

Abstract
Synovial sarcoma (SS) is a malignant soft-tissue tumor characterized by the recurrent chromosomal translocation SS18–SSX. Vascular endothelial growth factor (VEGF)-targeting anti-angiogenic therapy has been approved for soft-tissue sarcoma, including SS; however, the mechanism underlying the VEGF signal for sarcomagenesis in SS is unclear. Here, we show that SS18–SSX directs the VEGF signal outcome to cellular growth from differentiation. Synovial sarcoma cells secrete large amounts of VEGF under spheroid culture conditions in autocrine fashion. SS18–SSX knockdown altered the VEGF signaling outcome, from proliferation to tubular differentiation, without affecting VEGF secretion, suggesting that VEGF signaling promoted cell growth in the presence of SS18–SSX. Thus, VEGF inhibitors blocked both host angiogenesis and spheroid growth. Simultaneous treatment with VEGF and chemokine (C-X-C motif) (CXC) ligand 12 and CXC receptor 4 inhibitors and/or ifosfamide effectively suppressed tumor growth both in vitro and in vivo. SS18–SSX directs the VEGF signal outcome from endothelial differentiation to spheroid growth, and VEGF and CXC receptor 4 are critical therapeutic targets for SS.
Keywords: CXCR4, spheroid, SS18–SSX, synovial sarcoma, vascular endothelial growth factor
Synovial sarcoma (SS) is a malignant soft-tissue tumor characterized by a recurrent chromosomal translocation [t(X;18)(p11;q11)], which results in the formation of a pathogenic fusion protein, SS18–SSX.1–3 Comprising 8–10% all soft-tissue sarcomas, SS is commonly found in adolescents and young adults.4,5 To date, the standard treatment for SS has been wide surgical resection with high-dose neo-adjuvant chemotherapy, including ifosfamide (IFM) and/or radiotherapy. However, SS is frequently resistant to these therapies, resulting in disease recurrence followed by lung metastasis. The 5-year overall survival rate for patients with SS is between 68% and 81%; however, this rate in patients with lung metastasis at the initial visit is only 0–10%.4,5 Therefore, comprehensive understanding of the mechanism underlying SS sarcomagenesis is urgently required in order to improve patient prognosis.
The molecular mechanism underlying the sarcomagenesis mediated by the SS18–SSX fusion protein is partially understood. We have previously shown that SS cells express stem cell markers, such as OCT3/4, NANOG, and SOX2, and that the SS18–SSX fusion gene acts by dysregulation of cellular self-renewal and differentiation capacities.6 Garcia et al.7 reported that nuclear function of SS18–SSX and differentiation are required for sarcoma development and its maintenance beyond the initial growth phase. Su et al.8 showed that, epigenetically, SS18–SSX plays a critical role in oncogenesis by forming complexes with transducin-like enhancer of split 1 and activating transcription factor 2. Most recently, Kadoch and Crabtree showed that SS18–SSX formed complexes with mSWI/SNF (BAF).9 Upon SS18–SSX binding, the BAF complex undergoes a conformational change and binds to the SOX2 locus, thereby reversing polycomb-mediated repression and resulting in SOX2 activation.9
Various 3-D culture methods using normal and tumor cells have been regarded as an essential approach for eliciting the physiological properties of the cells by mimicking their in vivo state more accurately than can be achieved using conventional 2-D monolayer cultures.10–14 Recently, Chen et al.15 showed that 3-D culture using collagen scaffolds promoted secretion of vascular endothelial growth factor (VEGF) in MCF-7 cells. In normal healthy cells, under physiological conditions, VEGF is generally thought to play essential roles in vasculogenesis, angiogenesis, and vascular permeability.16–18 In tumor cells, VEGF functions in angiogenesis, cell proliferation, survival, invasion, and migration 19 in a paracrine and/or autocrine manner.20–22 Malignant tumors expressing clinically high levels of VEGF have been associated with poor prognosis.23 Therefore, VEGF has long been identified as an appropriate therapeutic target for several malignant tumors.23,24
In this study, we showed that human VEGF-A and VEGF receptor 2 (VEGFR2) were detected in the blood of nude mouse xenografting SS cells and clinical specimens. Therefore, we hypothesized that VEGF is a putative therapeutic target for SS because VEGF promoted cell growth in an autocrine manner. We also investigated the mechanism by which SS18–SSX is involved in the promotion of cell growth by the VEGF signal in SS.
Materials and Methods
Described in Data S1 (Supporting information).
Results
Vascular endothelial growth factor autocrine loop is enhanced upon spheroid growth of SS
We observed high expression levels of human VEGF-A in over 90% of clinical specimens from patients with SS (Figs1a,S1a). In addition, s.c. transplanted tumors of Yamato-SS, Aska-SS, and Suzak-SS cell lines showed aggressive angiogenesis, and elevated concentrations of human VEGF-A (Table S1) were found in the blood of mice bearing each of these SS cells. These results suggested that these SS cell lines secreted high levels of VEGF-A.
Fig 1.

Vascular endothelial growth factor (VEGF) autocrine loop is enhanced upon spheroid growth of synovial sarcoma (SS). (a) Immunohistochemical analysis of SS clinical specimen with VEGF-A. Nuclei were counterstained with hematoxylin. Scale bar = 100 μm. (b) Quantity of VEGF-A secreted by Yamato-SS under 2-D or spheroid (3-D) culture conditions, as determined by ELISA. Culture medium was collected every 24 h for 4 days (left panel, n = 3; *P < 0.005). Quantification of VEGFA mRNA of Yamato-SS by quantiative RT-PCR under 2-D or spheroid culture conditions from day 1 to day 7 (right panel, n = 3; *P < 0.05). A.U., arbitrary unit (the same l applies hereafter). (c) Immunoblot analysis of hypoxia-inducible factor-1α (HIF-1α) in Yamato-SS cells cultured under 2-D or spheroid culture conditions for 3 days. (d) Immunoblot analysis of VEGF receptor 2 (VEGFR2) in Yamato-SS cells cultured under 2-D or spheroid culture conditions, from day 1 to day 7. (e) Yamato-SS cells were cultured under 2-D conditions with or without recombinant human VEGF-A (rhVEGF-A; 10 ng/mL) or spheroid conditions for 5 days, followed by immunoprecipitation (IP) with anti-VEGFR2 and blotting for p-tyrosine (p-Tyr) and VEGFR2. (f) Immunofluorescent staining of Yamato-SS spheroids with proliferation marker proliferating cell nuclear antigen (PCNA; top panel), VEGF-A (middle panel), and VEGFR2 (bottom panel). Nuclei were counterstained with Hoechst33342 (blue). Scale bar = 100 μm. (g) Dose-dependence response to bevacizumab (Bev) treatment for 2 weeks, as seen with a soft agar assay. Colonies >200 μm are shown (n = 3; *P < 0.05, **P < 0.005). (h) Rescue experiments, using exogenously added rhVEGF-A (10 ng/mL), in Yamato-SS cells treated with Bev. Colonies >200 μm are shown (n = 3; *P < 0.005, **P < 0.05). M.W., molecular weight.
