

Abstract
Recent advances in improved force fields and sampling methods have made it possible for the accurate calculation of protein–ligand binding free energies. Alchemical free energy perturbation (FEP) using an explicit solvent model is one of the most rigorous methods to calculate relative binding free energies. However, for cases where there are high energy barriers separating the relevant conformations that are important for ligand binding, the calculated free energy may depend on the initial conformation used in the simulation due to the lack of complete sampling of all the important regions in phase space. This is particularly true for ligands with multiple possible binding modes separated by high energy barriers, making it difficult to sample all relevant binding modes even with modern enhanced sampling methods. In this paper, we apply a previously developed method that provides a corrected binding free energy for ligands with multiple binding modes by combining the free energy results from multiple alchemical FEP calculations starting from all enumerated poses, and the results are compared with Glide docking and MM-GBSA calculations. From these calculations, the dominant ligand binding mode can also be predicted. We apply this method to a series of ligands that bind to c-Jun N-terminal kinase-1 (JNK1) and obtain improved free energy results. The dominant ligand binding modes predicted by this method agree with the available crystallography, while both Glide docking and MM-GBSA calculations incorrectly predict the binding modes for some ligands. The method also helps separate the force field error from the ligand sampling error, such that deviations in the predicted binding free energy from the experimental values likely indicate possible inaccuracies in the force field. An error in the force field for a subset of the ligands studied was identified using this method, and improved free energy results were obtained by correcting the partial charges assigned to the ligands. This improved the root-mean-square error (RMSE) for the predicted binding free energy from 1.9 kcal/mol with the original partial charges to 1.3 kcal/mol with the corrected partial charges.
1. Introduction
Molecular dynamics (MD) simulations are widely used to study biological systems, such as protein–ligand complexes. Free energy calculations based on MD, such as Thermodynamic Integration (TI) and Free Energy Perturbation (FEP), use alchemical transformations to determine the free energy of going from one state to another.1,2 These calculations can be used to determine the relative binding free energy of two ligands by transforming one ligand to another while bound to a protein. This can be used to determine which ligand will bind with a greater affinity to the protein.
The accuracy of free energy calculations depends on the ability of the system to sample all relevant conformations as well as the accuracy of the underlying force field.3−5 For ligands with multiple possible binding poses separated by large barriers in the potential energy, it is very difficult to adequately sample all possible ligand poses,6 which is essential for the accurate calculation of the binding free energy. This causes the resulting free energy to be biased based on the initial conformation of the ligand.
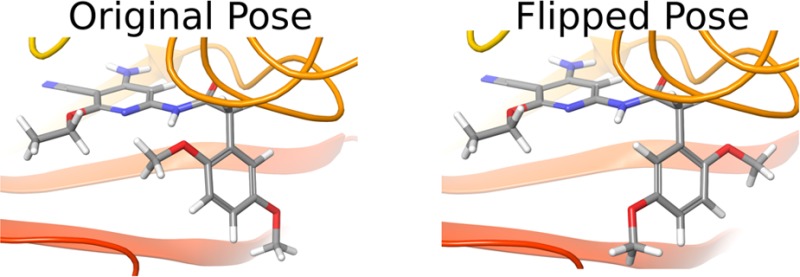
Many methods have been developed to overcome this sampling challenge in MD-based free energy calculations. The confine and release method and Umbrella Sampling (US) use harmonic restraints to force the system to sample certain states, and the free energy is determined incorporating all sampled states.7,8 Alternatively, Metadynamics modifies the potential energy along a set of collective variables to reduce the time spent sampling in potential wells, allowing the system to explore alternative conformations.9 These methods require prior knowledge about the important conformations of the system. Other methods, such as Replica Exchange with Solute Tempering (REST),10−13 Accelerated Adaptive Integration Method (AcclAIM),14 and accelerated MD (aMD),15−17 alter the underlying potential energy surface in a way that decreases the barriers between relevant conformations and recovers the equilibrium distribution by reweighting the sampled conformations. However, in some cases, the energy barriers separating the relevant conformations are very high so that even these enhanced sampling methods can not easily overcome the barrier. An example of this is a series of ligands that bind to c-Jun N-terminal kinase-1 (JNK1). These ligands have a phenyl ring with asymmetric substitutions, which cause the ligand to have two possible binding modes. The large size of the substituted phenyl ring and the steric restrictions of the protein environment make it very difficult to sample the two modes due to the large barrier between them. Even with these enhanced sampling techniques, the barrier between these conformations is too high to overcome, causing the free energy results to depend on the initial conformation of the phenyl ring.
Besides sampling, the accuracy of free energy calculations also depends on the accuracy of the force fields. The accuracy of the force fields can be assessed by comparing the potentials generated by the force fields with that from more accurate Quantum Mechanics (QM) calculations or by comparing the calculated physical properties with experimental data.5,18−20 In principle, the closer the fit between the force field potential and the QM potential, the more accurate the force field is, though there are limitations on the system size that can be studied directly using QM.
In this work, we describe a protocol to exhaustively sample the various enumerated binding poses in a systematic fashion, using rigorous FEP calculations. FEP calculations are run, alchemically transforming a common reference molecule to the ligands of interest. Separate simulations are run for the different possible ligand poses, explicitly sampling the fluctuations of the system associated with each pose. Then, the corrected free energy is calculated by combining the free energy results from these simulations. This protocol is applied to a series of JNK1 ligands, some of which have established binding modes as determined by crystallography. The ability for the method to accurately determine the favored ligand conformation is also investigated. For the ligands that do not have a known binding mode, the FEP calculated binding free energy is compared to the experimental value to determine the accuracy of the result. This protocol explicitly separates ligand sampling error from force field error, because the possible conformations for the ligand that are separated by large barriers are explicitly sampled in separate simulations—i.e., to the extent that we can exhaustively enumerate all of the regions of phase space that are separated by large energy barriers, convergence can be guaranteed, and the resulting error is likely due to the force field. Interestingly, the systematic deviations from the experimental affinities were seen for a subset of the JNK1 ligands suggesting a possible force field error. A modification to the calculation of the ligand partial charges improves the match between the force field potential and the QM potential, leading to improved free energy results as compared to experiment. This demonstrates how this protocol can be used to determine the presence of a possible inaccuracy in the force field. Further examination of the force field can then lead to improvement of the force field, thus improving the accuracy of the binding free energies.
