

Aurora B is a key regulator of cytokinetic abscission, yet the full range of cellular events and signaling factors that regulate Aurora B at this stage remains to be elucidated. Here it is shown that postmitotic genome surveillance is linked to the abscission checkpoint. This connection is dependent on ATR and a central Chk1-Aurora B signaling node.
Abstract
Aurora B regulates cytokinesis timing and plays a central role in the abscission checkpoint. Cellular events monitored by this checkpoint are beginning to be elucidated, yet signaling pathways upstream of Aurora B in this context remain poorly understood. Here we reveal a new connection between postmitotic genome surveillance and cytokinetic abscission. Underreplicated DNA lesions are known to be transmitted through mitosis and protected in newly formed nuclei by recruitment of 53BP1 and other proteins until repair takes place. We find that this genome surveillance initiates before completion of cytokinesis. Elevating replication stress increases this postmitotic process and delays cytokinetic abscission by keeping the abscission checkpoint active. We further find that ATR activity in midbody-stage cells links postmitotic genome surveillance to abscission timing and that Chk1 integrates this and other signals upstream of Aurora B to regulate when the final step in the physical separation of daughter cells occurs.
INTRODUCTION
Faithful replication and transmission of genetic information during cell division requires tight coordination between the process of DNA replication and cell cycle progression. Whereas checkpoints ensure that cells do not progress into mitosis until the vast majority of the genome has been duplicated, certain regions of the genome, including common fragile sites, are inherently difficult to replicate and can persist in an underreplicated state during cell cycle progression (Debatisse et al., 2012). It is becoming increasingly clear that mitotic entry despite a low level of incompletely replicated DNA is a frequent phenomenon and may be a fundamental aspect of cell division (Mankouri et al., 2013). Conditions that augment replication stress, such as low nucleotide pools or inhibition of DNA polymerases, further exacerbate this situation. When this occurs, regions of DNA are transmitted in a partially replicated state through mitosis and poised for repair in the subsequent G1/S phase by binding of the DNA damage response (DDR) protein 53BP1 (Harrigan et al., 2011; Lukas et al., 2011). 53BP1 is believed to play a key role in postmitotic genome surveillance by protecting vulnerable lesions until repair can occur (Harrigan et al., 2011; Lukas et al., 2011).
The presence of 53BP1 nuclear bodies (also known as OPT domains) in G1 nuclei occurs ∼1.5–2 h after the onset of mitosis (Harrigan et al., 2011; Lukas et al., 2011; Lee et al., 2014). At a mechanistic level, phosphorylation of T1609 and S1618 in 53BP1 prevents its association with chromatin during mitosis (Lee et al., 2014; Orthwein et al., 2014). PP4C/R3β subsequently dephosphorylates 53BP1 at these critical sites as cells emerge from mitosis, allowing its recruitment to chromatin and the formation of 53BP1 bodies (Lee et al., 2014). The final stages of cytokinesis occur with relatively similar timing as these events (Steigemann et al., 2009; Mackay et al., 2010a). At this juncture, cells are connected by an intercellular bridge containing a microtubule-based structure termed the midbody. Aurora B localizes to this structure, where it phosphorylates specific substrates in order to regulate events that lead to abscission (Steigemann et al., 2009; Capalbo et al., 2012; Carlton et al., 2012; Ferreira et al., 2013; Mathieu et al., 2013). Aurora B is well established to play an evolutionarily conserved role as a master regulator of cytokinesis progression and the abscission checkpoint (Steigemann and Gerlich, 2009). This checkpoint is responsive to a variety of events, including presence of a chromatin bridge (Steigemann et al., 2009), mechanical tension (Lafaurie-Janvore et al., 2013), and aberrant nuclear pore assembly (Mackay et al., 2010a).
Despite the emerging appreciation of conditions that affect the abscission checkpoint (Chen et al., 2012), the nature of signals that initiate and maintain Aurora B activity at the midbody is relatively uncharacterized. Here we report an unexpected connection between the presence of DNA lesions in nascent nuclei resulting from previous replication stress and progression through abscission. Increasing levels of replication stress result in an Aurora B–dependent delay of abscission. We further find that ATR and Chk1 participate in this cellular response, with Chk1 integrating additional signals upstream of Aurora B in the regulation of abscission timing. This cell cycle control mechanism is operational not only when extrinsic conditions augment replication stress, but also during unperturbed cell division. Together our results indicate that progression through cytokinesis is regulated by genomic surveillance mechanisms and for the first time identify a signaling pathway upstream of Aurora B at the time of abscission.