First, VEGF-A secretion and VEGFR2 expression were measured under conventional 2-D and spheroid culture conditions; the latter has previously been reported to better mimic tumor characteristics in vivo.10,15 Although only minimal VEGF-A was secreted in SS cell lines under 2-D culture conditions, the levels of VEGF-A showed a 51.0–717.2-fold increase in Yamato-SS (Fig.1b, left panel) and a 1.8–2.1-fold increase in Aska-SS (Fig. S2a) under spheroid culture conditions. Hypoxia-inducible factor-1α (HIF-1α) expression was upregulated in both SS cell lines under spheroid culture conditions (Fig.1c). However, there was no difference in HIF-1α expression between 3-day spheroid cultures of Yamato-SS and Aska-SS (Fig.1c). Thus, we speculated that differential VEGF expression noted between the two types of SS cells was not only due to differences in HIF-1α activity, but also due to other signals or factors. Although the reason for the different VEGF expression levels between the two SS cell lines was unclear, two possibilities are: (i) difference in origin or differentiation phase; and (ii) bias during establishing cell lines. Additionally, the increase in VEGF-A secretion correlated with spheroid growth (Fig.1b, left panel; Fig. S2a). Furthermore, VEGFA transcription levels also increased by 3.3–40.1-fold (Fig.1b, right panel). In addition, we observed higher expression levels of VEGF-A and VEGFR2 in clinical samples compared to these expressions in Yamato-SS cells under spheroid culture conditions (Fig. S1b).
The expression of VEGFR2 was then examined by immunoblot analysis. Three glycosylated protein bands were observed, corresponding to the predicted form (147 kDa), the immature form (200 kDa), and the mature form (230 kDa), which was glycosylated in two steps after translation 25 (Figs1d,S10a). Levels of all three VEGFR2 protein forms were elevated from day 1 to day 7 under spheroid culture, but not under 2-D culture, conditions. The immature and predicted VEGFR2 bands for day 7 of the 2-D culture were missing after receptor desensitization (Fig.1d). Tyrosine phosphorylation levels of VEGFR2 were upregulated in spheroid cultures compared to 2-D cultures (Fig.1e). These data suggested that the VEGF autocrine loop was enhanced under spheroid culture conditions. We observed that the inner region of spheroid was hypoxic (data not shown). It is known that cells in the region under the hypoxic state are not often proliferative.26 Consistent with that, although VEGF-A and VEGFR2 signals were observed in the surface region of the Yamato-SS spheroid, proliferative activity was observed only at a depth from the surface of approximately 0–100 μm (Fig.1f). Thus it was thought that proliferation activity of the cells located in the inner spheroid region was suppressed by hypoxia in spite of the existence of VEGF signaling. We also speculated that VEGFR2 expression was not upregulated only under hypoxic conditions in cancer cells, but also due to other signals or factors, including cell morphology or cell–cell contact.
The VEGF autocrine loop has been implicated in cell proliferation, migration, and stemness in normal and cancer cells.20–22 Subsequently, we investigated whether blocking the VEGF autocrine loop could suppress cell proliferation in the presence of either of two drugs, bevacizumab (Bev),27 a humanized anti-VEGF antibody, and pazopanib (Pazo),28,29 a VEGFR2-specific tyrosine kinase inhibitor. Neither drug inhibited proliferation of SS cells under conventional 2-D culture conditions (Fig. S2b,c).
To confirm that cell proliferation was blocked under spheroid culture conditions, the effect of Bev and Pazo on colony formation was examined using a soft agar assay. In Yamato-SS, both drugs inhibited colony formation, by 46.8–60.3% in the presence of Bev (Figs1g,S2d, left panel), and by 15.1–64.5% in the presence of Pazo (Fig. S2d, right panel; Fig. S2e). Similar results were obtained in Aska-SS; colony formation was inhibited by 40.4–53.9% in the presence of Bev (Fig. S2f, left panel) and by 6.5–63.4% in the presence of Pazo (Fig. S2f, right panel).
The inhibition of colony formation was not fully rescued by exogenous addition of VEGF-A by 21.2% in the presence of Bev (Fig.1h) and by 7.0% in the presence of Pazo (Fig. S2 g). These data suggested that the VEGF autocrine loop is required for colony formation in SS.
Taken together, these data suggested that the VEGF autocrine loop is involved in the surface growth of SS spheroids, and that VEGF inhibition had antitumor efficacy, at least in part, by inhibiting the VEGF autocrine loop.
Knockdown of the SS18–SSX fusion gene suppresses cell proliferation and induces endothelial differentiation
To determine the link between SS18–SSX and VEGF signaling, spheroid growth under SS18–SSX knockdown conditions was examined in Yamato-SS and Aska-SS. We have previously reported that SS cells display marked shape changes, from spherical to adherent, upon SS18–SSX knockdown.30 As cell morphological changes affect the protein and mRNA expression levels of VEGF-A and VEGFR2 (Figs1b,S2a), expression of the VEGF signal was examined under spheroid culture conditions with siRNA knockdown of SS18–SSX (Figs2a,S3a,S10b). SS18–SSX silencing did not affect protein and mRNA expression levels of VEGF-A or VEGFR2 (Figs2b,c,S3b).
Fig 2.

Knockdown of the SS18–SSX fusion gene suppresses cell proliferation and induces endothelial differentiation in synovial sarcoma (SS) cells. (a) Confirmation of SS18–SSX fusion gene knockdown by siRNA-A and siRNA-B with immunoblot analysis. Closed or open arrow heads represents SS18–SSX or SS18, respectively. M.W., molecular weight. (b) Amount of vascular endothelial growth factor-A (VEGF-A) secreted by Yamato-SS cells that had been treated with SS18–SSX siRNA under spheroid culture conditions, as determined by ELISA (n = 3). (c) VEGFA and VEGFR2 mRNA levels in Yamato-SS cells treated with SS18–SSX siRNA as determined by quantitative RT-PCR (n = 3). A.U., arbitrary unit (the same l applies hereafter). (d) Cell proliferation of Yamato-SS cells cultured in a tube formation assay treated with bevacizumab (Bev; 5 μg/mL) (left) or pazopanib (Pazo; 250 nM) (right) (n = 3; *P < 0.005, **P < 0.05). (e,f) Immunofluorescence analysis of CD31 (e), and von Willebrand factor (vWF) (f). Nuclei were counterstained with Hoechst33342 (blue). White arrowheads identify cells expressing CD31 or vWF, and phase contrast images of tube formation. Scale bar = 100 μm. (g) Quantitation of tubular formation assay in Yamato-SS cells treated with SS18–SSX siRNA, in the presence or absence of treatment with Bev (5 μg/mL, left) and Pazo (250 nM, right) (n = 2; *P < 0.005).
We have previously reported that SS cells possess a differentiation potential for mesenchymal lineages to develop into osteocytes and chondrocytes, and that SS cells display a much broader potential for differentiation into adiposites and macrophages upon SS18–SSX knockdown.6 Thus, we speculated that with SS18–SSX knockdown, the VEGF signal in SS cells induces endothelial differentiation. With the tube formation assay, cell proliferation was significantly inhibited in control samples (Bev, 43.9%; Pazo, 23.7%; Fig.2d) in Yamato-SS but not in Aska-SS (Fig. S3c). To confirm endothelial differentiation, the presence of the endothelial cell markers CD31 and von Willebrand factor was subsequently examined by immunofluorescence analysis. The expression of both proteins was detected under SS18–SSX knockdown conditions, but not under control siRNA conditions (Figs2e,f, S3d,e). SS18–SSX knockdown resulted in the promotion of tube formation (a 5.7–6.2-fold increase in the presence of Bev, or a 4.3–4.7-fold increase in the presence of Pazo), whereas the promoted tube formation was inhibited by treatment with Bev (53.1–59.7%; Fig.2g, left) and Pazo (58.3–59.7%; Fig.2g, right) with SS18–SSX knockdown in Yamato-SS. Similar results were obtained in Aska-SS; SS18–SSX knockdown resulted in the promotion of tube formation (a 3.8–3.9-fold increase in the presence of Bev, or a 2.3–3.8-fold increase in the presence of Pazo). This enhanced tube formation was inhibited by treatment with Bev (51.3–51.5%; Fig. S3f, upper) and Pazo (26.1–40.8%; Fig. S3f, lower) in the presence of SS18–SSX knockdown. Taken together, our results revealed that the SS18–SSX-generated signal was independent of the VEGF-dependent signal in tube endothelial differentiation, and that SS18–SSX indirectly deflected VEGF action in SS cells.