In addition to the FEP calculations, Glide docking and MM-GBSA calculations were also performed to predict the dominant ligand binding mode, and the results are compared with the FEP predictions. We found that only FEP correctly predicted the dominant binding mode for all the ligands studied here, while both MM-GBSA and Glide docking failed to predict the dominant binding mode for at least some of the ligands.
2. Theory and Methods
For a ligand that has multiple possible binding poses when bound to the protein receptor, if these poses are separated by high energy barriers, sampling all of the possible conformations in one simulation is very difficult even with enhanced sampling methods. For example, the JNK1 ligands shown in Figure 1 have two possible binding poses while bound to the JNK1 receptor, which differ by the flipping of the phenyl ring. When large branched functional groups are attached to the phenyl ring, flipping the ring requires the rearrangement of the surrounding protein residues, making it difficult to sample both poses even with an enhanced sampling method like REST.
Figure 1.
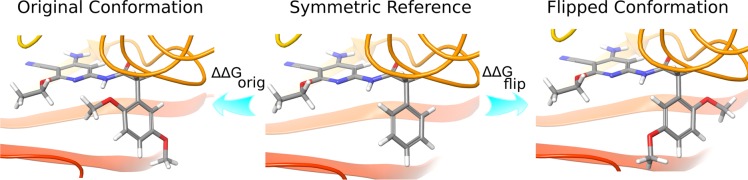
Free energies are calculated from the reference to the original conformation (ΔΔGorig) and to the flipped conformation (ΔΔGflip). Then, the corrected free energy can be calculated from these simulations using eq 8.
Here, rather than attempting to sample all of the possible poses in a single simulation, we adopt a method that combines the free energy results from multiple FEP calculations, each sampling one binding mode. Specifically, two alchemical FEP calculations are run from a common reference molecule that has an unbranched symmetric phenyl ring to the target molecule. The starting conformation of the target molecule corresponds to the two possible initial binding poses, one for each FEP calculation. Because the energy barrier is high enough that the flipping of the phenyl does not occur in the relatively short time scale of the FEP calculations, the relative binding free energy between the reference molecule and the target molecule can be calculated by separating the phase space into two components, each corresponding to one orientation of the phenyl ring. The derivation of the corrected free energy, which uses the results from multiple FEP calculations, is reviewed here, based on the description in previous work.8,21−23 The free energy for the isothermal–isobaric ensemble is related to the partition function Δ by
| 1 |
The partition function is calculated using the integral over phase space
| 2 |
where N is the number of atoms in the system, H is the Hamiltonian, p is the pressure, V is the volume, p is the momentum, and q are the atomic coordinates. The integrals can be separated into multiple components, as long as the sum includes all of phase space. In this work, the integral is separated into two, corresponding to each orientation of the phenyl ring
| 3 |
where the sum is over each ligand conformation i. Then using eq 1, this can be written as
| 4 |
where Gi is the free energy calculated over the phase space corresponding to the ith conformation.
Rather than calculate the absolute free energy, we calculate the relative binding free energy between two ligands A and B
| 5 |
where the subscript bound refers to the protein–ligand complex and the subscript free refers to the ligand free in solution. Only the ligands bound to the protein have difficulty sampling both phenyl ring orientations. Thus, eq 4 is only applied to the bound state free energies resulting in
| 6 |
where ΔGfreeA→B is the relative solvation free energy. The reference molecule, A, contains a phenyl ring, which has two degenerate states due to its symmetry. Then, the absolute free energies can be replaced by the relative free energies in the bound state giving
| 7 |
where the factor of 2 accounts for the two degenerate states of the reference molecule. Including the relative solvation free energy and labeling the two ligand conformations as “orig” and “flip” gives the corrected free energy
| 8 |
For the example shown here, the ligand only has two possible binding poses, but this method can be generalized for ligands with additional binding poses.
From these two separate FEP calculations, the dominant ligand binding mode can also be predicted—the pose with lower free energy is the dominant binding mode. Glide docking and MM-GBSA were also examined as possible methods to predict the more favorable binding mode. With Glide docking, both poses were docked into the receptor, with torsional restraints to prevent the ring from flipping. In some cases, docking was unable to generate a pose that matched the restrictions on the distance to the core and the torsional restraint. In these cases, the chosen conformation was the only conformation that was not filtered out due to these conditions. In cases where both poses were assigned a docking score, the pose with the lower docking score was chosen. With MM-GBSA, both poses were examined in separate calculations. The pose that had the lower MM-GBSA binding energy was chosen. Additional details of the Glide docking and MM-GBSA protocols are discussed later in Section 2.2.
The accuracy of the results was determined by comparing the predicted dominant binding mode to the available crystal structures and by comparing the predicted binding free energies to the experimental data. The Mean Unsigned Error (MUE) and Root Mean Squared Error (RMSE) were computed using the relative binding free energies. The predicted absolute binding free energy (Predicted dG) was also calculated by taking the experimental value for the reference molecule and adding it to the relative binding free energies. The coefficient of determination (R2) was computed using the absolute binding free energies.
The above protocol explicitly includes the major possible conformations for the ligand, so a deviation in the predicted free energy from the experimental value suggests a possible inaccuracy in the force field. Therefore, possible inaccuracies in the underlying force field were examined by comparing the corrected free energy to the experimental values.
2.1. JNK1 Series of Ligands Studied
JNK1 is an enzyme that has been implicated in the development of insulin resistance.24 Inhibitors of this enzyme have been explored to attempt to treat this path of insulin resistance.25,26 The ligands studied here are shown in Table 1, which also have been studied experimentally.26 Half of these ligands include an R1 methoxy group (red), which includes two ligands with a crystal structure (PDB 2H96(25) and 2GMX(26)). For this set of ligands, we can infer the dominant phenyl ring conformation based on these crystal structures. The second set of ligands does not have an R1 methoxy group (blue), and therefore the dominant ring conformation is not known.
Table 1. JNK1 Ligands Studieda.