RESULTS
53BP1 nuclear foci form before abscission
53BP1-mediated genomic surveillance and cytokinesis progression appear to occur in a similar window of time, but to date they have not been tracked simultaneously. We probed 53BP1 localization concurrently with tubulin and found that 53BP1 nuclear foci are present within a significant fraction (∼64%) of HeLa and U2OS cells that are connected by a midbody (midbody-stage cells; Figure 1A). We next tested whether elevated replication stress alters 53BP1 foci formation at the midbody stage of cell division by pretreating cells with low concentrations of either hydroxyurea (HU), to reduce nucleotide pools, or aphidicolin (APH), to impair DNA polymerase (Harrigan et al., 2011; Lukas et al., 2011). In both cases, an increase in the percentage of midbody-stage cells with 53BP1 foci (>80%) was observed. Moreover, after replication stress, we found an increase in both the number and size of 53BP1 foci in these cells (Figure 1, A and B). The prevalence of 53BP1 foci in nuclei of midbody-stage cells demonstrates that postmitotic genomic surveillance proceeds before abscission. Following replication stress, the proportion of midbody-stage cells also increases (Figure 1C). Previously 53BP1 foci were observed in G1 nuclei of BJ-Tert cells, skin fibroblasts immortalized by telomerase expression (Harrigan et al., 2011). Consistently, we found that 53BP1 foci form before abscission in BJ-Tert cells and that replication stress results in more cells at midbody stage (Supplemental Figure S1, A and B).
FIGURE 1:

53BP1 foci are present in nuclei before abscission, and their presence corresponds to longer time in midbody stage. (A) HeLa and U2OS cells were cultured for 24 h in the presence of DMSO (control), HU, or APH and analyzed for the presence of nuclear foci containing 53BP1 (green). Midbody-stage cells were identified with an antibody directed against α-tubulin (magenta), and DNA was stained with DAPI (blue). Total percentage of midbody-stage cells with 53BP1 foci after each treatment is indicated (mean and SD from four experiments). Scale bar, 10 μm. (B) Quantification of 53BP1 foci per daughter cell nucleus in midbody-stage cells from each cell line after the indicated treatments. Data are combined results from four experiments. (C) Quantification of the number of midbody-stage cells (HeLa) after the indicated treatments. Error bars are mean and SD from three experiments. *p < 0.05 (Mann–Whitney test). (D) Montages of HeLa cells expressing GFP-tubulin and mCherry-53BP1-FFR subjected to time-lapse imaging. Time from midbody formation (0 min) to midbody disassembly (white arrowheads) was quantified by tracking GFP-tubulin for all cells that passed through this stage, with or without 53BP1 foci. (E) Quantification of abscission timing as defined in D. Boxplot represents the 25th, median, and 75th percentile of values from the indicated treatments (n = number of cells per treatment, combined from four experiments). Whiskers represent the 10th and 90th percentiles. ***p < 0.0001.
We further probed the relative temporal formation of 53BP1 foci by time-lapse imaging using a reporter cell line expressing mCherry fused to the DNA damage foci–forming region (FFR) of 53BP1 (Dimitrova et al., 2008). Similar to previous reports for full-length 53BP1, the 53BP1-FFR was found to form foci 105 ±20 min after the onset of mitosis. These foci further mirror the previously characterized 53BP1-OPT domains in their colocalization with the DNA repair factor MDC1 (Supplemental Figure S1C). Tracking mCherry-53BP1-FFR shows that 79% (±5%) of cells that progressed through cytokinesis displayed nuclear 53BP1 foci before abscission. This is consistent with our previous quantification (Figure 1A); the somewhat larger proportion of foci-positive midbody-stage cells could be an effect of 53BP1-FFR expression, a difference in detection sensitivity, or an intrinsic distinction between cell sublines. Measurements of total midbody-stage duration (the time from midbody formation [0 min] until midbody disassembly) revealed that this stage was significantly longer in cells with 53BP1 nuclear foci than with those that were negative for foci formation (median time of 125 vs. 90 min; Figure 1, D and E). This increase in timing coordinate with 53BP1 foci formation, along with the overall increase in midbody-stage cells after replication stress, suggests that postmitotic genome surveillance may play a role in regulating progression through abscission.
Postmitotic genome surveillance is coordinated with abscission timing via the Aurora B–mediated abscission checkpoint
To address whether there is a functional connection between postmitotic genome surveillance and abscission timing, we first established a robust assay that would allow us to analyze the kinetics of midbody resolution and identify factors involved in regulation of abscission (Figure 2). Specifically, we performed time-lapse imaging of a HeLa cell line expressing green fluorescent protein (GFP)–tubulin and histone H2B-mCherry and then measured how long it took cells captured at any point in midbody stage in the first frame to complete abscission. This measurement is proportional to midbody-stage duration, but since cells have already entered this stage and progressed into it to various extents at the time of the first frame, the time measured is typically faster and has somewhat greater range. Control-treated cells completed abscission with a median time of 55 min, and >95% of those cells starting at midbody stage did so within 2 h (Figure 2, A–C). Using this assay, we asked whether heightened levels of replication stress affect the duration of the midbody stage by pretreating cells with low concentrations of either HU or APH for 24 h before imaging (Figure 3A). Indeed, under conditions of increased replication stress, median time to abscission was found to be prolonged from 55 to 75 min (Figure 3). This change in kinetics was not due to differences in cell density (Supplemental Figure S2), which can influence abscission timing (Lafaurie-Janvore et al., 2013). We also analyzed live-imaging movies for the distribution of cells entering mitosis and midbody stage, as well as the duration of mitosis, and found that none of these parameters was altered by replication stress (Supplemental Figure S3). This confirms the specificity of the prolonged time to abscission and indicates that this measurement was not biased by a preceding alteration in cell division.