Vascular endothelial growth factor-targeted therapy markedly suppresses tumor growth and angiogenesis in SS
As VEGF signal inhibition can block Yamato-SS cell proliferation and endothelial differentiation of SS cells, VEGF is a putative therapeutic target in SS, and VEGF-targeted therapy has already been validated as anti-angiogenic therapy for malignant tumors.31–33. Here, first, to confirm whether Bev possesses antitumor efficacy against SS in vivo, mice bearing Yamato-SS (Fig.3a, left) and Aska-SS (Fig. S4a) were treated with Bev. Tumors treated with Bev displayed a pale color, suggesting that Bev inhibited host angiogenesis (Fig.3a, right panel; Fig. S4a). Treatment with Bev markedly suppressed tumor growth in Yamato-SS (by 89.4%; Fig.3b) and in Aska-SS (by 70.4% of tumor volume and 80.7% of tumor weight, respectively; Fig. S4b,c).
Fig 3.

Vascular endothelial growth factor (VEGF)-targeted therapy significantly suppresses tumor growth and angiogenesis of synovial sarcoma (SS). (a) Mice bearing Yamato-SS were treated with bevacizumab (Bev) at a concentration of 100 μg/mouse, twice a week (n = 10). Control mice were treated with PBS (n = 8). Images show mice and tumors after 5 weeks of treatment. M.W., molecular weight. Scale bar = 1 cm. (b) Tumor volume was measured weekly (left; *P < 0.005). (c) Immunofluorescence analysis of tumor sections from (a) using anti-VEGF-A antibody (middle upper panel, red) and anti-VEGF receptor 2 (VEGFR2) antibody (middle lower panel, green). Nuclei were counterstained with Hoechst33342. Scale bar = 100 μm. (d) Quantification of human VEGFA (left panel) or human VEGFR2 (right panel) mRNA of Yamato-SS cells by quantitative RT-PCR under spheroid culture conditions for 7 days (n = 3) or tumor sections from mice bearing Yamato-SS, at 3 weeks (w) (n = 4). A.U., arbitrary unit (the same l applies hereafter). (e) Immunohistochemical analysis of tumor sections with mouse-specific anti-CD31 antibody from (a). White arrowheads identify vessels. Scale bar = 50 μm (upper, maginification, 100×; lower, maginification, 200×). Microvessel density is presented in the bar graph (*P < 0.005).
Fig 4.
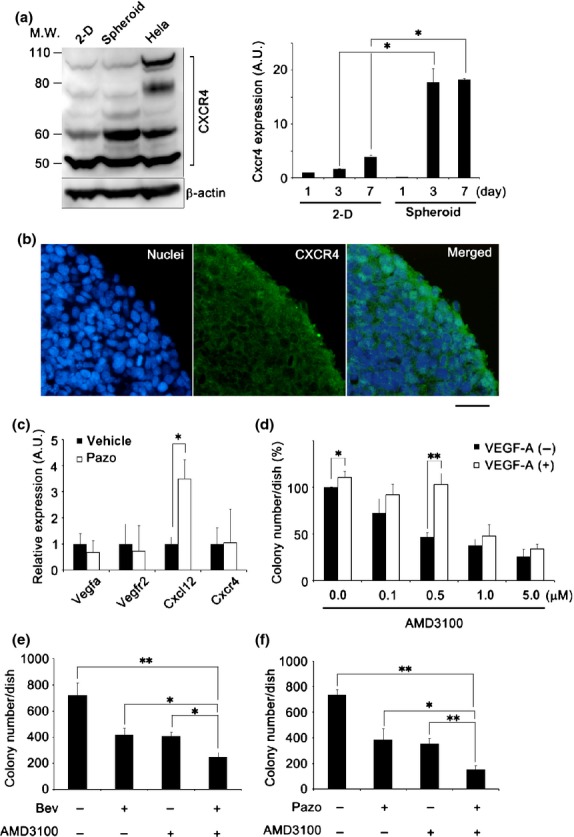
Blocking of vascular endothelial growth factor (VEGF) and chemokine (C-X-C motif) receptor 4 (CXCR4) signals synergistically suppresses cell proliferation in synovial sarcoma (SS). (a) Immun-oblot analysis of CXCR4 under 2-D or spheroid culture conditions (left), and quantification of CXCR4 mRNA by quantitative RT-PCR under 2-D or spheroid culture conditions from day 1 to day 7 (right panel) (n = 3; *P < 0.05). A.U., arbitrary unit (the same l applies hereafter); M.W., molecular weight. (b) Immunofluorescence staining of Yamato-SS spheroids with a CXCR4 antibody (green). Nuclei are counterstained with Hoechst33342 (blue). Scale bar = 100 μm. (c) Quantification of VEGFA, VEGFR2, CXCL12, and CXCR4 mRNA by quantitative RT-PCR in Yamato-SS spheroids treated with pazopanib (Pazo, 250 nM) for 24 h (n = 3; *P < 0.05). (d) Rescue experiments with exogenously added recombinant human VEGF-A (10 ng/mL) by soft agar assay after treatment with CXCR4-specific inhibitor AMD3100. Colonies >200 μm are shown. (n = 3; *P < 0.05, **P < 0.005). (e,f) Soft agar assay of Yamato-SS cells treated with bevacizumab (Bev, 0.2 μg/mL) (e) or Pazo (50 nM) (f) in combination with AMD3100 (0.5 μM). Colonies >200 μm were counted (n = 3; *P < 0.05, **P < 0.005).
Next, the antitumor effect of Bev in conjunction with another VEGF-targeted drug, Pazo, was investigated. Treatment with Pazo also inhibited tumor growth of Yamato-SS by 35.5–54.2% of the tumor volume, in a dose-dependent manner (Fig. S4d).
To verify the existence of a VEGF autocrine loop in SS, mouse tumor sections were examined histologically; expression of both VEGF-A and VEGFR2 was detected in the tumor (Fig.3c). Additionally, expression of human VEGF-A and VEGFR2 was upregulated compared to in vitro and the increase in the expression of these factors correlated with SS tumor growth (Fig.3d).
To examine the conventional anti-angiogenic effect of VEGF-targeted therapy, immunohistochemical analysis of SS cells, using anti-mouse CD31 antibody, was carried out. Microvessel density was markedly reduced with Bev treatment, by 80.9% in Yamato-SS (Fig.3e) and by 72.5% in Aska-SS (Fig. S4e). As no expression of human-specific CD31 was detected in the tumor sections (data not shown), we suspected that tumor angiogenesis was of mouse origin in this xenograft model.
These data suggested that inhibition of the VEGF paracrine signal resulted in the inhibition of host angiogenesis. Therefore, we suspected that tumor growth of SS was markedly suppressed, at least in part, by inhibition of host VEGF signaling.
Blocking of both VEGF and chemokine (C-X-C motif) receptor 4 signals synergistically suppresses cell proliferation in SS
Oda et al.34 also reported that chemokine (C-X-C motif) receptor 4 (CXCR4) expression correlated with VEGF expression as well as with poor survival in soft tissue sarcoma. Thus, we focused on CXCR4 signaling in SS in this study. We noticed that multiple bands of CXCR4 were detected in SS cell lysate by immunoblot analysis (Fig.4a, left panel; Fig. S5a, left panel, Fig. S10c). A 60-kDa ubiquitinated band was strongly expressed in Yamato-SS but slightly expressed in Aska-SS cells (Fig.4a, left panel; Fig. S5a, left panel), and the CXCR4 mRNA level was elevated from days 1 to 7 with (by 17.8- and 18.3-fold at days 3 and 7, respectively), only under spheroid culture conditions (Fig.4a, right panel). A similar tendency of elevation was observed in Aska-SS, but it was not statistically significant (Fig. S5a, right panel). Histologically, CXCR4 expression was exclusively observed in a one- to two-cell layer at the spheroid surface (Figs4b,S5b). CXCR4 is a receptor for chemokine (C-X-C motif) ligand 12 (CXCL12), and this cascade regulates cellular proliferation, invasion, migration, and metastasis.35,36 Furthermore, mesenchymal stem cells (MSCs) of putative SS origin strongly express CXCR4.35,36 We also observed various expression levels of CXCL12 and CXCR4 in clinical samples with SS patients (Fig. S5c).
