

Abstract
Contractility of the heart is impaired with advancing age via mechanical remodeling, as myocytes are lost through apoptosis and collagenous fibers accumulate. Exercise training confers protection against fibrosis and apoptosis in the aging heart, but the mechanisms remain poorly understood. We recently reported that exercise training elevates Mn isoform of superoxide dismutase (MnSOD) in the aging heart, concomitant with reduction in oxidative stress and fibrosis. Here, we tested the hypothesis that overexpression of MnSOD would be causal in protection against fibrosis and apoptosis in the aging heart. Hearts were extracted from young (8 months) wild-type, young mice overexpressing the Sod2 (MnSOD) gene, old (28 months) wild-type, and old transgenic mice. Left ventricle MnSOD protein levels were elevated in young mice overexpressing the Sod2 (MnSOD) gene and old transgenic mice. MnSODTg mice exhibited lower oxidative stress (total hydroperoxides, 4-hydroxynonenal, and 8-isoprostane) in the old group. Age-related cardiac remodeling and fibrosis was mitigated in MnSOD Tg mice with reductions in extramyocyte space (−65%), collagen-I, and transforming growth factor-β. Pro-apoptotic markers Bax (−38%) and caspase-3 cleavage (−41%) were reduced and apoptosis (terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling-positive nuclei, DNA laddering) was mitigated in MnSOD Tg hearts compared with old wild-type. We conclude that MnSOD elevation is indeed protective against oxidative stress, fibrosis, and apoptosis in the aging heart.
Key Words: Aging, Heart, Superoxide dismutase, Oxidative stress, Fibrosis.
Aging reduces cardiac function due to impaired Ca2+ homeostasis (1,2) and progressive structural remodeling. Age-related remodeling includes the loss of cardiac myocytes, a product of increased apoptosis and necrosis as well as reduced replacement by progenitor cells (3–5). Indeed, the loss of cardiac myocytes increases exponentially with advancing age in the heart, although the impact may be gender-dependent (5,6). For example, aging resulted in approximately 30% reduction in the number of cardiac myocytes in 70-year-old man (7). In addition, remaining cardiac myocytes experience reactive hypertrophy, and there is an accumulation of connective tissue (ie, fibrosis) (8). Increased fibrotic tissue reduces cardiac filling and enhances the risk of chronic heart failure (2).
Previously, we reported that treadmill exercise training reduced age-induced apoptosis and remodeling in the rat heart (4). In a follow-up study (5), we reported that exercise training reduced collagenous fibrosis that was associated with aging. Collagen-I and upstream regulatory proteins tissue inhibition of metalloproteinase-1 and transforming growth factor-β (TGF-β) were lower in the hearts of exercise trained rats. Treadmill exercise training also upregulated the mitochondrial cell protective Mn isoform of superoxide dismutase (MnSOD) and reduced oxidative stress in the aging heart (9). However, the causal relationship between elevation of MnSOD in the aging heart and reduction in fibrosis and apoptosis in the aging heart is unknown.
MnSOD is an essential antioxidant enzyme located in the mitochondrial matrix, protecting proteins, lipids, and DNA. MnSOD catalyzes the dismutation of superoxide anion (O2•−) produced in the mitochondrial inner membrane to hydrogen peroxide (H2O2) (10,11). MnSOD has been shown to confer cardioprotection in a number of models. For example, homozygous MnSOD (Sod2 −/−) knockout mice exhibit impaired mitochondrial enzyme and high susceptibility to oxidative mitochondrial injury (12,13). Furthermore, Sod2 −/− mice have enlarged hearts with dilated left ventricles (LV), leading to early postnatal mortality (12). In addition, the heterozygous MnSOD (Sod2 − /+) knockout mouse expresses a phenotype characterized by impaired mitochondrial enzyme activity and respiration (14). Moreover, the Sod2 −/+ mouse has a higher probability of permeability transition pore opening (15) and oxidative damage to mitochondria (16) than wild-types (WT). In contrast, overexpression of MnSOD in transgenic mice appears to protect against oxidative stress, mitochondrial oxidative capacity, apoptosis, and in liver, brain, and skeletal muscle (10,17–19).
Exercise enhances stress response including enhancement of MnSOD (20), heat-shock proteins (19), and insulin-like growth factor-1 (1,3). Indeed, upregulation of stress proteins may abrogate apoptosis and remodeling in the heart. Elevation of MnSOD is an attractive protection-mechanism of habitual exercise against fibrosis and apoptosis in the aging heart because inducibility of MnSOD by exercise training is retained with age (9). Exercise may also attenuate redox-sensitive inflammatory cytokines (21), which stimulate cardiac remodeling.
A primary source of elevated oxidative stress in the aging heart is believed to be complexes I and III of mitochondrial electron transport chain of the inner mitochondrial membrane (22). Furthermore, elevation of mitochondrial oxidative stress is associated with mitochondrial dysfunction (22,23). Indeed, age-related elevation of oxidative stress has been linked to pro-apoptotic proteins (eg, Bax, Bad, Bid) resulting in increased permeability transition pore opening and subsequent activation of caspases-9 and -3 (21).
Therefore, we tested the hypothesis that overexpression of MnSOD (Sod2 Tg) would attenuate age-related remodeling. Specifically, we postulated that old MnSOD Tg would exhibit significantly lower levels of cardiac oxidative stress, fibrotic markers (collagen-I, TGF-β), and apoptosis (Bax, caspase-3 cleavage, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling [TUNEL]-positive staining, DNA fragmentation) compared with old WT.
Methods
Animals
The Sod2 transgenic (Tg) mice overexpressing MnSOD used in this study were obtained from Dr. Epstein’s Laboratory (24). A 13-kb genomic Sod2 clone isolated from C57BL/6J mice, which encompassed 2kb of the native Sod2 promoter, was used to generate Sod2 Tg mice. Subsequent generations of Sod2 Tg mice were bred by mating male Sod2 Tg mice to female C57BL/6J WT, purchased from Jackson Laboratories (Bar Harbor, ME). Animals were cared for at the University of Texas Health Science Center at San Antonio in accordance with National Institutes of Health and the institutional animal care committee standards. An animal use protocol had been approved by the University of Texas Health Science Center at San Antonio for all procedures used in the study. Male WT and Sod2 Tg mice were housed four per cage in a temperature-controlled (23 ± 2°C) room with a 12:12-h light–dark cycle. Water and mouse chow were provided ad libitum. Young adult (8 months) and old (28 months) mice were divided into the following groups: young adult (8 months) WT (YWT; n = 10), young adult MnSOD Tg (YTg; n = 10), old (28 mo) WT (OWT; n = 10), and old MnSOD Tg (OTg; n = 10) mice.
