

Loss- and gain-of-function approaches coupled with binding studies show that the ER-resident NEFs Grp170 and Sil1 induce cholera toxin release from BiP in order to promote toxin retrotranslocation. Thus cholera toxin hijacks two ER-resident NEFs to cross the ER membrane and reach the cytosol in order to induce toxicity.
Abstract
Cholera toxin (CT) intoxicates cells by trafficking from the cell surface to the endoplasmic reticulum (ER), where the catalytic CTA1 subunit hijacks components of the ER-associated degradation (ERAD) machinery to retrotranslocate to the cytosol and induce toxicity. In the ER, CT targets to the ERAD machinery composed of the E3 ubiquitin ligase Hrd1-Sel1L complex, in part via the activity of the Sel1L-binding partner ERdj5. This J protein stimulates BiP's ATPase activity, allowing BiP to capture the toxin. Presumably, toxin release from BiP must occur before retrotranslocation. Here, using loss-and gain-of-function approaches coupled with binding studies, we demonstrate that the ER-resident nucleotide exchange factors (NEFs) Grp170 and Sil1 induce CT release from BiP in order to promote toxin retrotranslocation. In addition, we find that after NEF-dependent release from BiP, the toxin is transferred to protein disulfide isomerase; this ER redox chaperone is known to unfold CTA1, which allows the toxin to cross the Hrd1-Sel1L complex. Our data thus identify two NEFs that trigger toxin release from BiP to enable successful retrotranslocation and clarify the fate of the toxin after it disengages from BiP.
INTRODUCTION
Cholera toxin (CT) secreted by Vibrio cholera is a causative agent for massive secretory diarrhea. The CT holotoxin consists of catalytic (CTA) and receptor-binding (CTB) subunits. To initiate entry into intestinal epithelial cells, CTB binds to the ganglioside GM1 receptor, transporting the holotoxin in a retrograde manner to the endoplasmic reticulum (ER; Chinnapen et al., 2012). Here CT coopts an intrinsic quality control process called ER-associated degradation (ERAD), which normally ejects misfolded ER substrates to the cytosol, where they are degraded by the proteasome (Hazes and Read, 1997). When CT hijacks the ERAD pathway, CTA becomes reduced by a cellular reductase, generating the CTA1 subunit, which retrotranslocates to the cytosol. However, in contrast to the fate of a misfolded substrate, CTA1 evades proteasomal degradation in the cytosol and induces toxicity (Rodighiero et al., 2002). The molecular mechanism by which CT hijacks specific components of the ERAD machinery to reach the cytosol is slowly emerging.
The central ERAD machinery is composed of an E3 ubiquitin ligase complexed with different ER lumenal, membrane, and cytosolic adapters (Hirsch et al., 2009; Claessen et al., 2012); this complex acts coordinately to mobilize various misfolded substrates across the ER membrane. Our laboratory and others demonstrated that CT targets to the E3 ubiquitin ligase Hrd1 (Bernardi et al., 2010) and its membrane adapters Derlin-1 (Bernardi et al., 2008; Dixit et al., 2008) and Sel1L (Williams et al., 2013) to initiate retrotranslocation. By use of the CRISPR-mediated gene control strategy, the Hrd1-Sel1L complex was recently confirmed to be important for facilitating CT retrotranslocation (Gilbert et al., 2014). To help target CT for retrotranslocation, the Sel1L binding partner ERdj5, an ER-resident J-protein, stimulates the intrinsic ATPase activity of the Hsp70 chaperone BiP to form ADP-BiP. Because ADP-BiP exhibits a high affinity for substrates (Kampinga and Craig, 2010), it captures CT efficiently. By juxtaposing ERdj5 next to Hrd1 via ERdj5's interaction with Sel1L (Williams et al., 2013), capture of CT by BiP occurs proximal to the ERAD machinery. A previous study suggested that the BiP–toxin interaction functionally renders the toxin competent for retrotranslocation (Winkeler et al., 2003).
Because BiP is not retrotranslocated with the toxin, CTA must be released from BiP before the toxin reaches the cytosol. Substrate release from BiP normally occurs when ADP-BiP is converted to ATP-BiP, which displays low affinity for substrates. Because conversion of ADP-BiP to ATP-BiP is catalyzed by nucleotide exchange factors (NEFs; Kampinga and Craig, 2010; Otero et al., 2010), we hypothesize that NEFs facilitate CTA release from BiP to enable toxin retrotranslocation.
Grp170 and Sil1 are ER-resident NEFs (Weitzmann et al., 2007; Andréasson et al., 2010; Hale et al., 2010; Yan et al., 2011). Grp170 is considered a distinct member of the canonical Hsp70 protein family, whereas Sil1 is not. A unique feature of Grp170 is that, in addition to its recognized NEF activity, it contains a C-terminal holdase domain that functions to prevent protein aggregation (Easton et al., 2000; Park et al., 2003); Sil1 lacks this domain and activity. Of interest, a recent report found Grp170's holdase but not NEF activity to be responsible for promoting retrotranslocation of a sodium channel that acts as an ERAD substrate in mammalian cells (Buck et al., 2013). In the case of Sil1, although absence of SIL1 in yeast mildly disrupted ERAD (Travers et al., 2000), it is unclear whether Sil1's NEF activity is responsible for this phenotype. Thus whether the NEF activity of Grp170 or Sil1 exerts any role in CTA1 retrotranslocation is unknown.