FIGURE 2:

Assay to assess abscission timing. (A) HeLa cells expressing GFP-tubulin (green) and H2B-mCherry (magenta) were subjected to time-lapse imaging every 5 min for 2 h. Time to abscission (midbody disassembly; white arrowheads) was quantified for all cells in midbody stage at the beginning of each movie. Either DMSO (Control) or Aurora B inhibitor (AurBi) was added to cells immediately before imaging. Cells that were in mitosis (prophase–metaphase) at the time of AurBi exposure were prone to cytokinesis failure as illustrated. (B) Time to abscission was quantified for all midbody-stage cells. Boxplot represents the 25th, median, and 75th percentile of values from the indicated treatments (n = number of cells per treatment, combined from three to five independent experiments). Whiskers represent the 10th and 90th percentiles. ***p < 0.0001 (Mann–Whitney test). (C) Cumulative frequency plots of progression through abscission. Each point represents the percentage of all midbody cells analyzed in C that had proceeded through abscission over the time course of the experiment. Error bars represent the mean and SD from three to five independent experiments per treatment. (D) Quantification of failed cytokinesis in mitotic (prophase–metaphase) cells and midbody-stage cells in the presence or absence of Aurora B inhibitor.
FIGURE 3:

Replication stress leads to prolonged time to abscission. (A) Timeline of experimental procedure and montages of HeLa cells stably expressing GFP-tubulin (green) and histone H2B-mCherry (magenta) cultured in HU or APH. Scale bar, 20 μm. (B) Quantification of abscission timing using the assay described in Figure 2. ***p < 0.0001. (C) Cumulative frequency plots of progression through abscission. Each point represents the percentage of all midbody cells analyzed in B that had proceeded through abscission over the time course of the experiment. Error bars are mean and SD from three to five experiments.
Replication stress is also reported to increase the number of ultrafine DNA bridges (UFBs) in certain cells (Chan et al., 2009). These chromatin-free bridges typically connect segregating chromosomes at anaphase and, in yeast, have been found to correspond to delayed anaphase progression/nuclear division (Germann et al., 2014). Using antibodies to either the RecQ-like helicase BLM or the SNF2-related Plk1-interacting checkpoint helicase (PICH), we tracked UFBs in anaphase cells. We found no evidence for UFBs persisting into late telophase or midbody-stage cells, consistent with the notion that proteins such as PICH, as well as specific nucleases, typically resolve UFBs by late anaphase (Naim et al., 2013; Ying et al., 2013). Further, although we saw an increase in UFBs after treatment with the topoisomerase II inhibitor ICRF-159 as previously reported (Chan et al., 2007), UFBs did not increase in HeLa cells after aphidicolin treatment (Supplemental Figure S4, A and B). Because at some frequency UFBs collapse to form chromatin bridges between daughter cells (Ying et al., 2013) and in this state could result in delayed abscission (Steigemann et al., 2009), we next quantified chromatin bridges. This revealed a slight but not statistically significant increase in cells treated with aphidicolin (Supplemental Figure S4C). This small difference (2 vs. 4% of midbody-stage cells) is unlikely to account for the consistent shift toward longer time to abscission under these conditions (Figure 3). We also tested whether replication stress leads to alterations in nuclear assembly, since defects in this process also alter the timing of abscission (Mackay et al., 2010a), but found no obvious disturbance in the architecture of postmitotic nuclei (Supplemental Figure S4D).
Aurora B is a key regulator of abscission timing (Steigemann et al., 2009) but also functions during the metaphase–anaphase transition and midbody formation (Carmena et al., 2009). The short-term assay used here was chosen specifically because it is ideally suited to discriminate between these roles. By adding a small-molecule inhibitor and proceeding directly to imaging, an assessment of midbody-stage timing can be made without any confounding effects the inhibitor may have on earlier stages of cell division. Addition of an Aurora B inhibitor immediately before the onset of imaging resulted in acceleration of median time to abscission for cells at midbody stage in the first frame (35 vs. 55 min; Figure 2, A–C). In contrast, if we analyzed cells at early stages of mitosis (prophase–metaphase) in the first frame, the presence of Aurora B inhibitor resulted primarily in cytokinesis failure (Figure 2D), underscoring its important earlier roles. To test the role of Aurora B activity during the prolonged time to abscission seen after replication stress, we introduced an Aurora B inhibitor after a period of replication stress and specifically analyzed those cells already at midbody stage at the time of Aurora B inhibitor treatment after short-term imaging (Figure 3, B and C). This revealed a reduction of median time to abscission by more than half in the presence of the Aurora B inhibitor (35 vs. 75 min), indicating that the delay in abscission triggered by replication stress requires this kinase. Together these results indicate that cells coordinate postmitotic genome surveillance with the timing of abscission via the Aurora B–mediated abscission checkpoint.
