 2004;279:29797–804. doi: 10.1074/jbc.M313989200. [DOI] [PubMed] [Google Scholar]
- George RJ, Sturmoski MA, Anant S, Houchen CW. EP4 mediates PGE2 dependent cell survival through the PI3 kinase/AKT pathway. Prostaglandins Other Lipid Mediat. 2007;83:112–20. doi: 10.1016/j.prostaglandins.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res. 2010;8:569–77. doi: 10.1158/1541-7786.MCR-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Zhang J, Liu J. Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway. J Biol Chem. 2011;286:2111–20. doi: 10.1074/jbc.M110.154971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lala PK, Girish GV, Xin X, Majumder M, Tutunea-Fatan E, Nandi P. Breast cancer-associated lymphangiogenesis: Roles of PGE2 and EP4 receptor on lymphatic endothelial cells. Proc Amer Assoc Cancer Res. 2014;55 doi: 10.1186/s12885-016-3018-2. ; Late breaking abstract LB67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Huang Y, Porta R, et al. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006;66:9665–72. doi: 10.1158/0008-5472.CAN-06-1271. [DOI] [PubMed] [Google Scholar]
- Al HM, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Cook KR, Vincent L, Hall CS, Martin C, Lucci A. Role of COX-2 in tumorospheres derived from a breast cancer cell line. J Surg Res. 2011;168:e39–49. doi: 10.1016/j.jss.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Li S, Peng B, Ye Y, Deng X, Yao K. Embryonic stem cells markers SOX2, OCT4 and Nanog expression and their correlations with epithelial-mesenchymal transition in nasopharyngeal carcinoma. PLoS One. 2013;8:e56324. doi: 10.1371/journal.pone.0056324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley SJ, Gheordunescu E, Kakarala P, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–9. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinushi M, Chiba S, Yoshiyama H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci USA. 2011;108:12425–30. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashina T, Baghdadi M, Yoneda A, et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 2014;74:2698–709. doi: 10.1158/0008-5472.CAN-13-2169. [DOI] [PubMed] [Google Scholar]
- Nakao K, Murase A, Ohshiro H, et al. CJ-023, 423, a novel, potent and selective prostaglandin EP4 receptor antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2007;322:686–94. doi: 10.1124/jpet.107.122010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. We tested the effect of different COX-2 inhibitors (indomethacin, NS-398) and EP4 antagonists (CJ-0427994, GW627368X) on proliferation and migration of C3L5 cells.
Fig. S2. Therapy with RQ-15986 (EP4A) caused a reduction in proliferative cells and an increase in apoptotic cells.
Fig S3. Cyclooxygenase-2 and EP4 knockdown.