In this study, we used a cell-based, semipermeabilized system in the context of loss-and gain-of function approaches, coupled with biochemical strategies, to demonstrate that Grp170 and Sil1 regulate CTA release from BiP to facilitate toxin retrotranslocation. Moreover, after release from BiP, our data suggest that the toxin is handed off to protein disulfide isomerase (PDI), an ER redox chaperone known to unfold CTA1 that primes the toxin for translocation across the Hrd1-Sel1L complex. These findings thus reveal additional insights into how the toxin is delivered to the ERAD machinery.
RESULTS
Grp170 and Sil1 knockdown decreases CTA1 retrotranslocation
To evaluate the role of Grp170 and Sil1 in promoting CTA1 retrotranslocation, we used two distinct small interfering RNAs (siRNAs) against each protein (Grp170 siRNA #1 and #2, and Sil1 siRNA #1 and #2) to silence their expression in 293T cells. When compared with cells treated with a control siRNA (i.e., scrambled), Grp170 was efficiently down-regulated by Grp170 siRNA #1 and #2 (Figure 1A, top, compare lanes 2 and 3 to lanes 1, 4, and 5). Similarly, Sil1 was silenced effectively by Sil1 siRNA #1 and #2 (Figure 1A, second panel, compare lanes 4 and 5 to lanes 1–3). Sil1 expression was unperturbed by the Grp170-specific siRNAs (Figure 1A, second panel, compare lanes 2 and 3 to lane 1), whereas Grp170 expression was not disrupted by the Sil1-directed siRNAs (Figure 1A, top, compare lanes 4 and 5 to lane 1). Neither ER stress marker BiP nor PDI increased when Grp170 or Sil1 was knocked down (Figure 1A, third and fourth panels, compare lanes 2–5 to lane 1). Moreover, we found no evidence of XBP1 splicing (a sensitive indicator of ER stress induction) when Grp170 or Sil1 was silenced (Figure 1A, compare lanes 8–11 with lane 7), in contrast to cells treated with the chemical ER stress inducer dithiothreitol (DTT; Figure 1A, compare lane 6 with lane 7). Thus Grp170 and Sil1 can be specifically and efficiently down-regulated without causing massive ER stress.
FIGURE 1:

Grp170 and Sil1 knockdown decreases CTA1 retrotranslocation. (A) Lanes 1–5, WCEs derived from 293T cells transfected with the indicated siRNA were analyzed by SDS–PAGE and immunoblotted with the indicated antibodies and the film developed by using ECL. All immunoblots were developed using this method. Lanes 6–11, reverse-transcription PCR analysis of the unspliced (u) and spliced (s) forms of the XBP1 mRNA derived from cells transfected with the indicated siRNA and treated with or without DTT as indicated. (B) Cells were semipermeabilized with digitonin and subjected to centrifugation, and the generated supernatant and pellet fractions were analyzed for the presence of the cytosolic Hsp90 and the ER-resident PDI markers. This fractionation method is used in the retrotranslocation assay. (C) Cells transfected with the indicated siRNA were incubated with CT (10 nM) for 90 min and subjected to the retrotranslocation assay as presented in B. Both the supernatant and pellet fractions were analyzed by SDS–PAGE and immunoblotted with the indicated antibodies. (D) The intensity of the supernatant CTA1 band in C was quantified by ImageJ (National Institutes of Health, Bethesda, MD) using scans of films after ECL. Mean of three independent experiments. A two-tailed t test was used. Error bars, ± SD. (E) In the rescue experiment, cells transfected with scrambled or Grp170 siRNA #1 were transfected with either Grp170-FLAG* or FLAG-Sil1, as indicated. After intoxication with CT (10 nM) for 90 min, cells were processed and analyzed as in C. (F) In a rescue experiment similar to E, cells transfected with scrambled or Sil1 siRNA #2 were transfected with either Grp170-FLAG or FLAG-Sil1*, as indicated. After intoxication with CT (10 nM) for 90 min, cells were processed and analyzed as in C. (G) Cells were cotransfected with Δ F508 CFTR and a scrambled siRNA, Grp170 siRNA #2, or Sil1 siRNA #1. Δ F508 CFTR was immunoprecipitated from the resulting WCE, and samples were subjected to immunoblotting with an antibody against CFTR. The black line indicates that an intervening lane from the same immunoblot has been spliced out.
To determine whether silencing the NEFs affects CTA1 arrival to the cytosol from the ER, we relied on a previously established semipermeabilized retrotranslocation assay that monitors toxin ER-to-cytosol transport (Forster et al., 2006; Bernardi et al., 2010; Williams et al., 2013). This assay has been used by other laboratories to examine CTA1 retrotranslocation (Taylor et al., 2010; Wernick et al., 2010; Nery et al., 2011), as well as by us and other laboratories to investigate ER-to-cytosol transport of the viral pathogen SV40 (Inoue and Tsai, 2011; Geiger et al., 2011). Briefly, cells were intoxicated with CT for 90 min and treated with a low digitonin concentration to permeabilize the plasma membrane without affecting internal membranes. Subsequent high-speed centrifugation of the sample generates a supernatant and a pellet fraction. The supernatant fraction should harbor cytosolic proteins and any toxin that underwent retrotranslocation to the cytosol. In contrast, the pellet fraction should contain membranes, including the ER membrane, and any toxin that has reached the ER but has not retrotranslocated to the cytosol, as well as toxin present in other membranous compartments. Using this strategy, we detected the cytosolic Hsp90 in the supernatant fraction (Figure 1B, top, compare lane 1 with lane 2), whereas the ER-resident protein PDI fractionated to the pellet fraction (Figure 1B, bottom, compare lane 2 with lane 1). These results verify the integrity of the fractionation approach.