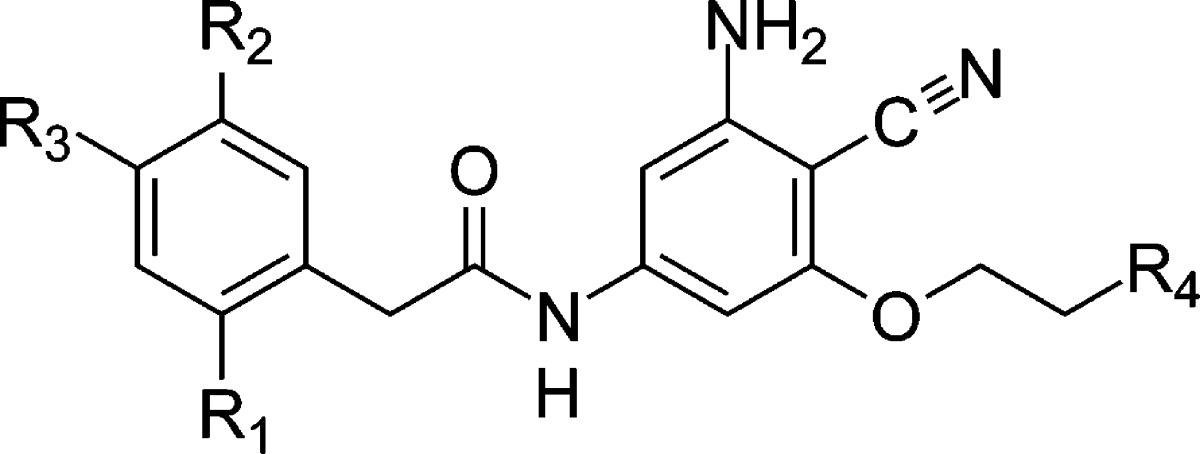

The reference ligand is shown in black, ligands with an R1 methoxy group are shown in red, and the additional ligands are shown in blue. Me is methyl, Ms is methanesulfonyl, and Ac is acetamide. Ref indicates the reference ligand, and Sym indicates the ligand is symmetric about rotation of the phenyl ring. These ligands have been studied experimentally.26 The crystal structure for 18634_2h96 is PDB 2H96(25) and the structure for 17124_2gmx is PDB 2GMX.26
The initial coordinates came from the crystal structure of ligand 17124 bound to JNK1 (PDB 2GMX(26)). The protein preparation wizard in Maestro 2014-3 was used to prepare the structure,27−32 by adding missing atoms. The bound complexes for the other ligands were based on this structure, with the ligand modified as needed. Both possible ring orientations were prepared as separate structures.
2.2. Simulation Details
Glide docking, MM-GBSA, and FEP calculations were examined to predict the dominant binding pose. These calculations were set up and run using Maestro 2014-3,28 unless otherwise noted. The ligands started in either of the two possible ring poses. Docking was run using Glide Extended Precision,33−36 with flexible ligand sampling. However, the core was restricted to 1 Å of the reference position, with the core shown in Figure S1 in the Supporting Information. Torsional restraints were also applied to the phenyl ring to prevent it from flipping to the alternate pose. MM-GBSA simulations were run using Prime,32,37,38 with the VSGB model39 for solvation, the OPLS2.118−20 force field, and minimization for sampling. As with Glide docking, both possible ring poses were run separately, with the best scoring pose used for further analysis.
FEP calculations were set up using the FEP Mapper in Maestro 2014-3.28 Perturbations were run between the reference molecule and the target molecule in each pose. Simulations were run using the OPLS2.118−20 force field, with production calculations run for 5 ns per λ window under constant pressure. In total, 12 λ windows were run, using the FEP/REST protocol.13,40,41 This protocol combines FEP with the REST to improve conformational sampling of the ligands. This is a Hamiltonian Replica Exchange method, where the potential energy of the ligand atoms near the region being perturbed are scaled at intermediate λ values, but not at λ = 0 or 1. This reduces the barrier separating the relevant ligand conformations, thereby improving ligand conformational sampling. However, when there is a large barrier separating the major ligand conformations, such as with the ligands studied in this work, FEP/REST will not be able to improve ligand conformational sampling and alternative methods, such as the corrected free energy (eq 8), need to be used. The Hamiltonian Replica Exchange method, where exchanges are attempted between different λ windows, is also called λ-hopping. Desmond18,42−44 was used to run the calculations.
The OPLS2.1 force field uses CM1A-BCC based partial charges for the ligands.45,46 In this method, charges are obtained from a combination of the Cramer-Truhlar CM1A charge model and specifically fit bond charge correction terms (BCC) to improve the accuracy of the resulting charges. The details about the OPLS2.1 force field is presented in a prior publication.47 The modified OPLS force field, labeled here as OPLS2.1 QM Charges, uses Jaguar48,49 to derive the partial charges for the ligands.
The ligand structure was modified so that the ring was halfway between the two possible poses, so that the charges were not biased to a particular ligand conformation. A single point energy calculation was performed, with the atomic electrostatic potential (ESP) fit to the atom centers. The QM calculation used HF/6-31*G for the ligands, with the exception of 17124. For this ligand, which contains Bromine atoms, HF/lacv3p* was used. These charges were then used for the FEP calculations, using a newer version of Desmond,50,51 which allows alternative charges to be used for the calculation.
3. Results and Discussion
3.1. Dominant Binding Poses Predicted by Different Methods
The dominant binding pose predicted by Glide docking, MM-GBSA, and FEP calculations for the ligands with an R1 methoxy group is shown in Figure 2. This shows the difference in the predicted score or free energy value between the crystal binding mode and the flipped conformation. Values below zero indicate that the crystal binding mode is predicted to be more favored, in agreement with experiment.
Figure 2.