Tissue Preparation
Before collecting the heart, mice were euthanized using a CO2 chamber followed by cervical dislocation. The LV was quickly extracted, weighed, and placed in ice-cold phosphate-buffered saline (PBS; pH = 7.4). The LV samples were then frozen in liquid nitrogen and stored at −80°C until analysis. We focused on the LV because it is the primary target in the aging heart with increased apoptosis and remodeling (5,9).
Homogenization Procedure
To measure markers of oxidative stress and the protein levels of apoptotic signaling, LV samples were minced into fine pieces and homogenized (20:1 w/v) in ice-cold (4°C) lysis buffer solution (pH = 7.4) containing the following: 20mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 350mM NaCl, 20% glycerol, 1% Igepal-CA630, 1mM MgCl2, 0.1mM dithiothreitol, 0.5mM ethylenediaminetetraacetic acid, 0.1mM ethylene glycol tetraacetic acid, and protease inhibitor cocktail (Roche Applied Science, Penzberg, Germany). Minced LV were homogenized using a ground glass on ground glass homogenizer (Bellco Biotechnology, Vineland, NJ) at 4°C, and then twice centrifuged (4°C) for 10 minutes at 10,000g with the supernatant extracted. Total protein was determined using a BCA assay kit (Pierce Biotechnology, Rockford, IL).
Histology and Immunofluorescence
In our histological experiments, cross-sections of the LV were cut (10 µm thick) in a cryostat (Thermo-Fisher, Shandon Cryotome FSE) at −15°C, placed on slides, and air-dried for 30 minutes. Hematoxylin and eosin staining was conducted at room temperature and utilized to localize cardiomyocyte nuclei, extramyocyte space, and geometry. Hematoxylin (QT) was also used as a counterstain for visualization at room temperature.
In order to conduct immunofluorescence staining, sections were first dried for 30 minutes and then fixed in acetone (−20°C) for 10 minutes. Sample slides were next blocked with 10% serum of the secondary antibody with Tris-buffered saline (100 μL) for 30 minutes after three washes with PBS with Tween-20. After air drying for 10 minutes, primary antibodies (1:100 dilution) for collagen-I (SC-25974; Santa Cruz Biotechnology, Santa Cruz, CA), TGF-β1 (MAB240; R&D systems, Minneapolis, MN), and laminin (L9393; Sigma, St. Louis, MO) were applied and incubated for 60 minutes in blocking buffer at room temperature. After three 10 minutes washes in PBS, sections were incubated in the appropriate secondary antibody (1:200 dilution) with a fluorochrome attached (eg, goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 594; Invitrogen, Carlsbad, CA) for 30 minutes at room temperature. Sections were then washed three times in PBS and air-dried for 20 minutes. Samples were covered with 1.2-mm cover slide with ProLong mounting medium (Invitrogen). Collagen-I, TGF-β1, and laminin for all groups were completed in single day runs under the same conditions. Images were captured on an Olympus inverted epifluorescence microscope (IX51) fitted with a ×20 objective (numerical aperture: 0.45). Microscope and camera settings were kept constant throughout the process for each protein assessed using the Q-capture system. Images were quantified using the NIH ImageJ (Version 1.46) analysis program.
Oxidative Stress Markers
We measured total hydroperoxides as a marker of oxidative stress using the xylenol orange technique with FeSO4 (250 µM) in sulfuric acid (25mM) reacting with xylenol orange (100 µM) (25). The principle relies on the oxidization Fe2+ to Fe3+ when hydroperoxides are reduced. Ionized sulfuric acid assists in the reduction and prevents spontaneous reaction of Fe2+ with xylenol orange, unless oxidized by hydroperoxides first. The Fe3+ then reacts with the xylenol orange to form a Fe3+–xylenol complex and changes to a purple color. Absorbance of the purple Fe3+–xylenol complex was measured at 580nm. Concentrations of hydroperoxides were quantified against a t-butyl hydroperoxide standard curve.
8-Isoprostane was determined using a Cayman EIA kit. 4-Hydroxynonenal was assessed using a modified Western blot analysis.
DNA Fragmentation
Histone-associated DNA fragmentation was assessed using a cell death detection ELISA (Roche Applied Science). About 20 µL from supernatant was transferred into a microplate and 80 µL of the immunoreagent was added to each well, followed by gentle shaking of the plate for 2 hour at 25°C. Then, 100 µL of ABTS solution was pipetted to each well, and the solution was incubated on a plated shaker at 250rpm until the color development was sufficient for a photometric analysis (10 minutes). Finally, we measured absorbance of the oxidized ABTS solution at 405nm (reference wavelength of 490nm), using a microplate reader (Spectra Max 190; Molecular Devices).
TUNEL-Positive Staining
TUNEL evaluation of myonuclei positive for DNA strand breaks was determined using fluorescence detection (Roche Applied Science). Cross-sections of LV were cut (10 µm) in cryostat (−15°C), and then fixed in 4% paraformaldehyde for 20 minutes at room temperature in a Coplar jar. Sections were then permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 20 minutes at 4°C. The TUNEL reaction mixture containing terminal deoxynucleotidyl transferase, fluorescein-dUTP was added to the sections in 50-µL drops and incubated for 60 minutes at 37°C in a humidified chamber in the dark. The sections were rinsed three times in PBS for 5 minutes each and mounted in Vector antifade media with 4,6-diamidino-2-phenylindole to tag all nuclei. Sections were then visualized with a fluorescence microscope (Axiophot2; Carl Zeiss, Thornwood, NY) with parallel positive control (DNase-I) and negative controls (label solution only). TUNEL-positive nuclei were then counted on multiple sections of the LV.