Of importance, we found that silencing Grp170 or Sil1 with either siRNA markedly decreased the CTA1 level in the supernatant when compared with the scrambled siRNA (Figure 1C, panel, compare lanes 2–5 to lane 1). We quantified these results from scans of films following enhanced chemiluminescence (ECL) as before (Forster et al., 2006; Bernardi et al., 2010; Williams et al., 2013) and found that knocking down Grp170 decreased CTA1 retrotranslocation by ∼50–60%, whereas down-regulating Sil1 impaired toxin retrotranslocation by 60–75% (Figure 1D, black bars). In a rescue experiment, expression of either a siRNA-resistant wild-type (WT) Grp170 FLAG-tagged construct (i.e., Grp170-FLAG*; Figure 1E, third and fifth panels, lane 3) or a WT FLAG-tagged Sil1 construct (i.e., FLAG-Sil1*; Figure 1E, fourth and fifth panels, lane 4) partially restored the CTA1 supernatant level derived from cells depleted of Grp170 (Figure 1E, top, compare lanes 3 and 4 to lane 2). Similarly, in Sil1-knockdown cells, expression of Grp170-FLAG (Figure 1F, third and fifth panels, lane 3) or a siRNA-resistant WT FLAG-tagged Sil1 construct (i.e., FLAG-Sil1*; Figure 1F, fourth and fifth panels, lane 4) partially restored the CTA1 level in the supernatant fraction (Figure 1F, top, compare lanes 3 and 4 with 2). These findings demonstrate that Grp170 and Sil1 are crucial for promoting CTA1 retrotranslocation and that their activities appear interchangeable during this process.
Silencing Grp170 or Sil1 does not appear to globally affect ERAD. This is because Grp170 or Sil1 knockdown had no effect on the steady-state level of the ERAD membrane substrate Δ F508 CFTR (cystic fibrosis transmembrane conductance regulator; Figure 1G, compare lanes 2 and 3 with lane 1), consistent with the yeast Grp170 homologue Lhs1p not controlling ERAD of CFTR (Buck et al., 2013). In addition, in the case of Sil1, we recently found that ER-to-cytosol transport of the viral pathogen SV40 is unaffected by Sil1 knockdown (Inoue and Tsai, 2015).
Grp170 and Sil1 overexpression stimulates CTA1 retrotranslocation
To complement the loss-of-function approach, we performed gain-of-function experiments to further probe a role of the NEFs in facilitating toxin retrotranslocation. When cells transfected with vector WT Grp170 containing a FLAG tag (i.e., Grp170-FLAG) or FLAG-Sil1 (Figure 2A, top, lanes 1–3) were intoxicated with CT for 90 min and subjected to the cell-based retrotranslocation assay, we found that overexpression of either Grp170 or Sil1 increased the CTA1 level in the supernatant fraction when compared with vector control (Figure 2B, top, compare lanes 2 and 3 to lane 1; quantified in Figure 2C, black bars). Overexpressing the NEFs also enhanced the CTA1 level in the supernatant when CT was intoxicated for 60 min, albeit more modestly (quantified in Figure 2C, gray bars). In contrast to overexpression of Grp170-FLAG, overexpression of a NEF-defective Grp170 construct (i.e., G41L Grp170-FLAG; Inoue and Tsai, 2015; Figure 2D, fifth panel, lane 3) did not enhance the CTA1 supernatant level when compared with the vector control (Figure 2D, top, compare lane 3 with lane 2); in fact, a moderate decrease in the toxin level in the supernatant fraction was reliably observed in cells expressing G41L Grp170-FLAG when compared with cells expressing the vector (Figure 2D, top, compare lane 3 with lane 1). Although a specific NEF-defective Sil1 mutant has yet to be shown, a yeast Sil1-Kar2p (homologue of mammalian BiP) complex structure suggests that Sil1 induces a conformational change in Kar2p that promotes release of ADP (Yan et al., 2011). Because the yeast H163A Sil1 mutant was found to be defective in Kar2p binding (Yan et al., 2011), it unlikely imparts NEF activity against Kar2p. Accordingly, we generated the corresponding mutant in human Sil1 (i.e., FLAG-H241A Sil1) and found that, by contrast to FLAG-Sil1, precipitation of FLAG-H241A Sil1 did not pull down endogenous BiP (Figure 2E, top, compare lane 3 with lane 2). Of importance, only overexpression of FLAG-Sil1 (Figure 2F, fifth panel, lane 2) but not FLAG-H241A Sil1 (Figure 2F, fifth panel, lane 3) stimulated toxin retrotranslocation (Figure 2F, top, compare lane 2 with lanes 1 and 3); again, a decrease in the toxin level in the supernatant fraction was observed in cells expressing FLAG-H241A Sil1 when compared with cells expressing the vector (Figure 2F, top, compare lane 3 with lane 1). Collectively these data reinforce a crucial function of Grp170 and Sil1 in mediating CTA1 ER-to-cytosol transport and suggest that the inherent NEF activities of these factors are responsible for driving this reaction.