The link between replication stress and the abscission checkpoint requires activity of ATR and Chk1
Although our results suggest that signaling mechanisms connect maintenance of genome integrity to the final stage of cell division, how DNA lesions that persist through, or are created during, mitosis after replication stress are recognized and the nature of signals they generate are not yet clear. We postulated that if DDR signaling pathways are involved in regulating abscission kinetics, they should be present and active in a cell population enriched at the midbody stage.
To test this possibility, we synchronized cells using a thymidine/nocodazole treatment protocol (Supplemental Figure S5). By 3–5 h after release from nocodazole, the mitotic marker phospho-Histone H3-S10 was down-regulated, and a majority of cells were in midbody stage (Figure 4A and Supplemental Figure S5). Coincident with the presence of midbody-stage cells, we found significant levels of Chk1 phosphorylated at S296 and the ATR target sites S317 and S345, modifications that indicate that both Chk1 and ATR are active (Zhao and Piwnica-Worms, 2001; Kasahara et al., 2010). Furthermore, we detected pChk2-T68, indicating that ATM is also active in midbody-stage cells (Figure 4A; Matsuoka et al., 2000). Conditions of this synchronization may enhance DDR signaling as side effects of blocking replication and/or prolonging mitosis. Nonetheless, activity of these kinase pathways appears to peak as cells progress through abscission. Moreover, previous reports also found that Chk1 (and pChk1-S317), Chk2 (and pChk2-T68), and pATM-S1403 localize to the midbody (Tsvetkov et al., 2003; Peddibhotla et al., 2009; Yang et al., 2011). These data indicate that both the ATR/Chk1 and ATM/Chk2 signaling pathways are active at the time and place to potentially play a role in regulating abscission timing.
FIGURE 4:
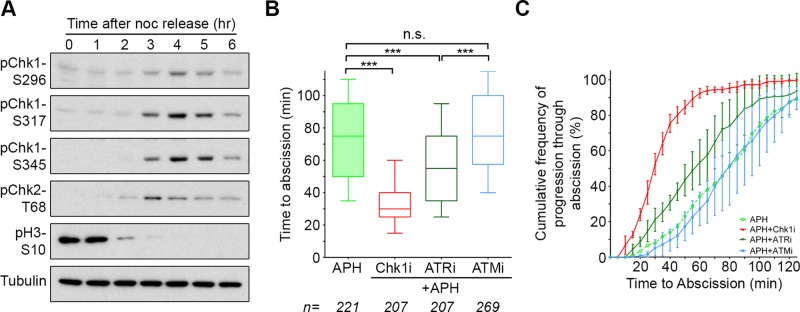
Prolonged time to abscission after replication stress requires activity of Chk1 and ATR. (A) Western blot analysis of HeLa cells after release from a thymidine/nocodazole synchronization (see Supplemental Figure S5). (B, C) Quantification of abscission using the assay in Figure 2. APH-only data are the same as that in Figure 3, B and C, and are included here for comparison. ***p < 0.0001; n.s., not significant.
We next determined the effect of inhibiting these two DDR pathways on abscission timing after replication stress. Of importance, this analysis assesses kinase inhibitors for their effect specifically at the time of cytokinesis (Figure 3). ATR inhibition reverted delayed abscission timing to that seen under control treatment (reduction of median time from 75 to 55 min), whereas Chk1 inhibition resulted in a more pronounced effect (Figure 4, B and C). In contrast, ATM inhibition had no significant effect on the timing of abscission. Different sensitivities to kinase inhibition did not relate to differential efficacy of inhibitors, as each was verified to be potent in the context of response to DNA damage (Supplemental Figure S6A). These results indicate that the altered timing in cytokinetic abscission that results from replication stress is mediated by the ATR/Chk1 pathway and further suggest that Chk1 has an additional, independent role in abscission timing.
ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with abscission timing
Underreplicated DNA lesions persist at some frequency during unperturbed cell division (Figure 1; Harrigan et al., 2011; Lukas et al., 2011; Bergoglio et al., 2013). This suggests that abscission timing may be responsive to similar—albeit lower-level—signals during normal cell division. Indeed, we found that inhibiting Chk1 in unperturbed midbody-stage cells accelerated median time to abscission, closely mirroring Aurora B inhibition (Figure 5, A and B). Of note, we found that exposure of mitotic cells to Chk1 inhibitor leads to failure of cytokinesis and consequent binucleation (Supplemental Figure S6B), as observed previously (Peddibhotla et al., 2009). This analysis clearly uncouples the novel role for Chk1 in controlling events of midbody resolution from its earlier cell cycle contributions. ATR inhibition had a lesser but significant effect during midbody stage, commensurate with a lower level of postmitotic genomic lesions during an unperturbed cell cycle (Figure 5, A and B, and Supplemental Figure S6C). In contrast, although ATM and Chk2 are present at the midbody, where they may be poised for action should cytokinesis proceed aberrantly (Janssen et al., 2011), they do not have an observable role in regulating abscission timing (Figure 5, A and B). To distinguish whether ATR and Chk1 contribute to the timing of abscission in all cells or only those in which DNA lesions are detected by postmitotic genome surveillance, we imaged cells expressing mCherry-53BP1-FFR in the absence or presence of specific inhibitors. Cells at midbody stage in the first frame were then binned into two categories, corresponding to whether or not postmitotic 53BP1 foci were detected, and time to abscission was tabulated. ATR inhibition did not have a statistically significant effect on foci-negative cells (Figure 5C). Foci-positive cells, as before, took longer to progress to abscission; median time to abscission was ∼10 min greater in cells with 53BP1 foci (vs. ∼25-min increase when the total duration of midbody stage was measured; Figure 1E). Significantly, in foci-positive cells, inhibition of ATR restored timing to that seen in foci-negative cells (Figure 5C). Chk1 activity was critical regardless of postmitotic 53BP1 foci. In the case of foci-positive cells, Chk1 inhibition had a robust effect, overcoming the longer time to abscission to reduce timing comparably to its effect on foci-negative cells. Together these data indicate that ATR activity is essential in cells with heightened levels of postmitotic genome surveillance, whereas Chk1 has a more central role in the regulation of abscission timing.
FIGURE 5:

ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with abscission timing. (A, B) Quantification of abscission timing using the time-lapse imaging assay in Figure 2. Control and AurBi data from Figure 2 are included for comparison. ***p < 0.0001. (C) HeLa cells expressing GFP-tubulin and mCherry-53BP1-FFR were subjected to time-lapse imaging, and abscission timing was quantified for cells with and without 53BP1 foci in the presence or absence of the indicated inhibitors. The difference in median time to abscission compared with cells negative for 53BP1 foci (which take a median time of 50 min) is indicated. **p < 0.01; ***p < 0.0001. (D) HeLa cells were treated for 15 min with the indicated inhibitors and analyzed for localization of Aurora B-pT232 (green) at the midbody (α-tubulin, magenta). Graph represents quantification of Aurora B-pT232 fluorescence intensity after each treatment compared with control. Error bars indicate SEM. Scale bar, 10 μm.
The similar effects of Chk1 and Aurora B inhibition with respect to abscission timing, coupled with evidence that Chk1 regulates Aurora B activity during prometaphase (Petsalaki et al., 2011), led us to test whether Chk1 is upstream of Aurora B in regulating abscission. To do so, we treated cells briefly with chemical inhibitors of Aurora B, Chk1, or ATR and analyzed immunolocalization of an indicator of Aurora B activity (AurB-pT232). As expected, Aurora B inhibition resulted in almost complete reduction of pAurB signal at the resolving midbody (Figure 5D). Chk1 inhibition also led to a lack of pAurB at the midbody, suggesting that the major role for Chk1 in regulating abscission timing is upstream of Aurora B. Similar brief inhibition of ATR had no measurable effect on pAurB midbody intensity, likely due to its specialized role in a subset of cells during unperturbed cell division. These data are consistent with a model in which ATR activity in midbody-stage cells links genomic surveillance to abscission timing but is only one of multiple upstream signals integrated by Chk1 and Aurora B to control timing of cytokinetic abscission (Figure 6).