Western Immunoblot
Protein levels for MnSOD, 4-hydroxynonenal adducts, Bax, and cleaved caspase-3 were determined by Western immunoblot analysis, in a manner similar to the outline mentioned previously (4). About 30 µg of LV homogenate were loaded on 10% polyacrylamide gels and electrophoresed using a Bio-Rad Protein III gel system. Briefly, solutions for separating gel (375mM Tris–HCl; pH = 8.8; 0.4% sodium dodecyl sulfate; 10% acrylamide) and stacking gel (125mM Tris–HCl; pH = 6.8; 0.4% sodium dodecyl sulfate; 10% acrylamide monomer) solutions were made, and polymerization was then initiated by tetramethylethylenediamine and ammonium persulfate. Separating and stacking gels were then quickly poured into a Bio-Rad Protein III gel-box (Bio-Rad, Hercules, CA). LV homogenates in sample buffer (Tris pH = 6.8 with 2% sodium dodecyl sulfate, 30 mM DTT, 25% glycerol) were then electrophoresed at 150 V for 50 minutes. The gels were then transferred at 30 V overnight onto a nitrocellulose membrane. Membranes were blocked in 5% nonfat milk in PBS with 0.1% Tween-20 for 6 hours. After blocking, membranes were incubated at room temperature in blocking buffer for overnight with the appropriate primary antibodies in blocking buffer: MnSOD (1:10,000, SC-7977; Santa Cruz Biotechnology), 4-hydroxynonenal (1:1,000, #393206; Calbiochem, San Diego, CA), Bax (1:200, SC-6236; Santa Cruz Biotechnology), and cleaved caspase-3 (1:500, #9661; Cell Signaling Technology, Beverly, MA). Following three membranes washings in PBS containing 0.1% Tween-20, membranes were then incubated in horseradish peroxidase–conjugated secondary antibodies for 1 hour. An enhanced chemiluminescence detection system (Amersham-Pharmacia Biotech., Piscataway, NJ) was used for visualization.
To ensure equal loading of protein, Ponceau-S staining was performed for each membrane. In addition, membranes were stripped and reprobed for glyceraldehyde-3-phosphate dehydrogenase (1:5,000, RGM2; Advanced Immunochemical, Long Beach, CA) to verify equal loading among lanes as an internal control. Densitometry (ie, area times grayscale relative to background) was performed using a Kodak film cartridge and film. NIH Image J analysis software was used for quantification of the blots.
Statistics
Data are presented as mean ± standard error of the mean. Two-way analysis of variance with Fishers-least significant difference used post hoc where appropriate were used to determine the existence of mean differences among test groups. Analysis of variance with Tukey’s post hoc test was used for analyzing collagen-I and TGF-β1 signal intensity. Significance level was set at 0.05.
Results
To demonstrate that MnSOD protein levels remained elevated in the old Tg mice, we measured protein abundance of LV from young (8 months) and old (28 months) WT and Sod2 Tg mice. As shown in Figure 1, the protein abundance of MnSOD in the LV was elevated by MnSOD genetic overexpression of YTg (+54%) compared with YWT mice. MnSOD protein abundance continued to be upregulated as well in the LV of OTg (+95%) compared with OWT. Total protein levels in the heart were not altered with age. Furthermore, we previously reported that MnSOD enzyme activity was increased approximately twofold in both young and old Tg mouse hearts (24). In summary, these data indicate that elevated MnSOD function was retained in Tg mice with advancing age.
Figure 1.
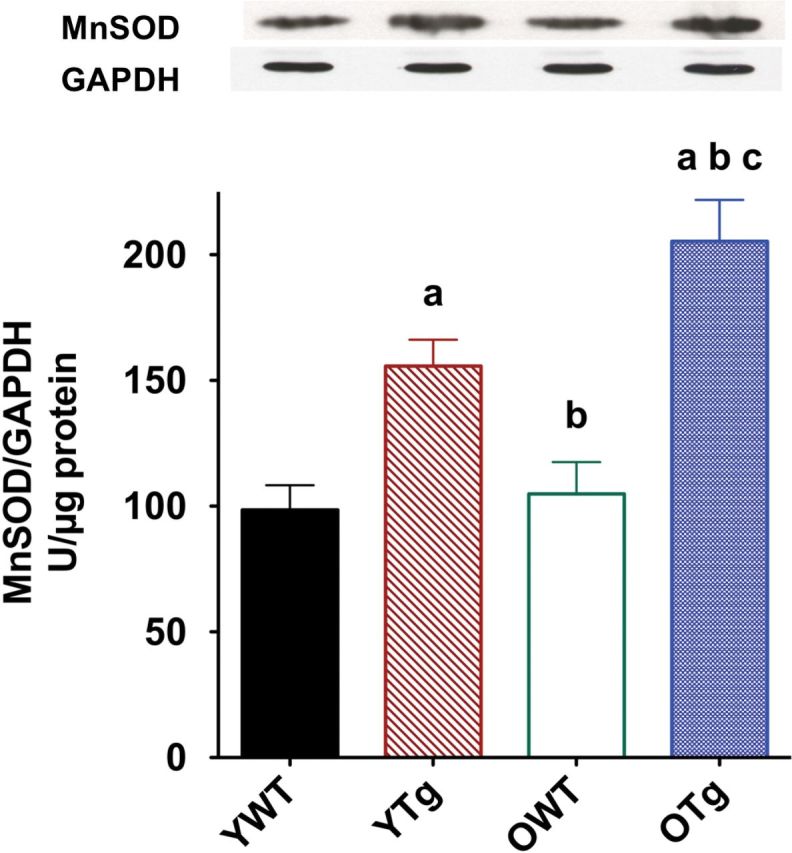
Mn isoform of superoxide dismutase (MnSOD) protein expression in left ventricle samples of wild-type (WT) and Sod2 transgenic (Tg) mice assigned to the following groups: young adult (8 mo) wild-type (YWT), young adult Sod2 Tg (YTg), old (28 mo) WT (OWT), and old Sod2 Tg (OTg). Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “b” indicates a significant difference vs YTg (p < .05). “c” indicates a significant difference vs OWT (p < .05).
We then tested the hypothesis that elevation of MnSOD protein levels reduced oxidative stress in the aging mouse heart. Indeed, hydroperoxide levels increased in the LV with age (+33%) but were decreased by MnSOD overexpression in the old (−29%) age group (Figure 2A). 8-Isoprostane was also measured as a marker of oxidative stress in heart from WT and MnSOD Tg mice. We found that 8-isoprostane was significantly elevated in the old WT, but not in the old Tg mice (Figure 2B). Similarly, 4-hydroxynonenal adducts, a marker of oxidative stress, were 20% higher in the LV of aging mice compared with young counterparts (Figure 2C). MnSOD overexpression significantly reduced 4-HNE by 31% in the LVs in the old age group (Figure 2C).