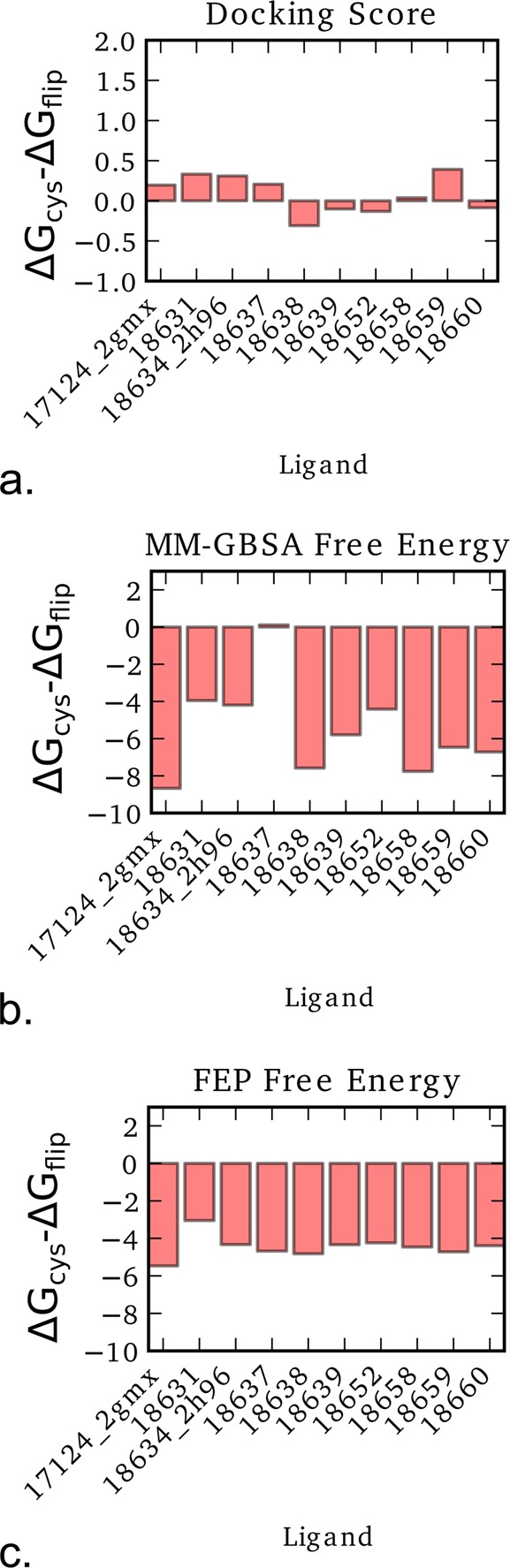
Difference in the docking score or predicted free energy values in kcal/mol for the R1 methoxy ligands in the crystal binding mode (ΔGcys) and the flipped conformation (ΔGflip). Results are shown for (a) Glide docking, (b) MM-GBSA, and (c) FEP calculated free energies. Values below zero indicate that the crystal binding mode is more favored, matching the expected result.
Glide docking is able to correctly predict the binding mode for only four of the ligands, indicating that for this set of partially solvent exposed R-groups, where the configurational entropy and solvent effects may play a significant role, Glide docking may not necessarily be a reliable method for correctly predicting the binding mode. MM-GBSA performed better, where only one ligand, 18637, had a predicted dominant pose not matching the experimental data. For this ligand, the MM-GBSA score was very close for both poses, with the difference in the predicted MM-GBSA binding free energy below 0.2 kcal/mol. The phenyl ring of the flipped pose is rotated such that the R2 methoxy group is close to the position occupied by the R1 methoxy group of the crystal-like conformation. This may be the reason for the small difference in the predicted MM-GBSA binding free energy. These results indicate that MM-GBSA is more reliable than Glide docking for predicting the binding modes of the studied R-groups for this set of ligands. Interestingly, using FEP, the binding mode for all ligands in this set is correctly predicted. Therefore, at least for this admittedly very limited testing, FEP is the most reliable for correctly predicting the binding mode of such R-groups.
3.2. Free Energy Results
Next, the free energy results for the ligands with an R1 methoxy group are examined. The Glide docking score and the FEP calculated free energy from the docking pose is shown in Figure 3a,c, respectively. The Glide docking score predicts the ligands to bind more favorably than the reference shown in black, which is consistent with the experimental trend. However, there is little distinction among the ligands, with many ligands having nearly the same score. As discussed above, only four of these ligands are predicted to have a binding mode matching that of the crystal structure. When using the Glide docking predicted dominant poses as the initial structure for FEP calculations, the predicted free energies have a large root mean squared error (RMSE) as seen in Figure 3c.
Figure 3.

Results for the ligands with an R1 methoxy group. The black mark indicates the reference ligand, and the red marks indicate ligands containing an R1 methoxy group. The upper row shows (a) the Glide docking score and (b) the MM-GBSA binding free energy for the more favorable pose compared to the experimental binding free energy. The lower row shows the FEP calculated free energy for the more favorable pose which was predicted using (c) Glide docking and (d) MM-GBSA. Equation 8 was used to calculate (e) the corrected free energy. The solid black line represents a perfect fit to the experimental free energies. The dashed lines represent a 1 kcal/mol deviation from experiment, and the dotted lines represent a 2 kcal/mol deviation. The predicted dG values were computed by adding the predicted relative binding free energy between the reference and each ligand to the experimental absolute binding free energy for the reference.
The MM-GBSA predicted free energy and the FEP calculated free energy from the MM-GBSA pose is shown in Figure 3b,d, respectively. According to the MM-GBSA binding free energy, the ligands are predicted to bind more favorably than the reference ligand. This is consistent with the experimental free energy, even though the incorrect binding mode is predicted for one of the ligands. This suggests that the MM-GBSA binding free energy is able to capture the experimental trend, despite the incorrect prediction of the binding mode for one ligand. The FEP calculated free energy from the dominant MM-GBSA pose reflects the more accurate prediction of the initial binding mode, with an improved RMSE compared to the docking results. Thus, the MM-GBSA method seems to work reasonably well in predicting the binding mode for this set of ligands, though improvements could be made so that the correct binding mode is predicted for all of the ligands.
The corrected free energy calculated using eq 8 is shown in Figure 3e. These calculations favored the pose matching the crystal structure for all of these ligands, leading to a small reduction in the RMSE. This method to determine the corrected free energy is theoretically rigorous to account for the lack of proper sampling in the simulations.8,22,23 However, even with the corrected free energy, there still is a significant deviation from experiment for these ligands. The RMSE is just under 1.9 kcal/mol. The corrected free energy takes into consideration the different possible ligand poses, so the large RMSE suggests a possible inaccuracy in the force field, which is studied in detail in next section.
3.3. Force Field Error
The predicted free energy for adding the R1 methoxy group to the phenyl ring is more favorable using the default OPLS2.1 force field than is observed experimentally. We are explicitly sampling both possible ring poses, so the error in the free energy suggests a possible error in the force field. This error could be caused by the potential energy for the interaction between the R1 methoxy group and the protein being too strong, the conformational potential energy for the two ring states making the crystal-like binding mode too favorable, or a combination of these factors.
To examine this problem further, we used a representative ligand to compare the force field potential to the Quantum Mechanics (QM) potential as shown in Figure 4. The structure of the representative ligand is shown in Figure 4, with R2 and R3 set to Hydrogen and the phenyl group attached to the amide nitrogen turned into a methyl group. The most favorable state in the potential energy corresponds to the crystal-like state shown in Figure 4c. The other potential well corresponds to the flipped conformation shown in Figure 4a.