FIGURE 6:
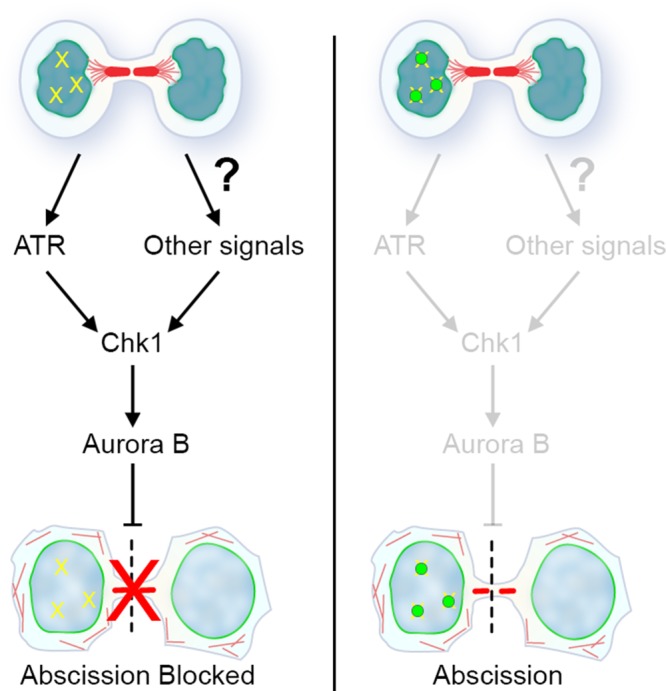
Model for abscission checkpoint regulation by ATR and Chk1. DNA lesions in postmitotic cells (depicted schematically in one nucleus; yellow hatches) trigger activation of a signaling pathway via ATR. This feeds into the abscission checkpoint via the central regulatory node of Chk1 and the downstream kinase Aurora B to delay abscission. Recruitment of 53BP1 (green circles), and likely other factors that protect DNA lesions during genome surveillance, would eventually lead to suppression of this signaling path when these lesions are properly poised for repair. Although the molecular network is not yet defined, other signals (e.g., those triggered by tension forces) feed independently into the Chk1-Aurora B pathway to further control abscission timing.
DISCUSSION
Aurora B–mediated control of progression through cytokinesis during both normal and aberrant division has been demonstrated in a variety of species (Norden et al., 2006; Steigemann et al., 2009; Mackay et al., 2010a; Bembenek et al., 2013; Lafaurie-Janvore et al., 2013; Mathieu et al., 2013). Most research has focused on the role of the Aurora B–mediated checkpoint in protecting cells against chromosome instability by coordinating chromosome segregation with cytokinesis progression. The data presented here provide evidence that Aurora B is also sensitive to the presence of damaged DNA resulting from previous replication stress. These observations are consistent with an additional role for the Aurora B–mediated abscission checkpoint in coordinating completion of cell division with recognition and shielding of such genomic lesions (Figure 6). Surveillance of genomic DNA in newly formed nuclei has been described as a G1 event (Harrigan et al., 2011; Lukas et al., 2011). Our findings here that this process initiates before cytokinetic abscission and influences the timing of abscission integrates well with the recent description of cytokinetic abscission as a G1 event based on tracking cell division with molecular markers (Gershony et al., 2014).
We further found that abscission timing is particularly dependent on Chk1 activity, whereas inhibition of ATR restores cells to baseline timing but does not further accelerate the process. Given the appearance at midbody stage of Chk1 phosphorylation at residues characterized to be targets of ATR (Figure 4A), we propose that ATR mediates a specific link between genome surveillance and Chk1 activity to regulate abscission timing. Additional signals emanating from separate pathways of quality control may feed into Chk1 activity at this time as well. It is also formally possible that noncanonical (ATR-independent) signaling provides additional connections between genome surveillance and Chk1 activity. In either case, this raises the question of what other upstream factors regulate Chk1 during cytokinesis. Although other kinases have been implicated upstream of Chk1 at mitosis (Shiromizu et al., 2006), further studies will be needed to uncover relevant upstream signaling pathways involved in the regulation of abscission timing. The assay used here, which combines short-term treatment with small-molecule inhibitors and short-term live imaging, will be a useful strategy to map out signaling pathways at this cell cycle stage.
Checkpoints at other times in the cell cycle are well characterized to respond to DNA damage, yet postmitotic genome surveillance is important because damage can be acquired during mitosis. Underreplicated regions of the genome are one source of such damage, as these go on to either be processed or break at the time of mitotic chromosome condensation and segregation (Naim et al., 2013; Ying et al., 2013; Germann et al., 2014). Although protecting and repairing these lesions is clearly imperative, the connection back to abscission timing is unexpected. The interconnection of these events may ensure that dividing cells coping with potential genomic instability retain the option of cleavage furrow regression as a final quality control mechanism. Under normal circumstances, the resulting tetraploid cell would be removed from the population in the subsequent cell cycle (Ganem et al., 2014).
In summary, we found a new role for the abscission checkpoint in coordinating completion of cell division with postmitotic genome surveillance. We found that cells respond to elevated replication stress by sustaining the activation of this checkpoint. This study also places ATR and Chk1 in a molecular pathway that connects to Aurora B activity at the midbody stage of cell division. The abscission checkpoint is likely to be frequently engaged due to the consequences of replication stress, as certain regions of the genome are inherently difficult to replicate and prone to underreplication. Various conditions, including oncogene expression, exacerbate replication stress (Neelsen et al., 2013), and replication stress can be a contributing factor in the early stages of tumorigenesis (Gorgoulis et al., 2005), further underscoring the importance of checkpoints sensitive to these defects. Other conditions that are linked to the abscission checkpoint, such as lagging chromosomes (Steigemann et al., 2009; Bembenek et al., 2013) and disruptions in postmitotic nuclear assembly (Mackay et al., 2010a), are rare events but become more likely under pathological circumstances. Whereas these events are distinct in origin from those described in this study, it will be of interest to determine whether they share commonalities in the molecular pathway that leads to Aurora B regulation. Of interest, there are recently recognized connections between Nup153 and DNA damage response (Lemaitre et al., 2012; Moudry et al., 2012; Wan et al., 2013), suggesting that an underlying molecular defect that feeds into the abscission checkpoint when the role of Nup153 is impaired could be a deficiency in genomic surveillance.