Figure 2.

Total hydroperoxides (A), 8-isoprostane (B), and 4-hydroxynonenal adducts (C) in left ventricle samples of young adult wild-type (YWT), young adult Sod2 Tg (YTg), old WT (OWT), and old Sod2 Tg (OTg). Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “c” indicates a significant difference vs OWT (p < .05).
We then assessed whether overexpression of the MnSOD gene and elevation of MnSOD protein levels protected against age-associated cardiac remodeling in mouse LV. Hematoxylin and eosin staining revealed significant structural changes in the LVs from old mice (Figure 3A and B). Extramyocyte space was dramatically greater in LVs from OWT than LVs from YWT (Figure 3C). In addition, the number of myonuclei per cross-sectional area from hematoxylin-stained sections was significantly decreased in the LVs from OWT compared with YWT (Figure 3D). These observations mirrored previous aging studies using the Fischer-344 strain of rat (4) and Fischer-344 × Brown Norway F1 hybrid strain of rat (21). In contrast, LV cross-sections from OTg exhibited less age-related cardiac remodeling than WTs, suggesting overexpression of MnSOD reduced cardiac remodeling in the aging LVs (Figure 3A–D). MnSOD overexpression and enhancement of protein levels also reduced extramyocyte space (−65%) in the OTg compared with OWT (Figure 3C). Furthermore, increasing MnSOD protein levels also enhanced the number of myonuclei (+191%) per cross-sectional area (Figure 3D).
Figure 3.
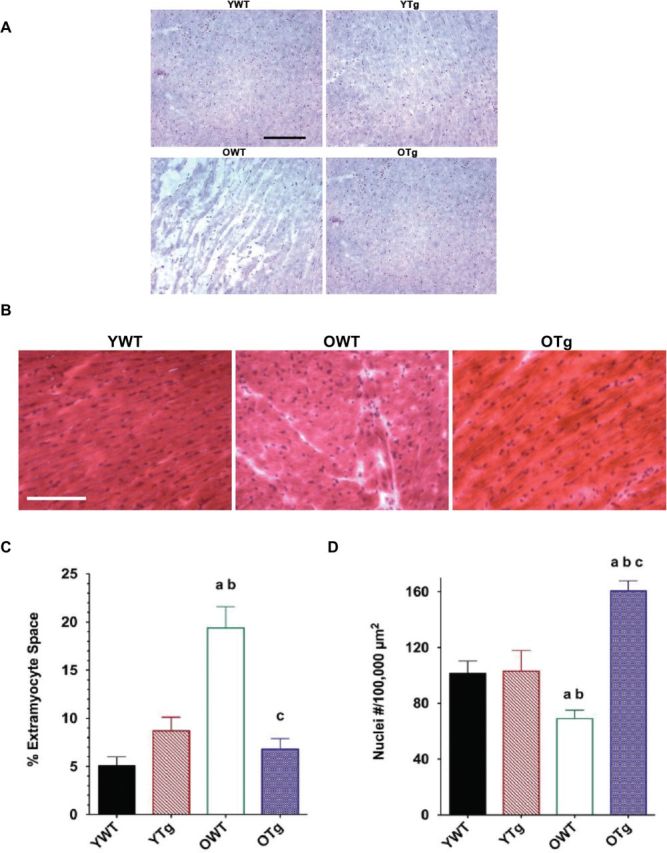
Histologic cross-sections stained with hematoxylin (×16; A), hematoxylin and eosin (×20; B), percent (%) extramyocyte space (C), and number (D) of myonuclei per 100,000 µm2 area in left ventricle samples of young adult wild-type (YWT), young adult Sod2 Tg (YTg), old WT (OWT), and old Sod2 Tg (OTg). Unstained areas indicate extramyocyte space for hematoxylin staining. Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “b” indicates a significant difference vs YTg (p < .05). “c” indicates a significant difference vs OWT (p < .05). Stage micrometer indicates 100 μm.
To determine whether age and MnSOD overexpression-induced differences in extramyocyte space could be explained by changes in connective tissue, we measured collagen-I localization or positive staining. Consistent with extramyocyte space described previously, we found that significant collagen-I positive staining was visualized in the LV samples from OWT compared with YWT. In contrast, overexpression of MnSOD resulted in limited collagen-I positive staining (Figure 4A and B), consistent with the notion that MnSOD protection against extracellular matrix proliferation with old age was linked to reduced levels of collagen-I. In healthy, young hearts, a thin, linear layer of connective tissue surrounds “pods” of myocardial cells. Collagen-I and hematoxylin and eosin staining revealed a less linear and thicker layers of connective tissue surrounding cardiomyocytes.
Figure 4.
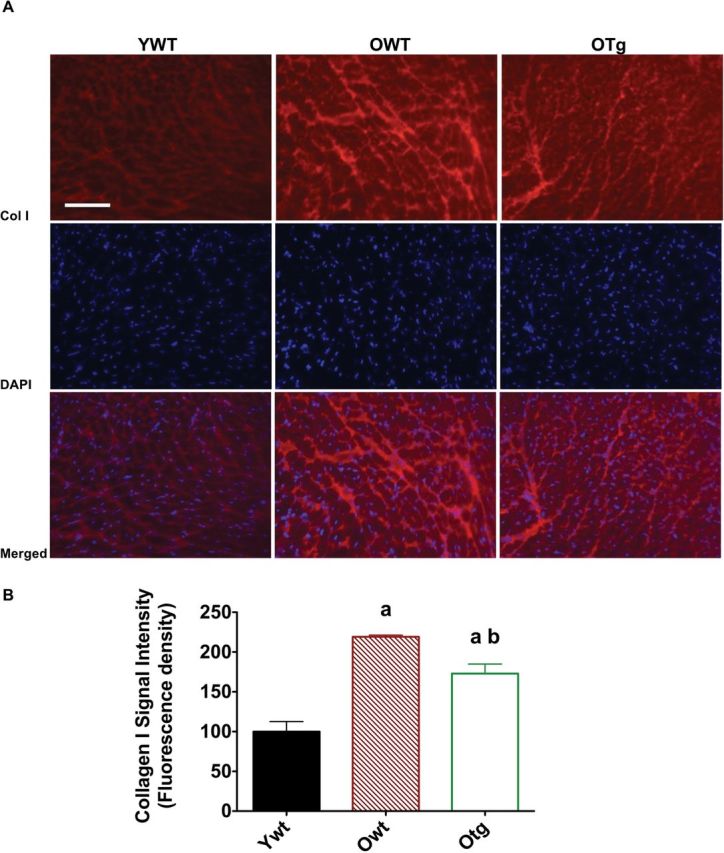
Collagen type-I (top), 4,6-diamidino-2-phenylindole DAPI (middle), and merged (bottom) are visualized by immunofluorescence staining (A; ×16), and collagen type-I signal intensity is shown (B) in left ventricle samples of young adult wild-type (YWT), old WT (OWT), and old Sod2 Tg (OTg). Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “b” indicates a significant difference vs OWT (p < .05).