FIGURE 2:

Grp170 and Sil1 overexpression stimulates CTA1 retrotranslocation. (A) WCEs derived from 293T cells intoxicated with CT for 90 min and transfected with the indicated construct were analyzed by SDS–PAGE and immunoblotted with the indicated antibodies. (B) Cells intoxicated with CT for 90 min and expressing the constructs as in A were subjected to the retrotranslocation assay and analyzed as in Figure 1C. (C) The supernatant CTA1 band intensity in B was quantified (black bars) as in Figure 1D. In addition, quantification of the supernatant CTA1 band intensity derived from cells treated with CT for 60 min is presented in the gray bars. Mean of three independent experiments. A two-tailed t test is shown for the 90-min intoxication experiments. Error bars, ± SD. (D) As in B, except cells were transfected with the indicated construct. (E) WCEs derived from 293T cells transfected with the indicated constructs were subjected to immunoprecipitation using FLAG antibody–conjugated beads. The precipitated samples were analyzed by SDS–PAGE and immunoblotted with the indicated antibodies. (F) As in B, except that cells were transfected with the indicated construct.
Knockdown of Grp170 or Sil1 decreases toxin release from BiP
We reasoned that, if Grp170 and Sil1 exert their NEF functions to promote release of CTA from BiP, altering their expression level should concomitantly affect the extent of the toxin–BiP interaction. To decrease the NEF expression level, we silenced Grp170 or Sil1 using Grp170 siRNA #1 or Sil1 siRNA #1 and incubated the knocked-down cells with or without CT. As before (Figure 1A), Grp170 siRNA #1 efficiently down-regulated Grp170 without disrupting the Sil1 level (Figure 3A, compare top to second panel), whereas Sil1 siRNA #1 silenced Sil1 markedly without perturbing Grp170's level (Figure 3A, compare the second panel to the top panel). Under these conditions, the whole-cell extracts (WCEs) derived from cells were incubated with an antibody against CTA and the immunoprecipitates subjected to SDS–PAGE, followed by immunoblotting with either a BiP or CTA antibody. BiP was only found in the immunoprecipitates from CT-intoxicated cells (Figure 3B, top and second panels; compare lanes 4–6 with lanes 1–3), indicating that BiP specifically engages CTA, as previously observed (Williams et al., 2013). (Only CTA1 appears in the immunoblot because the sample buffer contained the reducing agent β-mercaptoethanol to efficiently detect the toxin.) Of importance, silencing Grp170 increased the level of BiP that coprecipitated with CTA (Figure 3B, top, compare lane 5 with lane 4). A modestly higher level of BiP consistently coprecipitated with CTA in cells lacking Sil1 (Figure 3B, top, compare lane 6 with lane 4). These findings indicate that decreasing the levels of Grp170 and Sil1 preferentially trapped the toxin on BiP, consistent with the posited role of these NEFs in releasing CTA from BiP. It is important to note that the less efficient release of CTA from BiP is consistent with a decrease in toxin retrotranslocation found under NEF-knockdown conditions (Figure 1).
FIGURE 3:
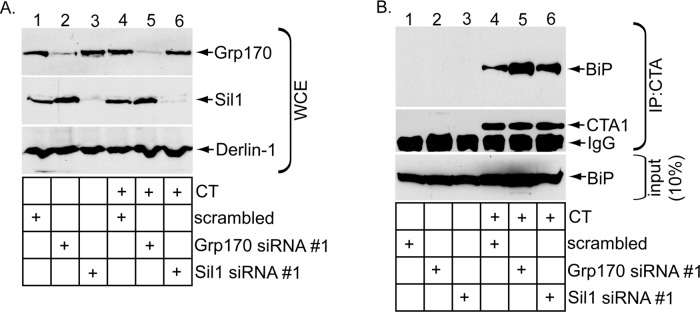
Knockdown of Grp170 or Sil1 decreases toxin release from BiP. (A) WCEs derived from 293T cells transfected with the indicated siRNA and intoxicated with or without CT were analyzed by SDS–PAGE and immunoblotted with the indicated antibodies. (B) WCEs derived from cells in A were subjected to immunoprecipitation using an antibody against CTA. The immunoprecipitates were analyzed by reducing SDS–PAGE and immunoblotted against the indicated antibodies.
Overexpressing Grp170 or Sil1 markedly stimulates toxin release from BiP
Because knockdown of the NEFs traps the toxin on BiP, we asked whether overexpressing the NEFs (Figure 4A, top) might stimulate CTA release from BiP and found that it did (Figure 4B, top, compare lanes 5 and 6 to lane 4). The enhanced release of toxin from BiP likely explains why toxin retrotranslocation increased under the overexpression conditions (Figure 2). As a control, expressing G41L Grp170-FLAG or FLAG-H214A Sil1 did not enhance toxin release from BiP (Figure 4C, top, compare lanes 2 and 3 with lane 1), in line with the observation that their overexpression did not stimulate toxin retrotranslocation (Figure 2). In fact, overexpressing mutant Sil1 modestly trapped the toxin to BiP (Figure 4C, top, compare lane 3 with lane 1), consistent with the ability of this construct to inhibit CTA1 retrotranslocation when overexpressed (Figure 2F, top, compare lane 3 with lane 1). Because overexpressing either WT Grp170 or Sil1 increased toxin disengagement from BiP and retrotranslocation, Grp170 and Sil1 likely use their NEF activities to promote toxin release from BiP in order to promote the toxin for retrotranslocation.
FIGURE 4:

Overexpressing Grp170 or Sil1 markedly stimulates toxin release from BiP. (A) WCEs derived from 293T cells transfected with the indicated construct and intoxicated with or without CT were analyzed by SDS–PAGE and immunoblotted with the indicated antibodies. (B) WCEs derived from cells in A were subjected to immunoprecipitation using an antibody against CTA. The immunoprecipitates were analyzed by reducing SDS–PAGE and immunoblotted against the indicated antibodies. (C) As in B, except that G41L Grp170-FLAG or FLAG-H214A Sil1 was transfected instead of the respective WT counterpart.