We investigated the expression of several factors that have been reported to be elevated upon VEGF-targeted therapy, such as interleukin-6, interleukin-8, tumor necrosis factor-α, epidermal growth factor, and CXCL12.37 The mRNA transcription of CXCL12, but not that of the other factors, was elevated by treatment with Bev (1.6-fold increase; Fig. S5d, left panel; Fig. S5e) and with Pazo (3.5-fold increase; Figs4c,S5d, right panel) in Yamato-SS. We observed not only elevation of CXCL12 but also VEGFa, VEGFR2, and CXCR4 expression by treatment with Pazo in Aska-SS (Fig. S5f). Furthermore, we also checked the expression of other chemokines. The mRNA of CXCL12, but not that of the other chemokines, was elevated by the treatment with Bev and Pazo in Yamato-SS (Fig. S6a). By contrast, we did not observed such a tendency in Aska-SS (Fig. S6b). We thus suspected that the CXCL12/CXCR4 axis contributes to the sarcomagenesis in some SS by collaboration with VEGF signaling.
To verify that the CXCL12/CXCR4 autocrine loop was involved in cell proliferation, a soft agar assay was carried out using treatment with the CXCR4-specific inhibitor, AMD3100. AMD3100 inhibited colony formation in a dose-dependent manner, by 27.5–73.8% in Yamato-SS (Fig.4d), or by 31.2–67.7% in Aska-SS (Fig. S7a). In order to investigate whether the activity of the VEGF autocrine loop was enhanced by blockade of the CXCL12/CXCR4 autocrine loop, mRNA expression was examined using treatment with AMD3100 (Fig. S7b). AMD3100 was not observed to affect VEGF signaling. Finally, an attempt to rescue the inhibition of colony formation with AMD3100 was made by adding exogenous VEGF-A; VEGF-A treatment partially restored (by 7.8–55.9%) the inhibition of colony formation (Fig.4d).
To assess whether simultaneous blockage of VEGF and CXCL12/CXCR4 signaling had any marked antitumor efficacy in SS, we validated the antitumor effects of VEGF signal inhibitors in combination with CXCL12/CXCR4 signal inhibitors on two SS cells. At a low dose, the combination treatment effectively inhibited colony formation in the soft agar assay; colony formation relative to that in the control was reduced by 41.7–65.3% in the presence of Bev (Fig.4e) and by 45.7–79.2% in the presence of Pazo (Fig.4f) in Yamato-SS. The combination treatment also inhibited colony formation by 29.0–56.0% in the presence of Bev (Fig. S5c, left panel) and by 38.5–60.1% in the presence of Pazo (Fig. S5c, right panel) in Aska-SS. These results suggested that dual inhibition of VEGF and CXCL12/CXCR4 signaling displayed more effective antitumor efficacy than monotherapy in SS.
Vascular endothelial growth factor-targeted therapy in combination with IFM and/or a CXCR4 inhibitor effectively suppresses tumor growth
High-dose neo-adjuvant chemotherapy, a standard treatment for SS, causes several serious side-effects, including increased risk for the emergence of a second cancer.4,5 To reduce the side-effects and to improve prognosis, a VEGF-targeted therapy was designed by combining Bev with low-dose IFM, a first-line chemotherapy drug against SS; this regimen was approved by the Institute of the Japanese Musculoskeletal Oncology Group. Treatment with IFM results in the alkylation of DNA, thereby inhibiting DNA replication and resulting in reduced cellular proliferation and induced cell death. Treatment with activated IFM (aIFM) inhibited colony formation in a soft agar assay in a dose-dependent manner, by 16.2–88.7% in Yamato-SS (Fig.5a) and by 31.2–63.9% in Aska-SS (Fig. S8a). Combinatorial treatment with a low dose of Bev and aIFM additively inhibited colony formation in both cell lines, by 15.3–62.6% in Yamato-SS (Fig.5b) and by 21.8–67.7% in Aska-SS (Fig. S8b), as compared to control. Treatment with aIFM at 0.125–2 μg/mL reduced the colony size, and at 20 μg/mL it induced cell death (Fig.5c).
Fig 5.
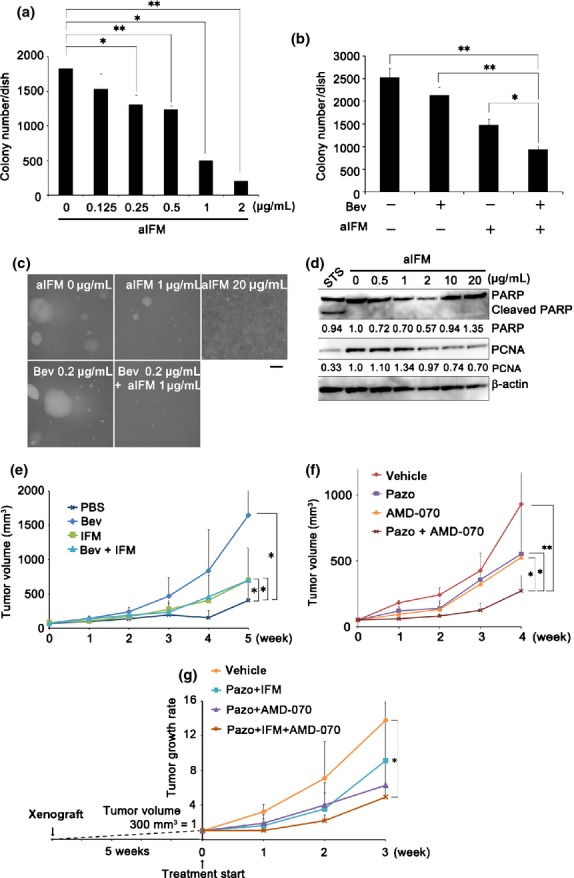
Vascular endothelial growth factor (VEGF)-targeted therapy in combination with ifosfamide (IFM) and/or a chemokine (C-X-C motif) receptor 4 (CXCR4) inhibitor effectively suppresses synovial sarcoma (SS) tumor growth. (a) Soft agar assay of Yamato-SS cells treated with activated IFM (aIFM). Colonies >200 μm are shown (n = 3; *P < 0.05, **P < 0.005). (b) Soft agar assay of Yamato-SS cells treated with bevacizumab (Bev; 0.2 μg/mL) and/or aIFM (1 μg/mL). Colonies >200 μm are shown (n = 3; *P < 0.05, **P < 0.005). (c) Phase-contrast images of soft agar assay of Yamato-SS cells treated with aIFM and/or Bev. Scale bar = 200 μm. (d) Immunoblot analysis of poly(ADP-ribose) polymerase (PARP) and proliferating cell nuclear antigen (PCNA) signals in Yamato-SS treated with aIFM for 24 h. STS, 0.5 μM staurosporine for 6 h. (e) Mice bearing Yamato-SS were treated with Bev (25 μg/mouse, twice per week) and/or IFM (2 mg/mouse, 3 days, every 3 weeks). Tumor volume was measured weekly for mice treated with PBS (n = 6), Bev (n = 7), IFM (n = 6), and Bev + IFM (n = 7). *P < 0.05. (f) Mice bearing Yamato-SS were treated with pazopanib (Pazo, 30 mg/kg) and/or CXCR4 inhibitor AMD-070 (10 mg/kg). The tumor volumes were measured every week for mice treated with vehicle (n = 5), Pazo (n = 8), AMD-070 (n = 7), and Pazo + AMD-070 (n = 8; *P < 0.05, **P < 0.005). (g) Mice bearing Yamato-SS were treated with Pazo (30 mg/kg) and IFM (2 mg/mouse, 3 days), Pazo (30 mg/kg) and AMD-070 (10 mg/kg), or Pazo (30 mg/kg), IFM (2 mg/mouse, 3 days) and AMD-070 (10 mg/kg). The tumor volumes were measured every week for mice treated with vehicle (n = 6), Pazo + IFM (n = 7), Pazo + AMD-070 (n = 7), and Pazo + AMD-070 (n = 7), and Pazo + IFM + AMD-070 (n = 7; *P < 0.05).