In order to determine potential upstream mechanisms for protection against fibrosis in old hearts where MnSOD was overexpressed, we determined positive staining and protein abundance of the ECM-regulating cytokine TGF-β1, a consensus upstream regulator of fibrosis (26) and deposition of collagen-I and laminin. We used immunofluorescence staining with TGF-β1 and laminin-specific antibodies to visualize their location near the myocyte membrane and extramyocyte space. We found greater positive staining for TGF-β1 in LVs from the OWT mice compared with that from YWT (Figure 5A). Similar to the hematoxylin and eosin staining, a more web-like and alinear pattern was observed for TGF-β1 in the extramyocyte area (Figure 5B), as indicated by laminin-positive (Figure 5B) in LVs from OWT compared with YWT. Remarkably, MnSOD overexpression substantially mitigated age-induced elevation in TGF-β1 staining (Figure 5A and B), suggesting that MnSOD indeed protects against age-related fibrosis in the murine LVs.
Figure 5.

Transforming growth factor-β1 (TGF-β1; top) and laminin (bottom; A) are visualized by immunofluorescence staining (×20), and TGF-β1 signal intensity is shown (B) in left ventricle samples of young adult wild-type (YWT), old WT (OWT), and old Sod2 Tg (OTg). Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “b” indicates a significant difference vs OWT (p < .05).
Given that MnSOD is a mitochondrial-specific antioxidant enzyme, we tested the protective role of MnSOD overexpression against mitochondrial pro-apoptotic signaling (Bcl-2 pathway). Protein levels for Bax and cleaved (ie, active) caspase-3 were assessed in the LVs. No significant changes in Bax protein levels were found in the OWT compared with YWT. However, LVs from OTg exhibited significantly lower protein levels (−38%) for Bax by MnSOD overexpression (Figure 6A). Levels of cleaved caspase-3 (a marker of caspase-3 activation), downstream of the mitochondrial Bax (4), were significantly increased in the LVs of OWT compared with YWT (Figure 6B). Conversely, MnSOD overexpression resulted in a significant decrease (−41%) of cleaved caspase-3 protein levels in the LVs of old groups (Figure 6B).
Figure 6.
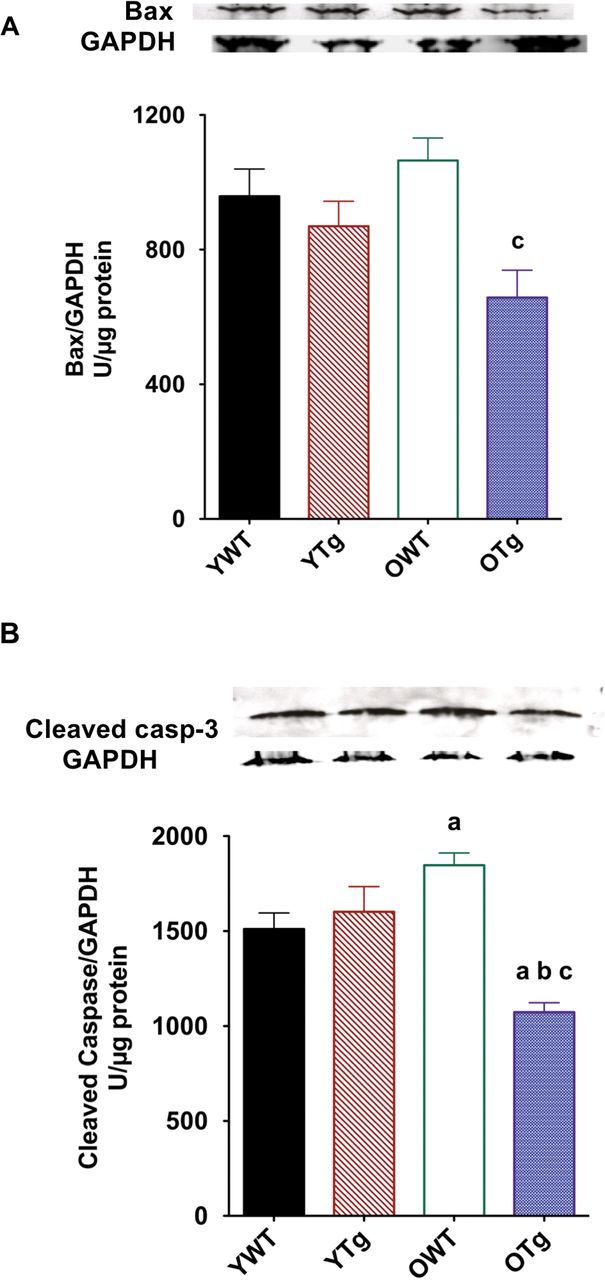
Protein expression for Bax (A) and cleaved caspase-3 (B) using Western immunoblotting in left ventricle samples of young adult wild-type (YWT), young adult Sod2 Tg (YTg), old WT (OWT), and old Sod2 Tg (OTg). Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “b” indicates a significant difference vs YTg (p < .05). “c” indicates a significant difference vs OWT (p < .05).
Given that enhancement of MnSOD levels provides significant protection against pro-apoptotic signaling (eg, Bax, cleaved caspase-3) in the aging heart, we tested the hypothesis that MnSOD overexpression would also mitigate age-induced elevation of apoptosis in the LV. Histone-associated DNA fragmentation and TUNEL-positive staining were used as direct markers of apoptosis and visualized using the TUNEL method with fluorescence microscopy. Positive controls revealed excellent co-localization with 4,6-diamidino-2-phenylindole and TUNEL-positive nuclei (Figure 7A). Representative TUNEL-positive nuclei and 4,6-diamidino-2-phenylindole fluorescence micrographs are shown in Figure 7A for LVs from YWT, OWT, and OTg groups, respectively. Aging increased TUNEL-positive nuclei in the LVs of WT mice. Conversely, MnSOD overexpression greatly reduced both markers of apoptosis in old Tg mice versus age-matched WTs. In addition, aging significantly increased histone-associated DNA fragmentation (Figure 7B) in the OWT LVs compared with YWT. MnSOD overexpression also attenuated DNA fragmentation in the LV in the old age group when compared with old WTs. In summary, these data indicate that the induction of MnSOD via genetic overexpression ameliorates the apoptotic process of myocytes in the LV of aging hearts.