CTA is transferred to PDI upon NEF-dependent release from BiP
We previously hypothesized that, once the toxin is captured by and released from BiP, it is transferred to PDI (Williams et al., 2013), which unfolds the toxin (Tsai et al., 2001; Forster et al., 2009; Wernick et al., 2010), a reaction required for its retrotranslocation (Forster et al., 2006). To test this possibility, we knocked down Grp170 (using Grp170 siRNA #1) or Sil1 (using Sil1 siRNA #1) to decrease toxin release from BiP (Figure 3B). Under this condition, PDI binds to the toxin less efficiently (Figure 5A, top, compare lanes 2 and 3 with lane 1), suggesting that the toxin–PDI interaction occurs downstream of toxin release from BiP. Conversely, when PDI was down-regulated (Figure 5B, fourth panel, compare lane 2 with lane 1), the toxin accumulates on BiP (Figure 5B, top, compare lane 2 with lane 1). Presumably, when the PDI level is lowered, the released toxin inefficiently transfers to PDI and as a consequence reengages BiP via the action of the J-protein ERdj5. These data are consistent with a scenario in which the toxin is handed off to PDI upon release from BiP.
FIGURE 5:

CTA is transferred to PDI upon NEF-dependent release from BiP. (A) WCEs derived from cells expressing the indicated siRNA were incubated with an antibody against CTA. The immunoprecipitates were subjected to reducing SDS–PAGE and immunoblotted with the indicated antibodies. (B) As in A, except that a siRNA against PDI was used, and the immunoblot was probed using a BiP antibody. (C) Model depicting the role of the ER-resident NEFs Grp170 and Sil1 during CTA1 retrotranslocation. In the ER, CT targets to the ERAD machinery formed by the Hrd1/Sel1L/Derlin-1 complex partly due to the activity of the Sel1L-binding partner ERdj5. This J-protein enables BiP to capture CT by stimulating BiP's intrinsic ATPase activity. Grp170 and Sil1 impose their NEF activities on BiP, triggering CTA to disengage from BiP (arrow). Next CTA is delivered to PDI, which in turn unfolds the toxin. The identity of an ER reductase that generates the CTA1 peptide from CTA is unknown. In the final step, unfolded CTA1 translocates across the Hrd1/Sel1L/Derlin-1 complex to reach the cytosol.
DISCUSSION
CT is postulated to disguise as a misfolded protein when it reaches the ER, coopting the ERAD pathway to enter the cytosol and induce toxicity (Hazes and Read, 1997). Not surprisingly, CT engages BiP (Winkeler et al., 2003; Williams et al., 2013), an ER-resident chaperone that normally binds to misfolded clients to prevent them from aggregation (Nishikawa et al., 2001; Kabani et al., 2003). Once the toxin binds to BiP, it must be released from BiP in order to cross the retrotranslocation machinery on the ER membrane to access the cytosol. The identity of the release factors that trigger toxin release from BiP is unknown.
In this study, we pinpoint two NEFs that promote toxin release from BiP to facilitate retrotranslocation. The major conclusions of our findings are summarized in Figure 5C. When CT reaches the ER from the cell surface, it targets to the Hrd1/Sel1L/Derlin-1 membrane complex, in part due to the activity of the Sel1L-interacting protein ERdj5 (Williams et al., 2013). This J-protein stimulates the BiP ATPase activity, enabling ADP-BiP to capture the CT holotoxin (Williams et al., 2013). To release the captured toxin, this study demonstrates that the ER-resident NEFs Grp170 and Sil1 cause CTA to be released from BiP, presumably by converting ADP-BiP to ATP-BiP (Figure 5C, arrow). The released CTA is then transferred to PDI, which unfolds the toxin. Precisely when an ER reductase reduces CTA to generate the CTA1 peptide is unclear. Regardless, the unfolded CTA1 peptide is transferred to the Hrd1/Sel1L/Derlin-1 complex for translocation across the ER membrane. The driving force that in turn pulls the toxin into the cytosol is unknown but may involve an unidentified GTPase (Moore et al., 2013).
The present study suggests that the NEF activities of both Grp170 and Sil1 are hijacked during retrotranslocation of CTA1. This conclusion is based on three lines of evidence. First, silencing Grp170 or Sil1, well-established NEFs of BiP (Weitzmann et al., 2007; Andréasson et al., 2010; Hale et al., 2010; Yan et al., 2011), decreases CTA1 retrotranslocation. Second, overexpression of WT but not a NEF-defective Grp170 stimulates toxin retrotranslocation. Similarly, overexpression of WT but not of a Sil1 mutant that cannot bind to BiP (and thus unlikely to impart NEF activity to BiP) enhances CTA1 retrotranslocation. Third, altering the levels of Grp170 or Sil1 affected toxin–BiP binding, in line with the posited role of NEFs in regulating substrate–BiP interaction. In fact, overexpressing the mutant Grp170 or Sil1 construct decreased CTA1 retrotranslocation, although a precise molecular explanation for these observations is unclear. It is possible that these NEFs normally interact with ERAD components to position the release reaction proximal to the retrotranslocation site. In this scenario, the NEF mutants would act in a dominant-negative manner simply by binding to the ERAD components, thereby preventing the endogenous WT counterparts from properly engaging the ERAD machinery. We note that down-regulating either Grp170 or Sil1 individually did not fully block CTA1 arrival to the cytosol, consistent with the notion that the toxin hijacks multiple NEFs to disengage from BiP. We envision that CT's ability to use both NEFs maximizes the efficiency by which it can be released from BiP. This likely leads to more efficient retrotranslocation. Silencing Grp170 and Sil1 simultaneously compromised cellular integrity, thereby precluding us from assessing whether double knockdown of Grp170 and Sil1 perturbed CTA1 retrotranslocation more drastically than the single-knockdown condition.