MATERIALS AND METHODS
Cell lines, culture, and treatments
Cells were grown in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) at 37ºC/5% CO2. The stable HeLa cell line (mCherry-53BP1-FFR) used in Figure 1 was generated by transfecting a plasmid containing mCherry-53BP11220-1711 (Addgene, Cambridge, MA; Dimitrova et al., 2008) into a cell line stably expressing GFP-tubulin (Mackay et al., 2010a) using Lipofectamine LTX (Life Technologies) and selecting with 0.5 mg/ml G418 plus 0.5 μg/ml puromycin. Generation of the HeLa cell line stably expressing GFP-tubulin and histone H2B-mCherry was previously described (Mackay et al., 2010a).
To induce replication stress, cells were treated for 24 h with either 50 μM hydroxyurea (MP Biomedicals, Santa Ana, CA) or 0.4 μM aphidicolin (Fisher Bioreagents). Where indicated, inhibitors were used at the following concentrations: AurBi (ZM447439; Biotechne, Minneapolis, MN), 2 μM; Chk1i (AZD7762; Selleck Chemicals, Houston, TX), 2 μM; ATRi (NU6027; EMD Millipore, Billerica, MA), 10 μM; ATMi (KU55933; Selleck Chemicals), 10 μM; and Chk2i (Chk2 inhibitor II; Millipore), 10 μM. As a control for the identification of DNA ultrafine bridges, ICRF-159 (Sigma-Aldrich, St. Louis, MO) was used at a concentration of 10 μM (Chan et al., 2007).
Immunofluorescence analysis and antibodies
In general, cells were fixed for immunofluorescence analysis by incubation in −20ºC methanol for 10 min. For visualization of DNA ultrafine bridges, cells were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) for 20 min at room temperature, followed by incubation in −20°C methanol for 10 min. Antibody incubations were at room temperature for 2 h or at 4ºC overnight in blocking solution (3% FBS plus 0.05% Triton in PBS). The following antibodies were used: 53BP1 (sc-22760, 1:1000; Santa Cruz Biotechnology, Dallas, TX); α-tubulin (YL1/2, 1:1000; Accurate Chemical & Scientific Corp., Westbury, NY); α-tubulin (ab18251, 1:2500; Abcam, Cambridge, MA); BLM (ab2179, 1:100; Abcam); PICH (#04-1540, 1:250; Millipore); MDC1 (#05-1572, 1:500; Millipore); Nup153 (SA1; provided by B. Burke, Institute of Medical Biology, Singapore); Tpr (IHC-00099, 1:1000; Bethyl Laboratories, Montgomery, TX); lamin B2 (ab8983, 1:500; Abcam); and Aurora B-pT232 (1:500; Rockland Immunochemicals, Limerick, PA). Secondary antibodies were from Invitrogen. Coverslips were mounted in Prolong Gold plus 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies). Sytox Green (Life Technologies) was used to stain DNA in Supplemental Figure S4. Images were acquired with a Zeiss Axioskop2 microscope equipped with a 63× PlanApo (numerical aperture [NA] 1.4) objective. Fluorescence intensity measurements in Figure 5D were performed using ImageJ (National Institutes of Health, Bethesda, MD).
Cell synchronization
For Figure 4A, cells were synchronized by incubation in 2 mM thymidine for 24 h, washed several times with PBS, and released into fresh medium containing 50 ng/ml nocodazole for 16 h. Mitotic cells were harvested by shake-off, washed several times with PBS, and released into fresh medium. Samples were harvested every hour for 6 h postrelease, lysed, and analyzed by Western blot analysis. For Supplemental Figure S5, HeLa cells stably expressing GFP-tubulin were synchronized, and mitotic cells were released onto fibronectin-coated coverslips, which were removed at 1-h time points, fixed in −20°C methanol, and analyzed for GFP expression.