To observe the effect of aIFM, the expression of cleaved poly (ADP-ribose) polymerase was investigated; however, its expression was not detected at 0.5–20 μg/mL aIFM (Fig.5d). In addition, proliferating cell nuclear antigen (PCNA) expression was reduced at 2–20 μg/mL aIFM (Figs5d, S10d), suggesting that aIFM inhibited cellular proliferation without inducing apoptosis. Combinatorial treatment with Bev and low-dose IFM markedly suppressed Yamato-SS (Bev, 63.1%; IFM, 66.3%; Bev + IFM, 73.5%; Fig.5e) and Aska-SS (Bev, 73.4%; IFM, 42.0%; Bev + IFM, 86.4%; Fig. S8c) tumor growth in vivo. These data suggested that use of a low-dose VEGF signal inhibitor, in combination with low-dose IFM, suppresses tumor growth more effectively than monotherapy.
Next, we determined the effectiveness of simultaneous inhibition of VEGF and CXCR4 signals in the xenograft model. Combination of Pazo with the available oral CXCR4 inhibitor AMD-070 achieved greater tumor growth suppression (70.9%) than monotherapy (Pazo, 40.7%; AMD-070, 43.3%, relative to tumor growth reduction in the control; Fig.5f).
Finally, we treated SS tumors after the tumor volume reached approximately 300 mm3, using a combination of Pazo and AMD-070 and/or IFM. The triple drug combination significantly suppressed tumor growth (31.0%) compared to a combination of Pazo + IFM (50.0%) or Pazo + AMD-070 (56.4%), relative to tumor growth reduction in the control (Fig.5g).
Discussion
The present results show that SS18–SSX contributes to sarcomagenesis in SS by directing the VEGF signal outcome from endothelial differentiation to spheroid growth. We surmise that the VEGF signal had a dual effect on SS tumor growth, namely, on proliferation in an autocrine loop and on host angiogenesis in a paracrine manner; thus, VEGF-targeted therapy showed marked antitumor efficacy in SS. We also developed a novel combinatorial therapy involving a VEGF inhibitor and a current chemotherapy drug (IFM); this regimen constitutes a VEGF-/CXCR4-targeted therapy against SS. Pàez-Ribes reported that anti-VEGF-treated tumors can be aggressive and show increased metastasis.38 However, it has also been reported that VEGF-targeted therapy improved the prognosis of patients with several types of malignant tumors. We considered that anti-VEGF treatment against SS could increase lung metastasis, but inhibit tumor growth, by combining it with IFM and/or CXCR4 inhibitors. A summary of the proposed model is depicted in Fig.6.
Fig 6.
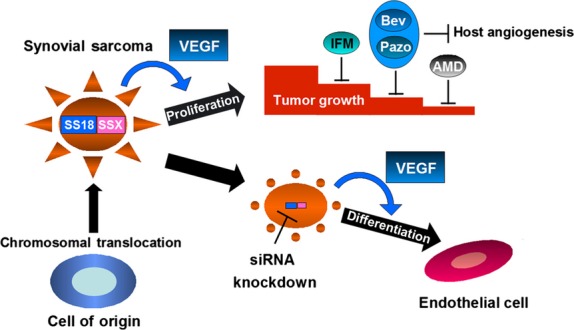
Proposed model based on this study. Synovial sarcoma (SS) cells show cell proliferation, depending on the vascular endothelial growth factor (VEGF) autocrine loop under the influence of SS18–SSX. However, after silencing of SS18–SSX, SS cells can differentiate into endothelial cells depending on VEGF signaling. Therefore, the action of VEGF in SS cells was deflected by SS18–SSX. In addition, VEGF-targeted therapy combined with ifosfamide (IFM) and/or chemokine (C-X-C motif) receptor 4 (CXCR4)-targeted therapy effectively suppresses tumor growth in SS. AMD, CXCR4 inhibitor; Bev, bevacizumab; Pazo, pazopanib.
Our results show that SS18–SSX altered the VEGF-mediated cellular response—from tubular differentiation to proliferation—in SS, probably through epigenetically regulated chromatin remodelling of SS18–SSX. Thus, SS18–SSX is an oncogene that plays central roles in SS. Several functions of SS18–SSX have previously been reported, such as maintaining stemness and inducing spherical morphology.6,8,9,30 Our findings indicated that stemness and morphology were not unrelated, and that secretion of VEGF and the cellular response to the VEGF signal was modified by this interaction under the direction of SS18–SSX. Our results also suggest that SS cells may differentiate into endothelial cells in tumors, as in endothelial–mesenchymal transition. 39 Ricci-Vitiani et al.40 reported that glioblastoma stem-like cells differentiate into endothelial cells in tumors. In our study, VEGF signal inhibition suppressed both tumor growth and endothelial differentiation in SS. We previously showed that MSCs are the cellular origin of SS cells, which thereby confers multipotency to these cells.6 SOX2 plays an important role in stemness and pluripotency in embryonic stem cells.41 Kadoch and Crabtree reported that SS18–SSX containing the BAF complex upregulates SOX2 expression.9 We thus speculated whether SOX2 suppression by SS18–SSX silencing allowed SS cells to differentiate into endothelial cells by VEGF stimuli under anchorage-independent culture conditions. In addition, CXCR4 expression in SS cells also displayed an MSC phenotype. Thus, SS likely originates from MSCs, which demonstrate a much broader differentiation potential than do SS cells, including endothelial differentiation; this view supports the findings in previous reports.6
Herein, we have developed a novel putative therapy for SS; however, the molecular mechanism underlying the therapeutic effect was not completely elucidated. To gain insights into the molecular mechanism of the antitumor efficacy of VEGF inhibition, we examined the expression of molecules acting downstream of VEGF signaling, after Bev and Pazo treatment, by using immunoblot analysis. With both VEGF inhibitors and CXCR4 inhibitors, the phosphorylation level of Akt was inhibited (Fig. S9a,b). Similar results were obtained after simultaneous inhibition of VEGF and CXCR4 signaling (Fig. S9c). In addition, RS6 kinase, which functions downstream of phosphoinositide-3-kinase (PI3K), was also effectively inhibited (Fig. S6c); PI3K has been implicated in cell survival of many malignant tumors.42 Treatment with IFM also inhibited the expression of PCNA (Fig.5d). Therefore, we speculate that PI3K supports SS tumor growth through the VEGF and CXCL12/CXCR4 signaling axes and that VEGF signaling inhibitors and IFM show collaborative antitumor effects.