Figure 7.

Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling-positive staining (A) is visualized by immunofluorescence staining (×40) in left ventricle (LV) samples of young adult wild-type (YWT), old WT (OWT), and old Sod2 Tg (OTg), and DNA fragmentation (B) was used as a marker of apoptosis through quantification of mononucleosomes and oligonucleosomes via ELISA in LV samples of YWT, young adult Sod2 Tg (YTg), OWT, and OTg. Data are expressed as means ± standard error of the mean. “a” indicates a significant difference vs YWT (p < .05). “b” indicates a significant difference vs YTg (p < .05).
Discussion
The goal of this study was to investigate the role of MnSOD in protection of fibrosis, apoptosis, and remodeling of the aging heart, by overexpressing the Sod2 gene. We found that Tg mice overexpressing MnSOD had reduced markers of oxidative stress (eg, total hydroperoxides, 4-HNE, 8-isoprostane) in the LV. MnSOD overexpression also attenuated age-associated elevation in extramyocyte space, collagen-I, and TGF-β1-positive staining in the LVs of old mice. In addition, enhancement of MnSOD proteins levels in Sod2 Tg mice protected against age-related increase of pro-apoptotic signaling (eg, Bax, cleaved caspase-3) and apoptosis (eg, TUNEL-positive nuclei, DNA fragmentation) in the LVs of mouse hearts. These results are consistent with the hypothesis that enhancement of MnSOD in the aging heart attenuates age-associated elevation in markers of oxidative stress and remodeling. Indeed, this is the first data to indicate that MnSOD overexpression has a protective effect against age-induced remodeling in the aging heart. A discussion of the primary findings follows.
MnSOD Overexpression and Oxidative Stress in Aging Heart
The MnSOD is a mitochondrial matrix enzyme that catalyzes the dismutation of superoxide anions (O2•−), produced from complexes I and III of the electron transport chain, to H2O2. H2O2 then diffuses out of mitochondrial matrix, or undergoes further reduction to water by other antioxidant enzymes such as glutathione peroxidase or catalase. MnSOD is thus an important first defense against mitochondrial oxidative stress and protection of mitochondrial integrity (24). In this study, we showed that Sod2 Tg mice significantly expressed higher MnSOD protein levels in the LVs of both young and old mice (Figure 1). Consistent with these observations, our previous report showed that the activity of MnSOD increased approximately twofold in heart of Sod2 Tg mice (24). Although overexpression of MnSOD does not increase life span, it does reduce pathology (24). Overexpression of catalase localized in the mitochondria enhances longevity (27,28), suggesting that mitochondrial damage via H2O2 reduces life span. In addition, the heterozygous knockout (SOD−/+) exhibits significant pathology (20). Thus, mitochondrial function is critical to both health and longevity, and may be disrupted by oxidative stress.
Consistent with previous observations, heterozygous knockout of the Sod2 gene demonstrates the importance of MnSOD role in the antioxidant defense system (11–15). Indeed, MnSOD Tg mice models with homozygous (Sod2 −/−) or heterozygous (Sod2 −/+) null mutations in the Sod2 gene display pathologic phenotypes. For example, Sod2 −/− knockout mice exhibited severe oxidative mitochondrial injury and cardiomyopathy (14,15). In addition, Sod2 −/+ mice also have decreased mitochondrial respiration and impaired activities of mitochondrial enzymes (13). Increased oxidative damage to mitochondria is typical of heterozygous knockout mice (11–13). Furthermore, Sod2 −/+ mice have a greater susceptibility to permeability transition pore opening and thus apoptosis (11,13). Therefore, protection against fibrosis and apoptosis observed in this study is consistent with the genetic ablation data for MnSOD. Moreover, it is possible that mitochondrial H2O2 may shorten life span, whereas elevated O2•− may enhance age-related pathology. However, additional study is warranted to test this hypothesis directly.
Indeed, aging is characterized by enhanced oxidative stress and mitochondrial dysfunction in the heart (9,22,24,29,30). There is emerging evidence that oxidative stress plays a central role in pathophysiology such as insulin resistance, apoptosis, activation of NF-κB, and proteolytic systems (eg, calpains, caspase-3, ubiquitin/proteasome) (31–33). In this study, we observed elevation of total hydroperoxides, 4-HNE, and 8-isoprostane as markers of oxidative stress in the hearts with advancing age (Figure 2). Previously, MnSOD overexpression in skeletal muscle reduced protein carbonyls and F2-isoprostanes indicators of protein oxidation and lipid peroxidation and oxidative stress that were elevated with aging (24). This study is the first direct evidence that overexpression of MnSOD attenuates oxidative stress in the LV of old mammals, as indicated by lower total hydroperoxides, 4-HNE, and 8-isoprostane adducts.
Although the mechanisms underlying reduced oxidative stress with MnSOD overexpression are not fully delineated, it is logical that reduced levels of mitochondrial superoxide in the old Tg mice may protect against lipid peroxide and radical formation via suppressing Haber-Weiss or Fenton reactions. Furthermore, our results also suggest an important role of mitochondrial superoxide in aging of the myocardium. Thus, MnSOD plays an important role in protecting the mitochondria from oxidative stress and damage by enzymatically scavenging superoxide anions generated as a by-product of the mitochondrial respiration. These results are consistent with previous findings, which indicated that cardiomyocytes of Sod2 −/+ mice were more sensitive to oxidative stress than those of Sod2 +/+ mice (14–16,20). MnSOD may protect against protonation of superoxide anions (O2•−), a highly reactive species, in old Tg mice. However, this hypothesis is untested.
MnSOD protein levels were overexpressed in the young, but markers of oxidative stress were not reduced. This suggests that overexpression of MnSOD may be more effective when (a) reactive oxygen species levels are elevated or (b) mice are old. We have noticed in previous studies that the effect of exercise training on oxidative stress and apoptosis is more pronounced in older rats (4). Therefore, MnSOD performs an important protective role against oxidative damage and remodeling primarily in the aging heart.