Figure 4.
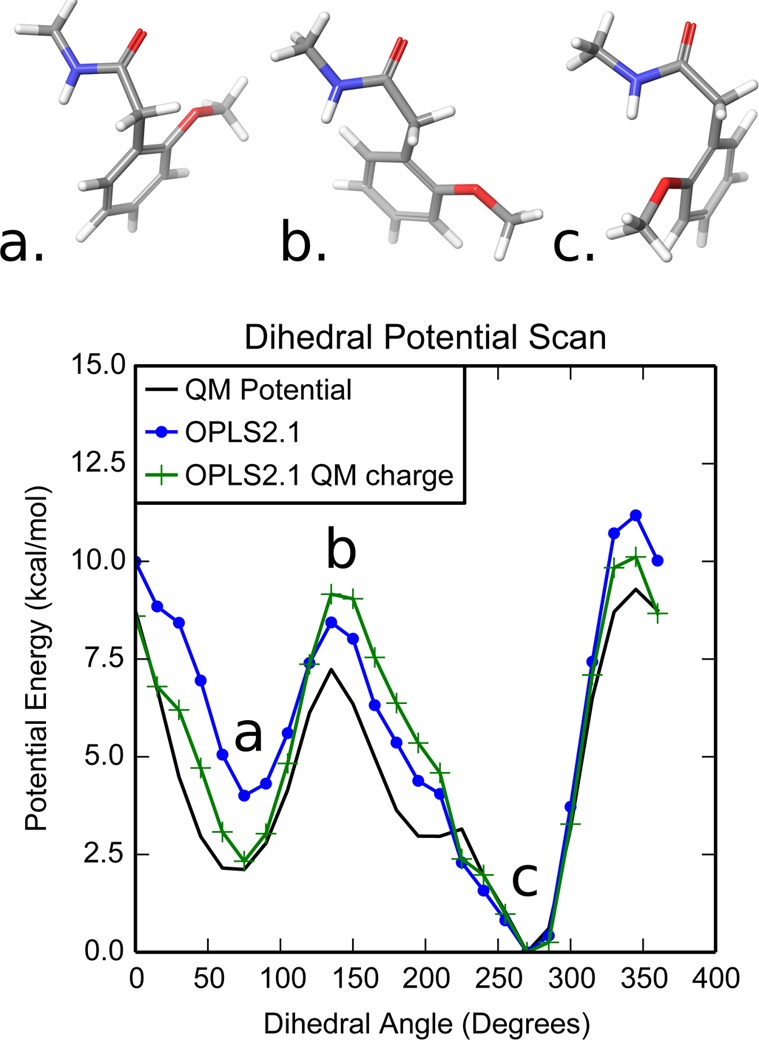
Representative molecule with an R1 methoxy group and the corresponding force field and QM potential energy as a function of the dihedral angle. The molecule is shown in (a) the flipped conformation, (b) the conformation halfway way between used to calculate the QM charges, and (c) the crystal-like binding mode. The locations of these conformations in the potential energy scan are labeled. The QM potential energy (black line) shows the crystal-like state is the minimum potential, while the flipped state is 2.1 kcal/mol higher in energy. With the original OPLS2.1 charges (blue circles), the flipped state is 4.0 kcal/mol higher in energy. Using the QM charges (green crosses), the flipped state is 2.3 kcal/mol higher in energy, which closely matches the QM potential.
With the default OPLS2.1 charges, the difference in the force field potential energy between the two states is 4.0 kcal/mol, while the QM potential shows a difference of only 2.1 kcal/mol. Thus, the error in the force field causes the crystal-like pose to be overstabilized relative to the flipped pose. To address the discrepancies between the QM potential and the force field potential, we refitted the partial charges assigned on the ligands atoms as described in the Theory and Methods section. As shown in the plot in Figure 4, using the QM derived charges more accurately models the conformational stability of the R1 methoxy group, compared with the original charge model. Using the QM charges, the difference in the potential energy between the two poses is reduced to 2.3 kcal/mol, which closely matches the QM potential.
3.4. Free Energy Results Using the QM Charges
The predicted free energy using the original and QM charges is shown in Figure 5. With the QM charges, the predicted free energies are now much closer to the experimental free energies, reducing the RMSE to 1.3 kcal/mol.
Figure 5.
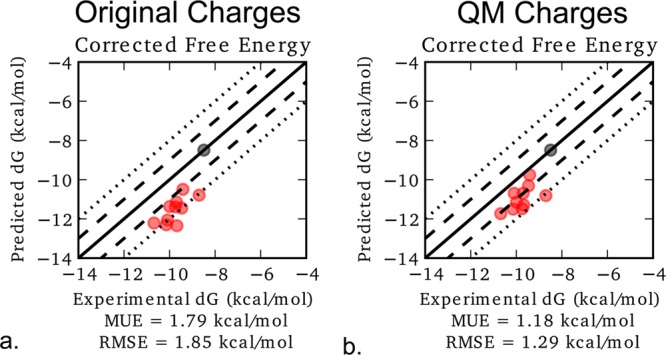
Predicted absolute binding free energy compared to the experimental values for (a) the original charges and (b) the QM charges. The black mark indicates the reference ligand, and the red marks indicate ligands with an R1 methoxy group. The solid black line represents a perfect fit to the experimental free energies. The dashed lines represent a 1 kcal/mol deviation from experiment, and the dotted lines represent a 2 kcal/mol deviation. The predicted dG values were computed by adding the predicted relative binding free energy between the reference and each ligand to the experimental absolute binding free energy for the reference.
Using the original charges, the largest deviation from the experimental free energy was 2.6 kcal/mol, which was seen for ligand 17124. Using the QM charges, the largest deviation is now 2.1 kcal/mol, which is seen for ligand 18660 (Table S5 in the Supporting Information).
Overall, these results show that by accurately modeling the conformational stability of the R1 methoxy group using QM derived charges, the accuracy of the free energy calculations improves significantly. These results indicate, perhaps unsurprisingly, that the reliable calculation of the protein–ligand binding free energy not only requires the accurate characterization of the interaction energy between the protein and the ligand but also requires correct characterization of the potential energy difference between the different ligand conformations. This also highlights how explicitly calculating the relative binding free energy from different conformations can be used to help distinguish sampling issues from force field issues. By doing so, improvements can be made to the force field, resulting in more accurate free energies.