After NEF-dependent release from BiP, our data also indicate that the toxin is transferred to PDI. We previously demonstrated that a fraction of PDI is localized proximal to the Hrd1 complex by virtue of PDI's interaction with Derlin-1 and Hrd1 (Bernardi et al., 2008; Moore et al., 2010). Using a redox-driven mechanism, PDI then unfolds CTA1 (Tsai et al., 2001; Forster et al., 2009; Wernick et al., 2010), a reaction required for toxin retrotranslocation in cells (Forster et al., 2006). Although a separate study found that PDI displaces CTA1 from CT (Taylor et al., 2011), this reaction likely also requires partial unfolding of CTA1 due to extensive interactions between CTA and CTB. There are conflicting results regarding whether PDI also acts as the cellular reductase for generating CTA1 from CTA (Orlandi, 1997; Majoul et al., 1997). Nonetheless, once the CTA1 peptide unfolds, it is primed to transport across a retrotranslocation channel. Previous findings implicated the E3 ligases Hrd1 and gp78 (Bernardi et al., 2010) and Derlin-1 (Bernardi et al., 2008; Dixit et al., 2008) in playing a critical function during toxin retrotranslocation, whereas a different study demonstrated that Derlin-1 is dispensable in this process (Saslowsky et al., 2010). Whether these membrane components serve as the actual channel for transferring CTA1 across the ER membrane is unknown, although yeast Hrd1 has been postulated as the protein conduit for canonical ERAD substrates (Carvalho et al., 2010; Stein et al., 2014). Another candidate for conducting CTA1 across the ER membrane is the Sec61 channel used during forward translocation (Schimitz et al., 2000). A reconstituted system using purified components should help to resolve this question.
Other toxins that employ the ERAD pathway can also use BiP and its cochaperones to promote their retrotranslocation into the cytosol. For instance, Shiga toxin uses BiP (Falguieres and Johannes, 2006) and the cochaperone ERdj3 (Yu and Haslam, 2005) to mobilize from the ER into the cytosol; this BiP cochaperone has also been found to be important for CTA1 retrotranslocation (Massey et al., 2011). In addition, the viral A/B toxin K28 uses BiP and its cochaperones to retrotranslocate into the cytosol during intoxication (Heiligenstein et al., 2006). The use of BiP and cochaperones by these toxins suggests that the toxins display structural properties inherent in a misfolded protein that are normally handled by this chaperone system. Whereas BiP promotes ER-to-cytosol transport of CT, Shiga toxin, and the K28 toxin, a recent report found that it antagonizes retrotranslocation of the plant toxin ricin (Gregers et al., 2013), despite the observation that Sel1L facilitates ricin retrotranslocation (Redmann et al., 2011). Hence how a toxin coopts the BiP/cochaperone system in the ER clearly varies and is dependent on the nature of the toxin.
MATERIALS AND METHODS
Materials
Polyclonal antibodies against PDI and Hsp90 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), polyclonal Grp170 and Sil1 antibodies from Genetex (Irvine, CA), monoclonal PDI antibodies from Abcam (Cambridge, MA), monoclonal BiP antibody from BD Biosciences (San Jose, CA), and monoclonal FLAG antibody and FLAG antibody–conjugated beads (M2 affinity gel) from Sigma-Aldrich (St. Louis, MO). Polyclonal CTA antibody was produced against denatured CTA purchased from EMD Biosciences (San Diego, CA). Purified CT was purchased from EMD Biosciences, protein G agarose from Thermo Fisher Scientific (Rockford, IL), and protein A agarose beads from Invitrogen (Carlsbad, CA). Polyclonal Derlin-1 antibody was a gift from T. Rapoport (Harvard University, Cambridge, MA).
Constructs
The Grp170-FLAG, G41L Grp170-FLAG, FLAG-Sil1, and GFP-FLAG constructs were generated as described in Inoue and Tsai (2015). To generate the FLAG-H241A Sil1 mutant, the corresponding mutation was introduced with an overlapping PCR method using FLAG-Sil1 as a template. For the siRNA-resistant Grp170-FLAG* construct, the following silent mutations were introduced into the construct by overlapping PCR: 2716-CTGAACAAAGCTAAATTC-2733. For the siRNA-resistant FLAG-Sil1* construct, the following silent mutations were introduced into the construct by overlapping PCR: 613-GCCCTGTTCGACCTGGAG-630. The underlines denote the introduced silent mutations.