MnSOD Overexpression and Fibrosis in Aging Heart
We also observed that MnSOD overexpression attenuated age-induced remodeling (eg, increased extramyocyte space, collagen-I) in the LVs of OTg (Figures 3 and 4). Previous data have demonstrated that cardiac remodeling occurs consistently with aging, characterized by a loss of cardiac myocytes, reactive hypertrophy of remaining cardiomyocytes, and excessive accumulation of extracellular matrix (4,8,34,35). Consistent with previous findings, old WT mice expressed higher level of pro-fibrotic proteins (collagen-I, TGF-β1) and resulted in a more web-like geometry of collagenous sheaths, with removal of more linear sheaths surrounding myocytes in the LV.
Interestingly, our findings in old MnSOD Tg mirror previous data that demonstrate exercise training upregulates MnSOD and reduces fibrosis and apoptosis in the aging rat heart (4,8,9). This supports the notion that interventions, including exercise, that upregulate MnSOD play a protective role against remodeling in the aging heart. In this study, overexpression of MnSOD reduced markers of fibrosis (eg, extramyocyte space, collagen-I, TGF-β1). Thus, we have established three tenants. First, oxidative stress is indeed causal in the age-associated fibrotic process. Second, mitochondrial reactive oxygen species may play a role in fibrosis in the aging heart. Third, at least part of the mechanism of exercise protection against age-associated fibrosis and remodeling in the aging heart may be attributed to inducibility of MnSOD.
Recent evidence has linked age-related cardiac fibrosis to elevated levels of TGF-β1, a potent stimulator of collagen synthesis via fibroblasts (8,36–39). Previously, we found that exercise training mitigated age-related upregulation of TGF-β1 in the FBNF1 rat (8). In this study, we observed that MnSOD overexpression substantially reduced levels of TGF-β1 (Figure 5) and collagen-I (Figure 4) in the aging LV of OTg mice versus age-matched WTs. These finding support the hypothesis that MnSOD is involved in inhibitory regulation of TGF-β1 and cardiac fibrosis. Further research is required to delineate the redox-sensitive mechanism(s) responsible for the link between MnSOD and TGF-β1 in ameliorating cardiac fibrosis in the aging heart.
MnSOD Overexpression and Apoptosis in Aging Heart
We expected that overexpression of MnSOD would reduce mitochondrial-mediated apoptotic signaling and apoptosis, as part of age-related remodeling. Oxidative stress can induce activation of the Bcl-2 family of pro-apoptotic proteins including Bax and downstream activation of caspase-3, a key promoter of apoptosis (21). Caspase-3 activation via cleavage of the pro-form is usually a consequence of an increase in mitochondrial permeability transition pore opening and cytochrome c release (21,40). In this study, Sod2 Tg mice displayed significantly lower levels of the pro-apoptotic protein Bax and cleaved caspase-3 in hearts of the old age group compared with WT of the same age (Figure 6A and B). In addition, MnSOD overexpression also reduced direct markers of apoptosis in the aging LV: TUNEL-positive myonuclei and DNA fragmentation (Figure 7). Therefore, the current sets of data indicate that MnSOD protects against age-related elevation of mitochondrial-mediated apoptosis in the aging mouse heart.
Our data are also consistent with previous studies establishing an inhibitory relationship between MnSOD and apoptosis. Previously, Van Remmen and colleagues(20) showed that heterozygous MnSOD (Sod2 −/+) knockout mice exhibited increased permeability transition pore and apoptosis in the heart. In addition, Majima and colleagues(41) reported that overexpression of MnSOD protected cells against cell death induced by alkaline condition in a mouse fibrosarcoma cell line. Lee and colleagues(42) recently demonstrated that exercise increased MnSOD levels and prevented ischemia–reperfusion-induced elevation of pro-apoptotic proteins in heart mitochondria. Therefore, emerging data, including our study, are consistent with the notion that upregulation of MnSOD via overexpression or exercise training (9) may indeed suppresses excess apoptosis in the aging heart (4).
Study Limitations
Unfortunately, we did not have enough frozen tissue remaining in the young Tg mice to complete TGF-β1 and collagen-I analyses, as well as Western blots on all four groups of mice. Thus, we may have missed a potential interactive effect of age and overexpression of MnSOD, and our conclusions are less robust. Because oxidative stress markers were not significantly depressed in the YTg group and collagen-I levels are low, we suspect a minimal effect in overexpressing MnSOD in the young group, but further study is warranted. TGF-β1-positive staining in the LV appears much different than both young and old WTs, suggesting a potential regulatory role of MnSOD on pro-fibrotic signaling. In addition, we were not able to measure in vivo cardiac function (echocardiography) or in an isolated heart preparation. Thus, we cannot be certain that blunting of fibrosis in remodeling in the OTg mice was accompanied by improved functional measures (eg, diastolic filling, shortening velocity, ejection fraction). Clearly, further study is warranted to elucidate the molecular and physiological mechanisms underlying remodeling in the aging heart.
In summary, MnSOD overexpression protected against oxidative stress, fibrosis, and apoptosis in the aging mouse heart. Age-related elevation of cardiac collagen-I and TGF-β were abrogated in the MnSOD Tg mouse. In addition, the old MnSOD Tg mouse experienced significant less pro-apoptotic signaling (Bax, cleaved caspase-3) than the old WT. We conclude that MnSOD is an important protective protein against fibrosis and remodeling in the aging heart. Our results are also consistent with the hypothesis that exercise-induced upregulation of MnSOD in the aging heart previously observed in our laboratory (9) may be cardioprotective against fibrosis (8) and apoptosis (4).
Funding
This study was supported by the American Heart Association (0555064Y and 0855158F), National Institutes of Health (AR054084), National Aeronautics and Space Administration (NASA; NNX12AR62G), and the Sydney and J. L. Huffines Institute for Sports Medicine.