3.5. Additional Ligands
Finally, we examine the results including the ligands without an R1 methoxy group. For consistency with the ligands with an R1 methoxy group, the QM charges were used for these free energy calculations. The Glide docking results are shown in Figure 6a,c. The Glide docking score predicts all of the ligands to bind more favorably than the reference ligand. However, the experimental results show that four of these ligands actually bind less favorably than the reference. Many of the ligands have nearly the same docking score, indicating that Glide docking has difficulty distinguishing between the different ligands. The FEP calculated free energy from the docking pose, shown in Figure 6c, has an RMSE of 1.4 kcal/mol, reflecting the fact that Glide docking incorrectly predicted the binding mode for some of the ligands.
Figure 6.

Results including the additional set of ligands using the QM charges. The black mark indicates the reference ligand, the red marks indicate ligands with an R1 methoxy group, and the blue marks indicate ligands without an R1 methoxy group. The upper row shows (a) the Glide docking score and (b) the MM-GBSA binding free energy for the more favorable pose compared to the experimental binding free energy. The lower row shows the FEP calculated free energy for the more favorable pose which was predicted using (c) Glide docking and (d) MM-GBSA. Equation 8 was used to calculate (e) the corrected free energy. The solid black line represents a perfect fit to the experimental free energies. The dashed lines represent a 1 kcal/mol deviation from experiment, and the dotted lines represent a 2 kcal/mol deviation. The predicted dG values were computed by adding the predicted relative binding free energy between the reference and each ligand to the experimental absolute binding free energy for the reference.
The MM-GBSA results are shown in Figure 6b,d. As with the Glide docking score, the MM-GBSA binding free energy predicts all of the ligands to bind more favorably than the reference ligand. However, there is more of a distinction between the R1 methoxy ligands and the additional ligands, where the latter are generally predicted to bind less favorably, in line with the experimental trend. Using the MM-GBSA pose, the FEP calculated free energy has an RMSE of 1.1 kcal/mol. However, the coefficient of determination R2 between the experimental and predicted absolute binding free energies is low, with a value of 0.41 (Table S8 in the Supporting Information), suggesting this method may have difficulty in rank ordering the ligands.
The corrected free energy results are shown in Figure 6e, where most of the ligands match closely to experiment, with an RMSE of 1.1 kcal/mol. Comparing to the FEP calculations using the MM-GBSA pose, using the corrected free energy improves R2 to 0.61. This shows that using the corrected free energy gives a better rank ordering for the ligands compared to using the MM-GBSA pose. The authors note that, for the same set of molecules, different perturbations can be performed to rank order their binding affinities, and the RMSE depends on the particular set of transformations studied. Recently, a study of the same set of ligands using the default OPLS2.1 force field and a different set of transformations was published, with an RMSE of 1.0 kcal/mol.47 Using this set of transformations, the RMSE error, including the correction to the force field discussed herein, drops to 0.9 kcal/mol. These results are a very good match to experiment and show how the corrected free energy along with the QM charges can be used to calculate accurate results, even when the dominant ligand pose is not known prior to the simulation.
4. Alternative Method To Deal with Multiple Binding Poses
The above proposed method involves running two separate transformations, from the reference molecule containing a phenyl ring to the target molecule in each of the two possible poses. Then, the corrected free energy is calculated using eq 8. This is necessary because large barriers separate the two possible ring conformations, so the predicted free energy is dependent on the initial ring conformation, as shown in Table 2. The average absolute difference in the free energy between the two initial poses is 1.7 kcal/mol. This value is large, as expected, because each simulation is sampling a different ring conformation.
Table 2. FEP Calculated Free Energy Given in kcal/mol for the Simulations Using the Original Reference with a Phenyl Groupa.

Errors were estimated using the bootstrap method. The free energy is given for simulations starting in the original conformation (Orig.) and the flipped conformation (Flip.). For the reference (ref) and symmetric (sym) ligands, the same value was used for both conformations. The absolute difference (|Diff.|) between these two values is also given. The average absolute difference (Avg. |Diff.|) is given in the last row.
However, an alternative reference molecule could be used instead to facilitate rotation of the substituted phenyl ring when it is decoupled and thus not interacting with the rest of the system. An example of this is replacing the unsubstituted phenyl ring of the reference with a methyl group. If both possible poses are properly sampled during the simulation, then the resulting free energy will be independent of the initial ring conformation. Using this alternative reference with the JNK1 series of ligands works well for most ligands, decreasing the average absolute difference to 0.7 kcal/mol, as shown in Table 3, column Methyl 5 ns. This improves sampling for the two poses, but there are still a few ligands where the free energy depends on the initial pose. Extending these simulations to 10 ns per replica improves the average absolute difference to 0.4 kcal/mol, as shown in Table 3, column Methyl 10 ns.
Table 3. FEP Calculated Free Energy Given in kcal/mol for the Simulations Using the the Alternative Reference with a Methyl Groupa.
