

Abstract
Segmental neurofibromatosis (SNF) is a rare type of neurofibromatosis (NF-1) resulting from post-zygotic somatic mutations in the neurofibromin gene that leads to mosaicism. Reported manifestations of SNF include neurofibromas, freckling, or café-au-lait spots limited to a single body region or limb.
We present a 5-month-old male referred to our clinic for evaluation of congenital excessive skin folds on the back. A mildly erythematous, poorly demarcated soft plaque was noted, consisting of excessive skin folds. A cluster of light brown hyperpigmented macules was seen overlying the plaque. A punch biopsy of the plaque confirmed a diagnosis of neurofibroma. Further investigation ruled out other manifestations of NF-1.
The early onset of our patient’s neurofibroma and its gross appearance with redundant skin folds are all unusual features. To our knowledge, congenital excessive skin folds found in a single tumor have not been previously described in the literature as a manifestation of SNF. Clinicians should be educated about the possibility of congenital localized skin folds in association with SNF in order to identify the disease in infancy and monitor any changes in neurofibroma pathology.
Keywords: neurofibromatosis, skin folds, congenital
Case presentation
A 5-month-old Israeli Jewish male of Lithuanian and Kazakh descent with no family history of neurocutaneous or dermatological disease was referred to our outpatient pediatric dermatology clinic for evaluation of skin folds on the lower back, which had been present since birth (Figure 1A).
Figure 1.

Clinical photographs of the patient at the age of 5 months (A) and at a follow-up visit at the age of 3 years (B, C) showing a large plaque consisting of congenital skin folds on an erythematous base with a cluster of café-au-lait spots on the right side of the plaque. [Copyright: ©2015 Helfand et al.]
On physical examination, a mildly erythematous, poorly demarcated soft plaque on the right side of the lower back was noted with sensory change. The plaque consisted of excessive skin folds. On the lateral border of the plaque, a 3 cm cluster of café-au-lait spots was present. A firm raised area on the scalp (present since birth, according to the patient’s parents) was also palpated.
Ultrasonographic examination of the lower back was negative for cysts, bony involvement, or muscular abnormalities. Ultrasound of the skull confirmed the presence of a benign bony prominence consistent with exostosis.
Punch biopsy of the soft plaque on the lower back revealed a proliferation of spindle cells in the dermis, arranged in fascicles. The majority of fascicles were wavy and surrounded by stroma consisting of thin, delicate connective tissue fibers (Figure 2A). Cells throughout the dermis stained positive for S-100 (Figure 2B) and negative for MelanA (not shown), compatible with the diagnosis of a neurofibroma.
Figure 2.
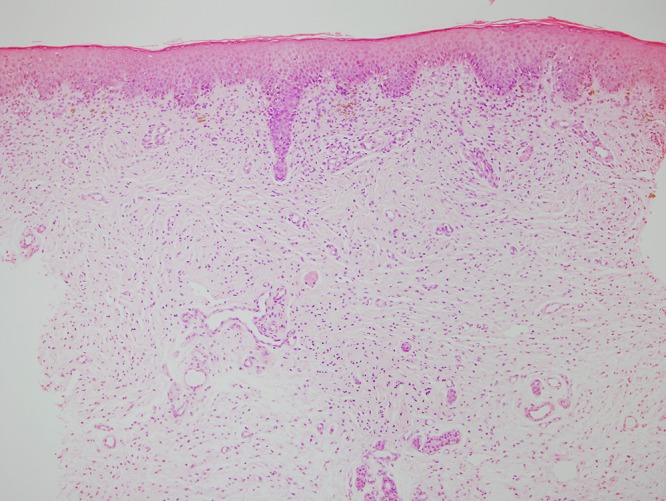
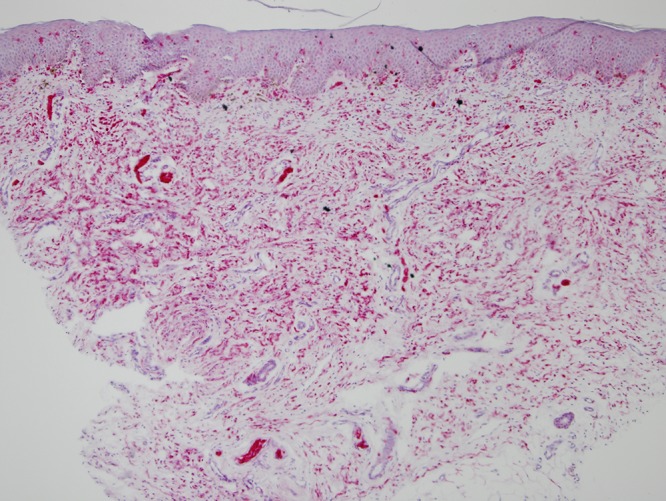
(A) H&E stained tissue section from SNF lesion on the lower back at 10× magnification, showing proliferation of spindle cells in the dermis, arranged in fascicles. The majority of fascicles are wavy and surrounded by stroma consisting of thin, delicate connective tissue fibers. (B) S100 stained tissue section from SNF lesion on the lower back at 10× magnification, with cells throughout the dermis staining positive for S100. [Copyright: ©2015 Helfand et al.]