Tissue culture, transfection and drug treatment
HEK 293T cells were cultured in DMEM with 10% fetal bovine serum and penicillin/streptomycin. The cells were grown to 30% confluency on a 6-or 10-cm dish before transfection with 1 μg/ml polyethylenimine. Cells were typically grown for an additional 24 or 48 h before experimentation. Where indicated, cells were treated with or without 10 nM CT in Hank's balanced salt solution (HBSS) for 90 min before experimentation.
siRNA knockdown of Grp170, Sil1, and PDI
Cells were grown to 30% confluency on a 6-, 10-, or 15-cm dish and transfected with the indicated siRNA construct for 48 h with the Lipofectamine RNAi MAX reagent (Invitrogen). The sequences of the siRNAs used in this study were Grp170 siRNA #1 (5′-GCUCAAUAAGGCCAAGUUUTddT-3′; Invitrogen), Grp170 siRNA #2 (5′-GCCUUUAAAGUGAAGCCAUdTdT-3′; Invitrogen), Sil1 siRNA #1 (5′-GCUGAUCAACAAGUUCAAUdTdT-3′; Invitrogen), Sil1 siRNA #2 (5′-GCGCUCUUUGAUCUUGAAUdTdT-3′; Invitrogen), and PDI siRNA (5′-CAACUUUGAAGGGGAGGUCUUdTdT-3'; Thermo Fisher Scientific).
XBP1 splicing and retrotranslocation assays
These were described previously in Williams et al. (2013).
Coimmunoprecipitation
293T cells were incubated with or without 10 nM CT in HBSS for 90 min, harvested, and lysed in buffer containing 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.5, 50 mM), NaCl (150 mM), sucrose (250 mM), MgCl2 (2 mM), N-ethylmaleimide (10 mM), 1% Triton X-100 or 1% deoxy Big Chap (Sigma-Aldrich), and protease inhibitors for 30 min at 4°C. Cells were centrifuged at 16,000 × g for 10 min, and the supernatant was used for immunoprecipitation. Where indicated, FLAG antibody–conjugated beads or a mixture of protein A/G agarose was added to the WCE and incubated at 4°C overnight. The immunocomplex was sedimented, washed, and subjected to SDS–PAGE, followed by immunoblotting with the appropriate antibody.
Δ F508 CFTR steady-state analysis
Cells reverse transfected with the indicated siRNA were further transfected with the Δ F508 CFTR construct, incubated for 24 h, harvested, and lysed in a buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride. The resulting WCE was incubated with a monoclonal CFTR antibody (M3A7) overnight, followed by capturing of the immune complex with Protein G magnetic beads (Life Technologies, Grand Island, NY). The bound proteins were eluted with SDS sample buffer and analyzed by immunoblotting with a CFTR antibody.
Acknowledgments
We thank Christopher Walczak (University of Michigan, Ann Arbor, MI) for critical review of the manuscript. B.T. was funded by the National Institutes of Health (RO1 083252-05). We also gratefully acknowledge partial support from the University of Michigan Fast Forward Protein Folding Diseases Initiative.
Abbreviations used:
- CFTR
cystic fibrosis transmembrane conductance regulator
- CT
cholera toxin
- CTA
cholera toxin catalytic A subunit
- CTB
cholera toxin catalytic B subunit
- DTT
dithiothreitol
- ECL
enhanced chemiluminescence
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- GFP
green fluorescent protein
- NEF
nucleotide exchange factor
- PDI
protein disulfide isomerase
- s
spliced
- siRNA
small interfering RNA
- u
unspliced
- WCE
whole cell extract
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-01-0014) on April 15, 2015.
REFERENCES
- Andréasson C, Rampelt H, Fiaux J, Druffel-Augustin S, Bukau B. The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J Biol Chem. 2010;285:12445–12453. doi: 10.1074/jbc.M109.096735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi KM, Forster ML, Lencer WI, Tsai B. Derlin-1 facilitates the retro-translocation of cholera toxin. Mol Biol Cell. 2008;19:877–884. doi: 10.1091/mbc.E07-08-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell. 2010;21:140–151. doi: 10.1091/mbc.E09-07-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Plavchak L, Roy A, Donnelly BF, Kashlan OB, Kleyman TR, Subramanya AR, Brodsky JL. The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum-associated degradation of the epithelial sodium channel. J Biol Chem. 2013;288:18366–18380. doi: 10.1074/jbc.M113.469882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnapen DJ, Hsieh WT, te Welscher YM, Saslowsky DE, Kaoutzani L, Brandsma E, D'Auria L, Park H, Wagner JS, Drake KR, et al. Lipid sorting by ceramide structure from plasma membrane to ER for the cholera toxin receptor ganglioside GM1. Dev Cell. 2012;23:573–586. doi: 10.1016/j.devcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit G, Mikoryak C, Hayslett T, Bhat A, Draper RK. Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp Biol Med (Maywood) 2008;233:163–175. doi: 10.3181/0705-RM-132. [DOI] [PubMed] [Google Scholar]
- Easton DP, Kaneko Y, Subjeck JR. The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones. 2000;5:276–290. doi: 10.1379/1466-1268(2000)005<0276:thagsp>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falguières T, Johannes L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol Cell. 2006;98:125–134. doi: 10.1042/BC20050001. [DOI] [PubMed] [Google Scholar]