In conclusion, our results clearly show that VEGF promotes cell growth in an autocrine fashion and that SS18–SSX directs the VEGF signal outcome from differentiation to cell growth in SS. Therefore, VEGF is an appropriate therapeutic target and VEGF-targeted therapy could be effective in SS patients. In addition, VEGF-targeted therapy, in combination with CXCR4-targeted therapy and/or IFM, could be an effective strategy for the treatment of SS. Our findings indicated that growth of SS tumors was due to the actions of VEGF under the influence of SS18–SSX, and that VEGF-targeted therapy combined with CXCR4-targeted therapy and IFM could lead to improvement of the particularly poor prognosis of SS, by blocking both the autocrine and paracrine loop of VEGF signaling.
Acknowledgments
Pazopanib was kindly provided as a gift from GlaxoSmithKline Co. We thank Drs. Hidetatsu Outani, Hirohiko Yasui, and Yoshinori Imura for helpful discussions. We are also grateful to Ms. Naoko Shimatani for technical assistance. This study was financially supported in part by the Program of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (Grant No. 23390372, to KI), a Grant-in Aid for Scientific Research (B) and for Challenging Exploratory Research (Grant No. 23659734, to KI), and a Grant-in-Aid for Young Scientists (B) (Grant No. 24791575, to SS) from MEXT, Japan.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. (a) Immunohistochemical analysis of synovial sarcoma (SS) clinical specimen with vascular endothelial growth factor-A (VEGF-A; n = 25). Nuclei were counterstained with hematoxylin. Scale bar = 100 μm. Staining intensity was scored as: −, negative; +, positive; and ++, strongly positive. (b) VEGFA and VEGFR2 mRNA levels in SS clinical samples (n = 6) relative to Yamato-SS in spheroid culture conditions, as determined by quantitative RT-PCR.
Fig. S2. (a) Quantity of vascular endothelial growth factor-A (VEGF-A) secreted by synovial sarcoma Aska-SS cells under 2-D or spheroid culture conditions, as determined by ELISA. Culture medium was collected every 24 h for 4 days (n = 3; *P < 0.05). (b) Growth of Yamato-SS cells under 2-D culture conditions, after treatment with bevacizumab (Bev; n = 3) or pazopanib (Pazo; n = 3). (c) Growth of Aska-SS cells under 2-D culture conditions upon treatment with Bev (n = 3) or Pazo (n = 3). (d) Soft agar assay of Yamato-SS cells treated with Bev (5 μg/mL) or Pazo (250 nM) for 2 weeks. Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3). (e) Dose-dependence of the response of Yamato-SS cells to 2 weeks of Pazo treatment, as determined by soft agar assay. Colonies >200 μm are shown (n = 3; *P < 0.05, **P < 0.005). (f) Dose-dependence of response of Aska-SS cells to treatment with Bev (left panel) or Pazo (right panel) for 4 weeks, as determined using a soft agar assay. Colonies >200 μm are shown (n = 3; *P < 0.005). (g) Rescue experiments with exogenously added recombinant human VEGF-A (10 ng/mL) in Yamato-SS cells treated with Pazo. Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3; *P < 0.05).
Fig. S3. (a) Confirmation of SS18–SSX fusion gene knockdown in Aska-SS synovial sarcoma cells by siRNA-A and siRNA-B, by immunoblot analysis. Closed or open arrowheads represent SS18–SSX or SS18, respectively. (b) VEGFA and VEGFR2 mRNA levels in Aska-SS cells treated with SS18–SSX siRNA, as determined by quantitative RT-PCR. (c) Cell proliferation of Aska-SS cells cultured in a tube formation assay and treated with bevacizumab (Bev, 5 μg/mL; left) or Pazo (250 nM; right) (n = 3). (d,e) Immunofluorescence analysis of CD31 (d), and von Willebrand factor (vWF) (e). Nuclei were counterstained with Hoechst33342 (blue). Scale bar = 100 μm. Phase contrast images of a tube formation. (f) Quantitation of tubular formation assay in Aska-SS cells treated with SS18–SSX siRNA, in the presence or absence of treatment with Bev (5 μg/mL; upper) and Pazo (250 nM; bottom) (n = 2; *P < 0.05, **P < 0.005).
Fig. S4. (a) Mice bearing Aska-SS synovial sarcoma cells were treated with bevacizumab (Bev, 100 μg/mouse, twice a week; n = 10). Control mice were treated with PBS (n = 9). The images of mice and tumors after 8 weeks of treatment are shown. Scale bar = 1 cm. (b) Tumor volume was measured weekly (*P < 0.005). (c) Tumor weight was measured after 8 weeks of treatment (*P < 0.005). (d) Mice bearing Yamato-SS were treated with pazopanib (Pazo, 30 mg/kg, n = 8 or 100 mg/kg, n = 7). Control mice were treated with the vehicle used for Pazo (n = 7). Tumor volume was measured weekly (*P < 0.005). (e) Immunohistochemical analysis of Aska-SS tumor sections in (a) with a mouse-specific anti-CD31 antibody. White arrowheads identify vessels. Scale bar = 50 μm. Microvessel density is shown in the bar graph (*P < 0.005).
Fig. S5. (a) Immunoblot analysis of chemokine (C-X-C motif) receptor 4 (CXCR4) under 2-D or spheroid culture conditions (left), and quantification of CXCR4 mRNA by quantitative RT-PCR (qRT-PCR) under 2-D or spheroid culture conditions from day 1 to day 7 in Aska-SS cells (right panel, n = 3). N.S., not significant. (The same l applies hereafter.) (b) Immunofluorescence staining of Aska-SS spheroids with a CXCR4 antibody (green). Nuclei were counterstained with Hoechst33342 (blue). Scale bar = 100 μm. (c) CXCL12 and CXCR4 mRNA levels as determined by qRT-PCR in SS clinical samples (n = 6) relative to Yamato-SS in spheroid culture conditions. (d) CXCL12, IL6, IL8, TNFα and EGF mRNA levels as determined by qRT-PCR in Yamato-SS spheroids treated with bevacizumab (Bev, 5 μg/mL, left panel) or pazopanib (Pazo, 250 nM, right panel) for 24 h (n = 3; *P < 0.05). (e) VEGFA, VEGFR2, and CXCR4 mRNA levels as determined by qRT-PCR in Yamato-SS spheroids treated with Bev (5 μg/mL, n = 3) for 24 h. (f) Quantification of VEGFA, VEGFR2, CXCL12, and CXCR4 mRNA by qRT-PCR in Aska-SS spheroids treated with Pazo (250 nM) for 24 h (n = 3; *P < 0.05).
Fig. S6. (a) mRNA levels of chemokines as determined by quantitative RT-PCR in Yamato-SS spheroids treated with bevacizumab (Bev, 5 μg/mL, upper panel) or pazopanib (Pazo, 250 nM, lower panel) for 24 h (n = 3; *P < 0.05). (b) mRNA levels of chemokines as determined by quantitative RT-PCR in Aska-SS spheroids treated with Bev (5 μg/mL, upper panel) or Pazo (250 nM, lower panel) for 24 h (n = 3; *P < 0.05).
Fig. S7. (a) Soft agar assay of Aska-SS synovial sarcoma cells after treatment with AMD3100 (0–5 μM). Colonies >200 μm are shown. (n = 3; *P < 0.05, **P < 0.005). (b) VEGFA, VEGFR2, CXCL12, and CXCR4 mRNA levels as determined by quantitative RT-PCR in Yamato-SS spheroids treated with AMD3100 (5 μM, n = 3) for 24 h. (c) Soft agar assay of Aska-SS cells after treatment with bevacizumab (Bev, 0.2 μg/mL, left panel) or pazopanib (Pazo, 50 nM, right panel) in combination with AMD3100 (0.5 μM). Colonies >200 μm were counted (n = 3; *P < 0.05, **P < 0.005).