As segmental neurofibromatosis (SNF) was suspected, a workup was ordered. The patient was found to have normal development and age-appropriate neurological function. He had no myopathy or muscle wasting. No Lisch nodules were seen and Doppler ultrasonography of the renal arteries detected no abnormalities.
At follow-up consultation at 3 years of age, the plaque appeared to have grown proportionally with the child to a width of 20 cm (Figure 1B, C). No axillary freckling was present. The parents were not interested in further genetic work-up. Yearly follow-up surveillance was recommended.
Conclusions
The first cases of segmental neurofibromatosis were reported in 1931 by Gammel [1]. It was postulated that the localization of the neurofibromatosis phenotype in these SNF patients signified a somatic mutation [2]. In 2000, Tinschert, et al confirmed that SNF was a consequence of a spontaneous post-zygotic somatic mutation in the neurofibromin tumor suppressor gene leading to a mosaic pattern of the disease [3]. Intragenic deletions in NF1 can occur early or late in fetal development, giving rise to somatic mosaicism that can manifest as highly localized neurofibromatosis (late mutation) or diffuse phenotypic findings (early mutation) difficult to differentiate clinically from germ-line NF-1 [4].
SNF has been divided into four subtypes: true segmental, localized with deep involvement, hereditary, and bilateral [5]. SNF can present with only pigmentary changes, only neurofibromas, isolated plexiform neurofibromas, or both pigmentary changes and neurofibromas, as in our patient [6,7].
The frequency of neurofibromatosis type 1 (NF-1) in Israeli Jews (1.04/1000) [8] has been reported to be 2–5 times that of the prevalence in the overall population, approximately 1/3000–1/5000 [9], though the prevalence of SNF in Israeli Jews has not been described. Moreover, the prevalence of NF-1 among Israeli Jews of Asian descent (like our patient) (0.95/1000) is 50% greater than in Jews of European or North American origin [8].
SNF has been estimated to be approximately 10–30 times rarer than NF-1. Ruggieri and Huson estimated SNF prevalence as 1/36,000 to 1/40,000 [4]. Kehrer-Sawatzki et al, however, found somatic mosaicism in 40% of patients with sporadic NF-1 [10], suggesting the possibility of much higher SNF prevalence. Further study into the true prevalence of SNF remains necessary.
Neurofibromatosis was suspected in our patient because of the café-au-lait spots found on the plaque. The other differential diagnoses of our patient’s cutaneous findings initially included nevus lipomatosus cutaneous superficialis, smooth muscle cell hamartoma, and nevus sebaceous. In addition, plexiform neurofibromas, sometimes present as loose, redundant skin folds [11,12]. However, histology findings precluded this diagnosis in our patient. The unusual appearance of excessive skin folds was mildly reminiscent of the congenital circumferential skin folds found in the ‘Michelin tire baby syndrome,’ although these folds were not circumferential.