- Forster ML, Mahn JJ, Tsai B. Generating an unfoldase from thioredoxin-like domains. J Biol Chem. 2009;284:13045–13056. doi: 10.1074/jbc.M808352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster ML, Sivick K, Park YN, Arvan P, Lencer WI, Tsai B. Protein disulfide isomerase-like proteins play opposing roles during retro-translocation. J Cell Biol. 2006;173:853–859. doi: 10.1083/jcb.200602046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol. 2011;13:1305–1314. doi: 10.1038/ncb2339. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregers TF, Skånland SS, Wälchli S, Bakke O, Sandvig K. BiP negatively affects ricin transport. Toxins (Basel) 2013;5:969–982. doi: 10.3390/toxins5050969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale SJ, Lovell SC, de Keyzer J, Stirling CJ. Interactions between Kar2p and its nucleotide exchange factors Sil1p and Lhs1p are mechanistically distinct. J Biol Chem. 2010;285:21600–21606. doi: 10.1074/jbc.M110.111211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- Heiligenstein S, Eisfeld K, Sendzik T, Jimenéz-Becker N, Breinig F, Schmitt MJ. Retrotranslocation of a viral A/B toxin from the yeast endoplasmic reticulum is independent of ubiquitination and ERAD. EMBO J. 2006;25:4717–4727. doi: 10.1038/sj.emboj.7601350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458:453–460. doi: 10.1038/nature07962. [DOI] [PubMed] [Google Scholar]
- Inoue T, Tsai B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011;7:e1002037. doi: 10.1371/journal.ppat.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Tsai B. A nucleotide exchange factor promotes ER-to-cytosol membrane penetration of the non-enveloped virus SV40. J Virol. 2015;89:4069–4079. doi: 10.1128/JVI.03552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM, Brodsky JL. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol Biol Cell. 2003;14:3437–3448. doi: 10.1091/mbc.E02-12-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoul I, Ferrari D, Söling HD. Reduction of protein disulfide bonds in an oxidizing environment. The disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett. 1997;401:104–108. doi: 10.1016/s0014-5793(96)01447-0. [DOI] [PubMed] [Google Scholar]
- Massey S, Burress H, Taylor M, Nemec KN, Ray S, Haslam DB, Teter K. Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect Immun. 2011;79:4739–4747. doi: 10.1128/IAI.05503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore P, Bernardi KM, Tsai B. The Ero1alpha-PDI redox cycle regulates retro-translocation of cholera toxin. Mol Biol Cell. 2010;21:1305–1313. doi: 10.1091/mbc.E09-09-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore P, He K, Tsai B. Establishment of an in vitro transport assay that reveals mechanistic differences in cytosolic events controlling cholera toxin and T-cell receptor α retro-translocation. PLoS One. 2013;8:e75801. doi: 10.1371/journal.pone.0075801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery FC, Armata IA, Farley JE, Cho JA, Yaqub U, Chen P, da Hora CC, Wang Q, Tagaya M, Klein C, et al. TosinA participates in endoplasmic reticulum-associated degradation. Nat Commun. 2011;2:393. doi: 10.1038/ncomms1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi PA. Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J Biol Chem. 1997;272:4591–4599. [PubMed] [Google Scholar]
- Otero JH, Lizák B, Hendershot LM. Life and death of a BiP substrate. Semin Cell Dev Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Easton DP, Chen X, MacDonald IJ, Wang XY, Subjeck JR. The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry. 2003;42:14893–14902. doi: 10.1021/bi030122e. [DOI] [PubMed] [Google Scholar]
- Redmann V, Oresic K, Tortorella LL, Cook JP, Lord M, Tortorella D. Dislocation of ricin toxin A chains in human cells utilizes selective cellular factors. J Biol Chem. 2011;286:21231–21238. doi: 10.1074/jbc.M111.234708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodighiero C, Tsai B, Rapoport TA, Lencer WI. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002;3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saslowsky DE, Cho JA, Chinnapen H, Massol RH, Chinnapen DJ, Wagner JS, De Luca HE, Kam W, Paw BH, Lencer WI. Intoxication of zebrafish and mammalian cells by cholera toxin depends on the flotillin/reggie proteins but not Derlin-1 or -2. J Clin Invest. 2010;120:4399–4409. doi: 10.1172/JCI42958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Herrgen H, Winkeler A, Herzog V. Cholera toxin is exported from microsomes by the Sec61p complex. J Cell Biol. 2000;148:1203–1212. doi: 10.1083/jcb.148.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M, Banerjee T, Ray S, Tatulian SA, Teter K. Protein-disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J Biol Chem. 2011;286:22090–22100. doi: 10.1074/jbc.M111.237966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M, Navarro-Garcia F, Huerta J, Burress H, Massey S, Ireton K, Teter K. Hsp90 is required for transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J Biol Chem. 2010;285:31261–31267. doi: 10.1074/jbc.M110.148981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- Weitzmann A, Baldes C, Dudek J, Zimmermann R. The heat shock protein 70 molecular chaperone network in the pancreatic endoplasmic reticulum—a quantitative approach. FEBS J. 2007;274:5175–5187. doi: 10.1111/j.1742-4658.2007.06039.x. [DOI] [PubMed] [Google Scholar]
- Wernick NL, De Luca H, Kam WR, Lencer WI. N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retrotranslocation from endoplasmic reticulum to cytosol. J Biol Chem. 2010;285:6145–6152. doi: 10.1074/jbc.M109.062067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JM, Inoue T, Banks L, Tsai B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell. 2013;24:785–795. doi: 10.1091/mbc.E12-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkeler A, Gödderz D, Herzog V, Schmitz A. BiP-dependent export of cholera toxin from endoplasmic reticulum-derived microsomes. FEBS Lett. 2003;554:439–442. doi: 10.1016/s0014-5793(03)01217-1. [DOI] [PubMed] [Google Scholar]
- Yan M, Li J, Sha B. Structural analysis of the Sil1-Bip complex reveals the mechanism for Sil1 to function as a nucleotide-exchange factor. Biochem J. 2011;438:447–455. doi: 10.1042/BJ20110500. [DOI] [PubMed] [Google Scholar]
- Yu M, Haslam DB. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun. 2005;73:2524–2532. doi: 10.1128/IAI.73.4.2524-2532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]