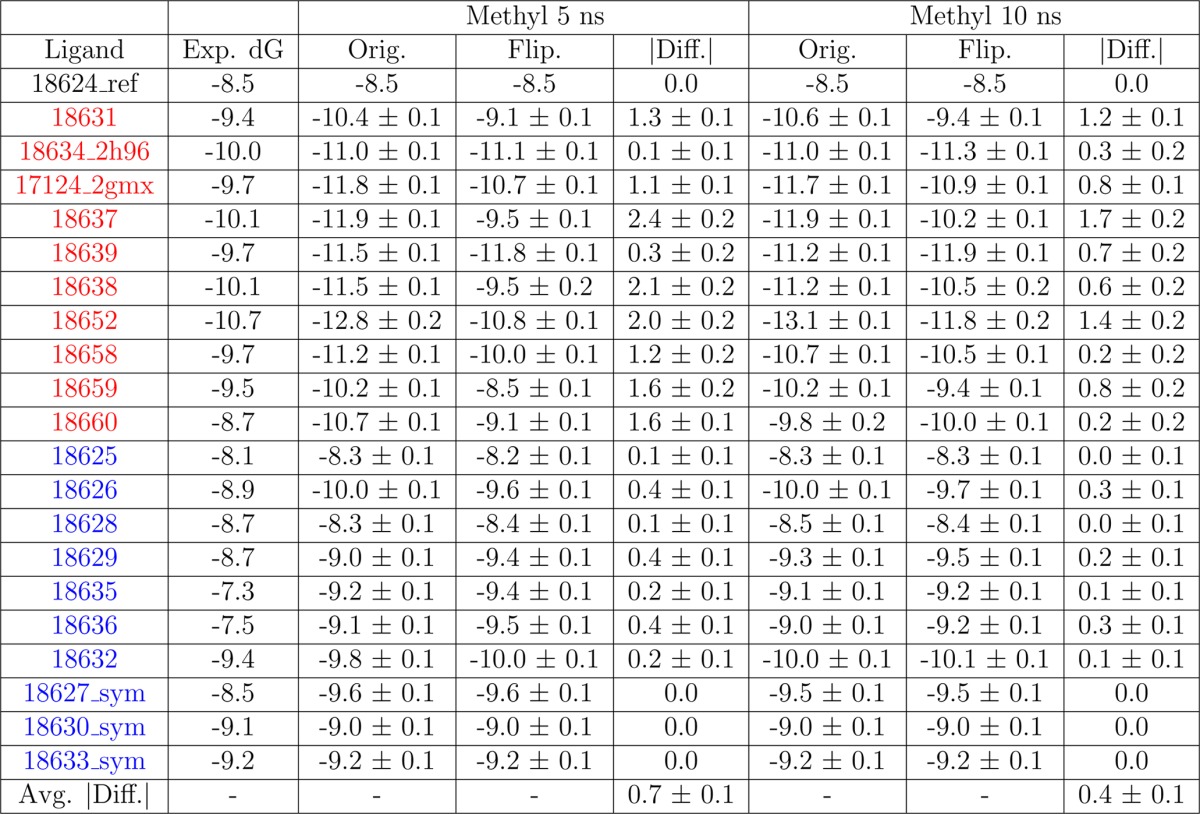
Errors were estimated using the bootstrap method. The results are also shown after extending the alternative reference simulations to 10 ns per replica. For these methyl reference simulations, the absolute predicted binding free energy was calculated using the experimental binding free energy for the phenyl reference, 18624_ref, adding the predicted relative binding free energy between 18624_ref and the methyl reference, and adding the predicted relative binding free energy between the methyl reference and the corresponding ligand. The free energy is given for simulations starting in the original conformation (Orig.) and the flipped conformation (Flip.). For the reference (ref) and symmetric (sym) ligands, the same value was used for both conformations. The absolute difference (|Diff.|) between these two values is also given. The average absolute difference (Avg. |Diff.|) is given in the last row.
Thus, for cases where each possible binding pose is known, the free energy can be calculated by explicitly sampling each ligand pose in a separate simulation and using eq 8 to determine the corrected free energy. For cases where these poses are not known, simulations may be started from a single pose, using a smaller intermediate reference ligand to improve conformational sampling. Running these simulations for an extended time will improve sampling and therefore the accuracy of the resulting free energy.
It may seem somewhat anomalous that large perturbations involving the annihilation and restoration of an aromatic ring with an attached R-group are found to be more convergent than the simpler strategy of leaving the aromatic ring untouched and just adding the R-group. This anomaly can be resolved by considering both the ease with which the relevant barriers can be traversed as well as the size of the perturbation. In this case, using the alternative reference molecule reduces the effective barrier associated with ring flipping, the slow degree of freedom, when it is decoupled from the system. While the size of the perturbation is large, there are still sufficient transitions between λ windows using the λ-hopping protocol such that conformations sampled in the decoupled state propagate to the other λ windows. An extremely large perturbation could prevent proper transitions between λ windows, indicating the need for additional λ windows or the use of multiple intermediate molecules. Similar results have been observed using the separated topologies approach, where a larger perturbation resulted in better convergence for ligands that can require reorientation in the binding site.23 Thus, if the nature of the sampling challenges are understood, one may be able to identify less obvious perturbation paths leading to more convergent simulations.
5. Conclusions
In this work, three methods for predicting the correct binding mode of a ligand were compared: Glide docking, MM-GBSA, and the corrected free energy from FEP calculations. Docking was not able to reliably predict the correct binding mode for the JNK1 ligands. MM-GBSA performed better, with only a single error in predicting the favored binding mode. The most rigorous corrected free energy correctly predicted the binding mode for all cases.
Using the corrected free energy, we compared the predictions to the experimental values and saw a systematic deviation for the subset of ligands with an R1 methoxy group, resulting in an RMSE of 1.9 kcal/mol. Because we are sampling both poses of the ligand, the error was attributed to an error in the force field. Comparison of the force field potential and the QM potential on a representative molecule showed the cause of the deviation was due to a difference in the relative stability of the two possible ring conformations. Calculating the partial charges using QM corrects this difference and significantly improves the match between the predicted free energy and the experimental values, resulting in an RMSE of 1.3 kcal/mol.
The protocol illustrates how multiple FEP calculation results can be combined to predict the binding free energies of ligands with multiple possible binding poses. Extensive sampling of all the possible ligand binding poses using the method makes deviations in the predicted binding free energy and experimental values indicative of a possible inaccuracy in the force field. We showed an example of how the force field error in the ligands is identified using this method, and improved free energy results are obtained when using an improved charge model for the ligands that better matches with the QM calculations.
We also explored an alternative protocol using a smaller reference molecule to improve sampling of the ring conformations, which may be useful if the possible ligand poses are not known prior to running the free energy calculations. We find that larger perturbations including more heavy atoms in the system can actually be more convergent than smaller perturbations if the motions of those perturbed heavy atoms have greater freedom in the decoupled state and there are sufficient transitions between λ windows using a λ-hopping protocol such that the resulting conformations can propagate into the other λ windows.
Acknowledgments
J.W.K., E.H., T.L., R.A., and L.W. are supported by Schrödinger. Additional support for J.W.K. and J.A.M. is provided by the NIH (NIH GM031749), the NIH Molecular Biophysics Training Program from the National Institute of General Medical Sciences (T32 GM08326), the NSF (MCB 1020765), the Howard Hughes Medical Institute, Achievement Rewards for College Scientists, and the National Biomedical Computation Resource.
Supporting Information Available
Core atoms used for docking, the structure of the representative molecule used to compare the OPLS2.1 and QM potential energy, and the calculated free energy values are available in the Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.5b00214.
The authors declare no competing financial interest.