The pathogenesis of such skin folds is unclear. Some hypothesize that growth factors secreted by the Schwann cells in the tumor, such as Vascular Endothelial Growth Factor, lead to overgrowth of the adjacent tissues [13]. The bony exostosis on the patient’s skull may be a random finding. Alternatively, it may represent mosaicism. Systemic NF-1 is much less probable, especially in light of the patient’s lack of systemic signs of neurofibromatosis. It should be noted, however, that Lisch nodules and axillary freckling occur at older ages and thus their absence in the age of three cannot exclude systemic NF-1. In compliance with the wishes of the patient’s family, genetic analysis was not performed. Nonetheless, the combination of localized neurofibroma and café-au-lait spots makes the diagnosis of SNF highly likely.
In summary, we report a case of SNF in a 5-month-old male manifesting in excessive skin folds limited to a large plaque on the lower back that has grown proportionally with the child. Café-au-lait spots are present on the right side of the plaque. The early onset of our patient’s neurofibroma and its gross appearance with redundant skin folds are all unusual features. To our knowledge, congenital excessive skin folds found in a single location on the lower back with rapid proportional growth throughout early childhood have not been previously described in the literature on mosaic SNF. While café-au-lait spots may be suggestive of the diagnosis, the alert clinician should be aware that segmental neurofibromatosis might present as localized excessive skin folds.
Footnotes
Funding: None.
Competing interests: The authors have no conflicts of interest to disclose.
All authors have contributed significantly to this publication.
References
- 1.Gammel JA. Localized neurofibromatosis. Arch Dermatol Syphilol. 1931;24:712–5. [Google Scholar]
- 2.Miller RM, Sparkes RS. Segmental neurofibromatosis. Arch Dermatol. 1977;113(6):837–8. [PubMed] [Google Scholar]
- 3.Tinschert S, Naumann I, Stegmann E, et al. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet. 2000;8(6):455–9. doi: 10.1038/sj.ejhg.5200493. [DOI] [PubMed] [Google Scholar]
- 4.Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56(11):1433–43. doi: 10.1212/wnl.56.11.1433. [DOI] [PubMed] [Google Scholar]
- 5.Roth RR, Martines R, James WD. Segmental neurofibromatosis. Arch Dermatol. 1987;123(7):917–20. [PubMed] [Google Scholar]
- 6.Gabhane SK, Kotwal MN. Segmental neurofibromatosis: a report of 3 cases. Indian J Dermatol. 2010;55(1):105–8. doi: 10.4103/0019-5154.60366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Listernick R, Mancini AJ, Charrow J. Segmental neurofibromatosis in childhood. Am J Med Genet A. 2003;121A(2):132–135. doi: 10.1002/ajmg.a.20183. [DOI] [PubMed] [Google Scholar]
- 8.Garty BZ, Laor A. Neurofibromatosis type 1 in Israel: survey of young adults. J Med Genet. 1994;31(11):853–7. doi: 10.1136/jmg.31.11.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89(1):1–6. [PubMed] [Google Scholar]
- 10.Kehrer-Sawatzki H, Kluwe L, Sandig C, et al. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet. 2004;75(3):410–23. doi: 10.1086/423624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korf BR. Plexiform Neurofibromas. Am J Med Genet. 1999;89(1):31–7. doi: 10.1002/(sici)1096-8628(19990326)89:1<31::aid-ajmg7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 12.Folpe AL, Goldblum JR, editors. Bone and Soft Tissue Pathology: A Volume in the Series Foundations in Diagnostic Pathology. 1st ed. Philadelphia, PA: Saunders; 2010. pp. 199–206. [Google Scholar]
- 13.Kawachi Y, Maruyama H, Ishitsuka Y, et al. NF1 gene silencing induces upregulation of vascular endothelial growth factor expression in both Schwann and non-Schwann cells. Exp Dermatol. 2013;22(4):262–5. doi: 10.1111/exd.12115. [DOI] [PubMed] [Google Scholar]