Fig. S8. (a) Soft agar assay of Aska-SS synovial sarcoma cells treated with activated ifosfamide (aIFM). Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3; *P < 0.05, **P < 0.005). (b) Soft agar assay of Aska-SS cells treated with bevacizumab (Bev, 0.2 μg/mL) and/or aIFM (1 μg/mL) for 4 weeks. Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3; *P < 0.05, **P < 0.005). (c) Mice bearing Aska-SS were treated with Bev (25 μg/mouse, twice a week) and/or IFM (2 mg/mouse, 3 days every 3 weeks). Tumor volume was measured weekly in mice treated with PBS (n = 5), Bev (n = 4), IFM (n = 5), and Bev + IFM (n = 5; *P < 0.05, **P < 0.005).
Fig. S9. (a) Immunoblot analysis of downstream phosphorylation in the vascular endothelial growth factor (VEGF) signaling pathway in Yamato-SS spheroids treated with various concentrations of bevacizumab (Bev) or pazopanib (Pazo) for 24 h. (b) Immunoblot analysis of downstream phosphorylation in the VEGF signaling pathway in Yamato-SS spheroids treated with various concentrations of AMD3100 for 24 h. (c) Immunoblot analysis of downstream phosphorylation in the VEGF signaling pathway of Yamato-SS spheroids treated with Bev (0.2 μg/mL) and Pazo (10 nM) in combination with AMD3100 (0.5 μM) for 24 h.
Fig. S10. Blue box frames were cropped for Figs 1(d) (a), 2(a) (b), 4(a) (c), and 5(d) (d).
Table S1. Concentrations of human vascular endothelial growth factor-A (VEGF-A) in s.c. transplanted tumors of synovial sarcoma cell lines in mice.
Table S2. Dilution rate and solution instructions for reagents and antibodies used in this study.
Table S3. Primers used in this study.
Data. S1. Materials and methods.
References
- De Leeuw B, Balemans M, Weghuis DO, van Kessel AG. Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum Mol Gene. 1995;4:1097–9. doi: 10.1093/hmg/4.6.1097. [DOI] [PubMed] [Google Scholar]
- Crew AJ, Clark J, Fisher C, et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995;14:2333–40. doi: 10.1002/j.1460-2075.1995.tb07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji S, Hisaoka M, Morimitsu Y, et al. Detection of SYT-SSX fusion transcripts in synovial sarcoma by reverse transcription-polymerase chain reaction using archival paraffin-embedded tissues. Am J Pathol. 1998;153:1807–12. doi: 10.1016/S0002-9440(10)65695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka S, Ueda T, Naka N, et al. Prognostic implication of SYT-SSX fusion type in synovial sarcoma: a multi-institutional retrospective analysis in Japan. Oncol Rep. 2008;19:467–76. [PubMed] [Google Scholar]
- Palmerini E, Staals EL, Alberghini M, et al. Synovial sarcoma. Cancer. 2009;115:2988–98. doi: 10.1002/cncr.24370. [DOI] [PubMed] [Google Scholar]
- Naka N, Takenaka S, Araki N, et al. Synovial sarcoma is a stem cell malignancy. Stem Cells. 2010;28:1119–31. doi: 10.1002/stem.452. [DOI] [PubMed] [Google Scholar]
- Garcia CB, Shaffer CM, Alfaro MP, et al. Reprogramming of mesenchymal stem cells by synovial sarcoma-associated oncogene SYT-SSX2. Oncogene. 2012;31:2323–34. doi: 10.1038/onc.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Sampaio AV, Jones KB, et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell. 2012;21:333–47. doi: 10.1016/j.ccr.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Brugge SJ. Modelling glandular epithelial cancer in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–88. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- Kenny AP, Lee GY, Myers CA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydarkhan-Hagvall S, Gluck JM, Delman C, et al. The effect of vironectin on the differentiation of embryonic stem cells in a 3D culture system. Biomaterials. 2012;33:2032–40. doi: 10.1016/j.biomaterials.2011.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J, Endo H, Okuyama H, et al. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. PNAS. 2011;108:6235–40. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Worster DT, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xiao Z, Meng Y, et al. The enhancement of cancer stem cell properties of MCF-7 cells in 3D collagen scaffolds for modeling of cancer and anti-cancer drugs. Biomaterials. 2012;33:1437–44. doi: 10.1016/j.biomaterials.2011.10.056. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signaling-in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signaling and therapeutic inhibition. Cell Signal. 2007;19:2003–12. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B, Driessens G, Goossens S, et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumors. Nature. 2011;478:399–403. doi: 10.1038/nature10525. [DOI] [PubMed] [Google Scholar]
- Lichtenberger BM, Tan PK, Niederleithner H, et al. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell. 2010;140:268–79. doi: 10.1016/j.cell.2009.12.046. [DOI] [PubMed] [Google Scholar]
- Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumor activity. Nat Rev Cancer. 2008;8:579–91. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- Potent M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–87. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Shibuya M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-γ pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene. 1997;14:2079–89. doi: 10.1038/sj.onc.1201047. [DOI] [PubMed] [Google Scholar]
- Harris AL. Hypoxia—a key regulatory factor in tumor growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a Novel and Potent Vascular Endothelial Growth Factor Receptor Inhibitor. J Med Chem. 2008;51:4632–40. doi: 10.1021/jm800566m. [DOI] [PubMed] [Google Scholar]
- Kumar R, Knick VB, Rudolph SK, et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther. 2011;6:2012–21. doi: 10.1158/1535-7163.MCT-07-0193. [DOI] [PubMed] [Google Scholar]
- Takenaka S, Naka N, Araki N, et al. Downregulation of SS18-SSX1 expression in synovial sarcoma by small interfering RNA enhances the focal adhesion pathway and inhibits anchorage-independent growth in vitro and tumor growth in vivo. Int J Oncol. 2010;36:823–31. doi: 10.3892/ijo_00000559. [DOI] [PubMed] [Google Scholar]
- Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Prac Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III Trial. J Clin Oncol. 2010;28:1061–8. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- Gray R, Bhattacharya S, Bowden C, Miller K, Comis RL. Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol. 2009;27:4966–72. doi: 10.1200/JCO.2008.21.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Tateishi N, Matono H, et al. Chemokine receptor CXCR4 expression is correlated with VEGF expression and poor survival in soft-tissue sarcoma. Int J Cancer. 2009;124:1852–9. doi: 10.1002/ijc.24128. [DOI] [PubMed] [Google Scholar]
- Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- Domanska UM. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219–30. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Azam F, Mehta S, Harris AL. Mechanism of resistance to antiangiogenesis therapy. Eur J Cancer. 2010;46:1323–32. doi: 10.1016/j.ejca.2010.02.020. [DOI] [PubMed] [Google Scholar]
- Pàez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–9. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, et al. Tumor vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–8. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Bareiss PM, Paczulla A, Wang H, et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res. 2013;73:5544–55. doi: 10.1158/0008-5472.CAN-12-4177. [DOI] [PubMed] [Google Scholar]
- Bianco I, Sawyers CL. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (a) Immunohistochemical analysis of synovial sarcoma (SS) clinical specimen with vascular endothelial growth factor-A (VEGF-A; n = 25). Nuclei were counterstained with hematoxylin. Scale bar = 100 μm. Staining intensity was scored as: −, negative; +, positive; and ++, strongly positive. (b) VEGFA and VEGFR2 mRNA levels in SS clinical samples (n = 6) relative to Yamato-SS in spheroid culture conditions, as determined by quantitative RT-PCR.