References
- 1. Li Q, Ren J. Influence of cardiac-specific overexpression of insulin-like growth factor 1 on lifespan and aging-associated changes in cardiac intracellular Ca2+ homeostasis, protein damage and apoptotic protein expression. Aging Cell. 2007;6:799–806. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y, Herman B. Ageing and apoptosis. Mech Ageing Dev. 2002;123:245–260. [DOI] [PubMed] [Google Scholar]
- 3. Torella D, Rota M, Nurzynska D, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–524. [DOI] [PubMed] [Google Scholar]
- 4. Kwak HB, Song W, Lawler JM. Exercise training attenuates age-induced elevation in Bax/Bcl-2 ratio, apoptosis, and remodeling in the rat heart. FASEB J. 2006;20:791–793. [DOI] [PubMed] [Google Scholar]
- 5. Kajstura J, Cheng W, Sarangarajan R, et al. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am J Physiol. 1996;271:H1215–H1228. [DOI] [PubMed] [Google Scholar]
- 6. Centurione L, Di Giulio C, Cacchio M, et al. Correlations between protein kinase C zeta signaling and morphological modifications during rat heart development and aging. Mech Ageing Dev. 2003;124:957–966. [DOI] [PubMed] [Google Scholar]
- 7. Higami Y, Shimokawa I. Apoptosis in the aging process. Cell Tissue Res. 2000;301:125–132. [DOI] [PubMed] [Google Scholar]
- 8. Kwak HB, Kim JH, Joshi K, Yeh A, Martinez DA, Lawler JM. Exercise training reduces fibrosis and matrix metalloproteinase dysregulation in the aging rat heart. FASEB J. 2011;25:1106–1117. 10.1096/fj.10-172924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawler JM, Kwak HB, Kim JH, Suk MH. Exercise training inducibility of MnSOD protein expression and activity is retained while reducing prooxidant signaling in the heart of senescent rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1496–R1502. 10.1152/ajpregu.90314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holley AK, Dhar SK, Xu Y, St Clair DK. Manganese superoxide dismutase: beyond life and death. Amino Acids. 2012;42:139–158. 10.1007/s00726-010-0600-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raha S, McEachern GE, Myint AT, Robinson BH. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med. 2000;29:170–180. [DOI] [PubMed] [Google Scholar]
- 12. Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. [DOI] [PubMed] [Google Scholar]
- 13. Lebovitz RM, Zhang H, Vogel H, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. [DOI] [PubMed] [Google Scholar]
- 15. Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98:2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strassburger M, Bloch W, Sulyok S, et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med. 2005;38:1458–1470. [DOI] [PubMed] [Google Scholar]
- 17. Silva JP, Shabalina IG, Dufour E, et al. SOD2 overexpression: enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J. 2005;24:4061–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Motoori S, Majima HJ, Ebara M, et al. Overexpression of mitochondrial manganese superoxide dismutase protects against radiation-induced cell death in the human hepatocellular carcinoma cell line HLE. Cancer Res. 2001;61:5382–5388. [PubMed] [Google Scholar]
- 19. Suzuki K, Murtuza B, Sammut IA, et al. Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia-reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation. 2002;106:I270–I276. [PubMed] [Google Scholar]
- 20. Van Remmen H, Williams MD, Guo Z, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;281:H1422–H1432. [DOI] [PubMed] [Google Scholar]
- 21. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. [DOI] [PubMed] [Google Scholar]
- 22. Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol. 2007;292:C1983–C1992. [DOI] [PubMed] [Google Scholar]
- 23. Xu X, Arriaga EA. Qualitative determination of superoxide release at both sides of the mitochondrial inner membrane by capillary electrophoretic analysis of the oxidation products of triphenylphosphonium hydroethidine. Free Radic Biol Med. 2009;46:905–913. 10.1016/j.freeradbiomed.2008.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang YC, Pérez VI, Song W, et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J Gerontol A Biol Sci Med Sci. 2009;64:1114–1125. 10.1093/gerona/glp100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hermes-Lima M, Willmore WG, Storey KB. Quantification of lipid peroxidation in tissue extracts based on Fe(III)xylenol orange complex formation. Free Radic Biol Med. 1995;19:271–280. [DOI] [PubMed] [Google Scholar]
- 26. Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819. [DOI] [PubMed] [Google Scholar]
- 27. Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. [DOI] [PubMed] [Google Scholar]
- 28. Dai DF, Santana LF, Vermulst M, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. 10.1161/CIRCULATIONAHA.108.822403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu JL, Wu QP, Yang XF, et al. L-malate reverses oxidative stress and antioxidative defenses in liver and heart of aged rats. Physiol Res. 2008;57:261–268. [DOI] [PubMed] [Google Scholar]
- 30. Judge S, Jang YM, Smith A, et al. Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1564–R1572. [DOI] [PubMed] [Google Scholar]
- 31. Reid MB. Response of the ubiquitin-proteasome pathway to changes in muscle activity. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1423–R1431. [DOI] [PubMed] [Google Scholar]
- 32. Powers SK, Kavazis AN, DeRuisseau KC. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288:R337–R344. [DOI] [PubMed] [Google Scholar]
- 33. Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. 10.1172/JCI37048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walker EM, Jr, Nillas MS, Mangiarua EI, et al. Age-associated changes in hearts of male Fischer 344/Brown Norway F1 rats. Ann Clin Lab Sci. 2006;36:427–438. [PubMed] [Google Scholar]
- 35. Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–1568. [DOI] [PubMed] [Google Scholar]
- 36. Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. 2004;9:43–51. [DOI] [PubMed] [Google Scholar]
- 37. Voloshenyuk TG, Landesman ES, Khoutorova E, Hart AD, Gardner JD. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine. 2011;55:90–97. 10.1016/j.cyto.2011.03.024 [DOI] [PubMed] [Google Scholar]
- 38. Chan EC, Peshavariya HM, Liu GS, Jiang F, Lim SY, Dusting GJ. Nox4 modulates collagen production stimulated by transforming growth factor β1 in vivo and in vitro. Biochem Biophys Res Commun. 2013;430:918–925. 10.1016/j.bbrc.2012.11.138 [DOI] [PubMed] [Google Scholar]
- 39. Pan X, Chen Z, Huang R, Yao Y, Ma G. Transforming growth factor β1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS One. 2013;8:e60335. 10.1371/journal.pone.0060335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. [DOI] [PubMed] [Google Scholar]
- 41. Majima HJ, Oberley TD, Furukawa K, et al. Prevention of mitochondrial injury by manganese superoxide dismutase reveals a primary mechanism for alkaline-induced cell death. J Biol Chem. 1998;273:8217–8224. [DOI] [PubMed] [Google Scholar]
- 42. Lee Y, Min K, Talbert EE, et al. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med Sci Sports Exerc. 2012;44:397–405. 10.1249/MSS.0b013e318231c037 [DOI] [PubMed] [Google Scholar]