Supplementary Material
References
- Straatsma T. P.; McCammon J. A. Annu. Rev. Phys. Chem. 1992, 43, 407–435. [Google Scholar]
- Straatsma T. P.; McCammon J. A. J. Chem. Phys. 1991, 95, 1175–1188. [Google Scholar]
- Wereszczynski J.; McCammon J. A. Q. Rev. Biophys. 2011, 45, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J. D.; Mobley D. L.; Shirts M. R.; Dixon R. W.; Branson K.; Pande V. S. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp K. A.; Lin Y.-S.; Das R.; Pande V. S. J. Chem. Theory Comput. 2012, 8, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley D. L.; Dill K. A. Structure 2009, 17, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souaille M.; Roux B. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar]
- Mobley D. L.; Chodera J. D.; Dill K. A. J. Chem. Theory Comput. 2007, 3, 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongelli V.; Bonomi M.; Parrinello M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 6358–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Kim B.; Friesner R. A.; Berne B. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13749–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Friesner R. A.; Berne B. J. J. Phys. Chem. B 2011, 115, 9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moors S. L. C.; Michielssens S.; Ceulemans A. J. Chem. Theory Comput. 2011, 7, 231–237. [DOI] [PubMed] [Google Scholar]
- Wang L.; Berne B. J.; Friesner R. A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaus J.; Arrar M.; McCammon J. A. J. Phys. Chem. B 2014, 118, 5109–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D.; Mongan J.; McCammon J. A. J. Chem. Phys. 2004, 120, 11919. [DOI] [PubMed] [Google Scholar]
- de Oliveira C.; Hamelberg D.; McCammon J. A. J. Chem. Theory Comput. 2008, 4, 1516–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrar M.; de Oliveira C. A. F.; Fajer M.; Sinko W.; McCammon J. A. J. Chem. Theory Comput. 2013, 9, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar D.; Williams J.; Wu Y.; Damm W.; Shelley J.; Sherman W. J. Chem. Theory Comput. 2010, 6, 1509–1519. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.; Maxwell D.; Tirado-Rives J. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar]
- Jorgensen W.; Tirado-Rives J. J. Am. Chem. Soc. 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- Straatsma T. P.; McCammon J. A. J. Chem. Phys. 1989, 90, 3300. [Google Scholar]
- Mobley D. L.; Chodera J. D.; Dill K. A. J. Chem. Phys. 2006, 125, 084902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocklin G. J.; Mobley D. L.; Dill K. A. J. Chem. Phys. 2013, 138, 085104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre V.; Uchida T.; Yenush L.; Davis R.; White M. F. J. Biol. Chem. 2000, 275, 9047–9054. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Serby M. D.; Xin Z.; Szczepankiewicz B. G.; Liu M.; Kosogof C.; Liu B.; Nelson L. T. J.; Johnson E. F.; Wang S.; Pederson T.; Gum R. J.; Clampit J. E.; Haasch D. L.; Abad-Zapatero C.; Fry E. H.; Rondinone C.; Trevillyan J. M.; Sham H. L.; Liu G. J. Med. Chem. 2006, 49, 4455–4458. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz B. G.; Kosogof C.; Nelson L. T. J.; Liu G.; Liu B.; Zhao H.; Serby M. D.; Xin Z.; Liu M.; Gum R. J.; Haasch D. L.; Wang S.; Clampit J. E.; Johnson E. F.; Lubben T. H.; Stashko M. A.; Olejniczak E. T.; Sun C.; Dorwin S. A.; Haskins K.; Abad-Zapatero C.; Fry E. H.; Hutchins C. W.; Sham H. L.; Rondinone C. M.; Trevillyan J. M. J. Med. Chem. 2006, 49, 3563–3580. [DOI] [PubMed] [Google Scholar]
- Sastry G.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Maestro, version 9.9; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Protein Preparation Wizard, release 2014-3; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Epik, version 2.9; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Impact, version 6.4; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Prime, version 3.7; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Friesner R.; Murphy R.; Repasky M.; Frye L.; Greenwood J.; Halgren T.; Sanschagrin P.; Mainz D. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shaw D. E.; Shelley M.; Perry J. K.; Francis P.; Shenkin P. S. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Glide, version 6.4; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J. F.; Honig B.; Shaw D. E.; Friesner R. A. Proteins: Struct Funct., Bioinf. 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Friesner R.; Xiang Z.; Honig B. J. Mol. Biol. 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
- Li J.; Abel R.; Zhu K.; Cao Y.; Zhao S.; Friesner R. A. Proteins: Struct Funct., Bioinf. 2011, 79, 2794–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Deng Y.; Knight J. L.; Wu Y.; Kim B.; Sherman W.; Shelley J.; Lin T.; Abel R. J. Chem. Theory Comput. 2013, 9, 1282–1293. [DOI] [PubMed] [Google Scholar]
- Cole D. J.; Tirado-Rives J.; Jorgensen W. L. J. Chem. Theory Comput. 2014, 10, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z.; Mohanty U.; Noehre J.; Sawyer T. K.; Sherman W.; Krilov G. Chem. Biol. Drug Des. 2010, 75, 348–359. [DOI] [PubMed] [Google Scholar]
- Maestro-Desmond Interoperability Tools, version 3.9; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Desmond Molecular Dynamics System, version 3.9; D. E. Shaw Research: New York, NY, 2014. [Google Scholar]
- Storer J.; Giesen D.; Cramer C.; Truhlar D. J. Comput.-Aided Mol. Des. 1995, 9, 87–110. [DOI] [PubMed] [Google Scholar]
- Jakalian A.; Jack D. B.; Bayly C. I. J. Comput. Chem. 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wu Y.; Deng Y.; Kim B.; Pierce L.; Krilov G.; Lupyan D.; Robinson S.; Dahlgren M. K.; Greenwood J.; Romero D. L.; Masse C.; Knight J. L.; Steinbrecher T.; Beuming T.; Damm W.; Harder E.; Sherman W.; Brewer M.; Wester R.; Murcko M.; Frye L.; Farid R.; Lin T.; Mobley D. L.; Jorgensen W. L.; Berne B. J.; Friesner R. A.; Abel R. J. Am. Chem. Soc. 2015, 137, 2695–2703. [DOI] [PubMed] [Google Scholar]
- Bochevarov A.; Harder E.; Hughes T.; Greenwood J.; Braden D.; Philipp D.; Rinaldo D.; Halls M.; Zhang J.; Friesner R. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar]
- Jaguar, version 8.5; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Maestro-Desmond Interoperability Tools, version 4.0; Schrödinger, Inc.: New York, NY, 2014. [Google Scholar]
- Desmond Molecular Dynamics System, version 4.0; D. E. Shaw Research: New York, NY, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.