Fig. S2. (a) Quantity of vascular endothelial growth factor-A (VEGF-A) secreted by synovial sarcoma Aska-SS cells under 2-D or spheroid culture conditions, as determined by ELISA. Culture medium was collected every 24 h for 4 days (n = 3; *P < 0.05). (b) Growth of Yamato-SS cells under 2-D culture conditions, after treatment with bevacizumab (Bev; n = 3) or pazopanib (Pazo; n = 3). (c) Growth of Aska-SS cells under 2-D culture conditions upon treatment with Bev (n = 3) or Pazo (n = 3). (d) Soft agar assay of Yamato-SS cells treated with Bev (5 μg/mL) or Pazo (250 nM) for 2 weeks. Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3). (e) Dose-dependence of the response of Yamato-SS cells to 2 weeks of Pazo treatment, as determined by soft agar assay. Colonies >200 μm are shown (n = 3; *P < 0.05, **P < 0.005). (f) Dose-dependence of response of Aska-SS cells to treatment with Bev (left panel) or Pazo (right panel) for 4 weeks, as determined using a soft agar assay. Colonies >200 μm are shown (n = 3; *P < 0.005). (g) Rescue experiments with exogenously added recombinant human VEGF-A (10 ng/mL) in Yamato-SS cells treated with Pazo. Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3; *P < 0.05).
Fig. S3. (a) Confirmation of SS18–SSX fusion gene knockdown in Aska-SS synovial sarcoma cells by siRNA-A and siRNA-B, by immunoblot analysis. Closed or open arrowheads represent SS18–SSX or SS18, respectively. (b) VEGFA and VEGFR2 mRNA levels in Aska-SS cells treated with SS18–SSX siRNA, as determined by quantitative RT-PCR. (c) Cell proliferation of Aska-SS cells cultured in a tube formation assay and treated with bevacizumab (Bev, 5 μg/mL; left) or Pazo (250 nM; right) (n = 3). (d,e) Immunofluorescence analysis of CD31 (d), and von Willebrand factor (vWF) (e). Nuclei were counterstained with Hoechst33342 (blue). Scale bar = 100 μm. Phase contrast images of a tube formation. (f) Quantitation of tubular formation assay in Aska-SS cells treated with SS18–SSX siRNA, in the presence or absence of treatment with Bev (5 μg/mL; upper) and Pazo (250 nM; bottom) (n = 2; *P < 0.05, **P < 0.005).
Fig. S4. (a) Mice bearing Aska-SS synovial sarcoma cells were treated with bevacizumab (Bev, 100 μg/mouse, twice a week; n = 10). Control mice were treated with PBS (n = 9). The images of mice and tumors after 8 weeks of treatment are shown. Scale bar = 1 cm. (b) Tumor volume was measured weekly (*P < 0.005). (c) Tumor weight was measured after 8 weeks of treatment (*P < 0.005). (d) Mice bearing Yamato-SS were treated with pazopanib (Pazo, 30 mg/kg, n = 8 or 100 mg/kg, n = 7). Control mice were treated with the vehicle used for Pazo (n = 7). Tumor volume was measured weekly (*P < 0.005). (e) Immunohistochemical analysis of Aska-SS tumor sections in (a) with a mouse-specific anti-CD31 antibody. White arrowheads identify vessels. Scale bar = 50 μm. Microvessel density is shown in the bar graph (*P < 0.005).
Fig. S5. (a) Immunoblot analysis of chemokine (C-X-C motif) receptor 4 (CXCR4) under 2-D or spheroid culture conditions (left), and quantification of CXCR4 mRNA by quantitative RT-PCR (qRT-PCR) under 2-D or spheroid culture conditions from day 1 to day 7 in Aska-SS cells (right panel, n = 3). N.S., not significant. (The same l applies hereafter.) (b) Immunofluorescence staining of Aska-SS spheroids with a CXCR4 antibody (green). Nuclei were counterstained with Hoechst33342 (blue). Scale bar = 100 μm. (c) CXCL12 and CXCR4 mRNA levels as determined by qRT-PCR in SS clinical samples (n = 6) relative to Yamato-SS in spheroid culture conditions. (d) CXCL12, IL6, IL8, TNFα and EGF mRNA levels as determined by qRT-PCR in Yamato-SS spheroids treated with bevacizumab (Bev, 5 μg/mL, left panel) or pazopanib (Pazo, 250 nM, right panel) for 24 h (n = 3; *P < 0.05). (e) VEGFA, VEGFR2, and CXCR4 mRNA levels as determined by qRT-PCR in Yamato-SS spheroids treated with Bev (5 μg/mL, n = 3) for 24 h. (f) Quantification of VEGFA, VEGFR2, CXCL12, and CXCR4 mRNA by qRT-PCR in Aska-SS spheroids treated with Pazo (250 nM) for 24 h (n = 3; *P < 0.05).
Fig. S6. (a) mRNA levels of chemokines as determined by quantitative RT-PCR in Yamato-SS spheroids treated with bevacizumab (Bev, 5 μg/mL, upper panel) or pazopanib (Pazo, 250 nM, lower panel) for 24 h (n = 3; *P < 0.05). (b) mRNA levels of chemokines as determined by quantitative RT-PCR in Aska-SS spheroids treated with Bev (5 μg/mL, upper panel) or Pazo (250 nM, lower panel) for 24 h (n = 3; *P < 0.05).
Fig. S7. (a) Soft agar assay of Aska-SS synovial sarcoma cells after treatment with AMD3100 (0–5 μM). Colonies >200 μm are shown. (n = 3; *P < 0.05, **P < 0.005). (b) VEGFA, VEGFR2, CXCL12, and CXCR4 mRNA levels as determined by quantitative RT-PCR in Yamato-SS spheroids treated with AMD3100 (5 μM, n = 3) for 24 h. (c) Soft agar assay of Aska-SS cells after treatment with bevacizumab (Bev, 0.2 μg/mL, left panel) or pazopanib (Pazo, 50 nM, right panel) in combination with AMD3100 (0.5 μM). Colonies >200 μm were counted (n = 3; *P < 0.05, **P < 0.005).
Fig. S8. (a) Soft agar assay of Aska-SS synovial sarcoma cells treated with activated ifosfamide (aIFM). Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3; *P < 0.05, **P < 0.005). (b) Soft agar assay of Aska-SS cells treated with bevacizumab (Bev, 0.2 μg/mL) and/or aIFM (1 μg/mL) for 4 weeks. Colonies of different sizes (<100, 100–200, >200 μm) are shown (n = 3; *P < 0.05, **P < 0.005). (c) Mice bearing Aska-SS were treated with Bev (25 μg/mouse, twice a week) and/or IFM (2 mg/mouse, 3 days every 3 weeks). Tumor volume was measured weekly in mice treated with PBS (n = 5), Bev (n = 4), IFM (n = 5), and Bev + IFM (n = 5; *P < 0.05, **P < 0.005).
Fig. S9. (a) Immunoblot analysis of downstream phosphorylation in the vascular endothelial growth factor (VEGF) signaling pathway in Yamato-SS spheroids treated with various concentrations of bevacizumab (Bev) or pazopanib (Pazo) for 24 h. (b) Immunoblot analysis of downstream phosphorylation in the VEGF signaling pathway in Yamato-SS spheroids treated with various concentrations of AMD3100 for 24 h. (c) Immunoblot analysis of downstream phosphorylation in the VEGF signaling pathway of Yamato-SS spheroids treated with Bev (0.2 μg/mL) and Pazo (10 nM) in combination with AMD3100 (0.5 μM) for 24 h.
Fig. S10. Blue box frames were cropped for Figs 1(d) (a), 2(a) (b), 4(a) (c), and 5(d) (d).
Table S1. Concentrations of human vascular endothelial growth factor-A (VEGF-A) in s.c. transplanted tumors of synovial sarcoma cell lines in mice.
Table S2. Dilution rate and solution instructions for reagents and antibodies used in this study.
Table S3. Primers used in this study.
Data. S1. Materials and methods.