Western blot analysis
Samples for Western blot were lysed in NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 15 mM MgCl2, 1% Nonidet P-40, 60 mM β-glycerophosphate, 1 mM dithiothreitol, 0.1 mM sodium vanadate, 100 μM phenylmethylsulfonyl fluoride, 0.1 mM NaF, and 1× Complete Protease Inhibitor Cocktail [Roche, Indianapolis, IN]). Cleared lysates were then separated by SDS–PAGE and transferred to polyvinylidene fluoride membrane. Membranes were blocked with 5% milk in Tris-buffered saline (TBS) plus 0.1% Tween-20 and blotted with primary antibodies diluted in either 5% milk or 5% bovine serum albumin in TBS plus 0.1% Tween-20. The following antibodies were used: Chk1-pS296 (#2349, 1:1000; Cell Signaling Technology, Danvers, MA); Chk1-pS317 (#8191, 1:1000; Cell Signaling Technology); Chk1-pS345 (#2348, 1:1000; Cell Signaling Technology); Chk2-pT68 (#2661, 1:1000; Cell Signaling Technology), and Histone H3-pS10 (#06-570, 1:4000; Millipore). After incubation with horseradish peroxidase–conjugated secondary antibodies, protein levels were detected using the Western Lightning Plus ECL reagent (Perkin Elmer, Waltham, MA). The peak of midbody-stage enrichment was determined by immunofluorescence analysis with α-tubulin antibodies to identify midbodies.
Live-cell imaging and analysis
Stable HeLa cell lines expressing either GFP-tubulin and mCherry-53BP1-FFR (amino acids 1220–1711) or GFP-tubulin and H2B-mCherry were grown in 35-mm glass-bottom dishes coated with fibronectin (MatTek, Ashland, MA). Where indicated, cells were treated with dimethyl sulfoxide (DMSO), HU, or APH as described for 24 h. Just before imaging, the culture medium was replaced with Imaging Medium (DMEM/F-12, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, no phenol red [Life Technologies], supplemented with 10% FBS). Time-lapse imaging of multiple X-Y stage positions was performed essentially as described, with a few modifications (Mackay et al., 2010b). Indicated cell lines were imaged at 37ºC using either a DeltaVision RT imaging system equipped with an Olympus IX-71 microscope and a 20× UPlanSApo (NA 0.75) objective or a Nikon wide-field imaging system with a heated stage and 20× objective. To determine duration of midbody stage (Figure 1), images were acquired (2 × 2 binning) every 5 min for 6 h, and time from midbody formation until midbody disassembly was calculated for all cells that progressed through this stage during the time-lapse acquisition. For Supplemental Figure S3, images were acquired every 5 min for 3 h, and the following parameters were recorded: time from the start of time lapse until cells enter mitosis, time from start until cells enter midbody stage, and total time from onset of mitosis until midbody formation. To determine time to abscission (Figures 2–5 and Supplemental Figures S3 and S6), images were acquired every 5 min for 2 h. For kinase inhibitor studies, compounds were added just before the first frame. Time to abscission was determined for all cells connected with a midbody in the first frame by manually quantifying the time until the midbody was no longer visible between the two daughter cells. Quantification and statistical analysis were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgments
We thank Zhongsheng You, Trudy Oliver, Srividya Bhaskara, Jody Rosenblatt, and members of the Ullman and Rosenblatt labs for helpful discussions. This work was supported by National Institutes of Health Grant R01 GM61275 (K.U.) and 5T32 HD07491 Developmental Biology Training Grant (D.M.). Shared resources used in this project are supported in part by P30 CA042014 awarded to the Huntsman Cancer Institute.
Abbreviations used:
- APH
aphidicolin
- 53BP1-FFR
53BP1 foci-forming region
- DDR
DNA damage response
- HU
hydroxyurea
- UFB
ultrafine bridge.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-11-1563) on April 22, 2015.
REFERENCES
- Bembenek JN, Verbrugghe KJ, Khanikar J, Csankovszki G, Chan RC. Condensin and the spindle midzone prevent cytokinesis failure induced by chromatin bridges in C. elegans embryos. Curr Biol. 2013;23:937–946. doi: 10.1016/j.cub.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoglio V, Boyer AS, Walsh E, Naim V, Legube G, Lee MY, Rey L, Rosselli F, Cazaux C, Eckert KA, Hoffmann JS. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol. 2013;201:395–408. doi: 10.1083/jcb.201207066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capalbo L, Montembault E, Takeda T, Bassi ZI, Glover DM, D'Avino PP. The chromosomal passenger complex controls the function of endosomal sorting complex required for transport-III Snf7 proteins during cytokinesis. Open Biol. 2012;2:120070. doi: 10.1098/rsob.120070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JG, Caballe A, Agromayor M, Kloc M, Martin-Serrano J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science. 2012;336:220–225. doi: 10.1126/science.1217180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol. 2009;21:796–805. doi: 10.1016/j.ceb.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. doi: 10.1038/sj.emboj.7601777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- Chen CT, Hehnly H, Doxsey SJ. Orchestrating vesicle transport, ESCRTs and kinase surveillance during abscission. Nat Rev Mol Cell Biol. 2012;13:483–488. doi: 10.1038/nrm3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. Common fragile sites: mechanisms of instability revisited. Trends Genet. 2012;28:22–32. doi: 10.1016/j.tig.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JG, Pereira AJ, Akhmanova A, Maiato H. Aurora B spatially regulates EB3 phosphorylation to coordinate daughter cell adhesion with cytokinesis. J Cell Biol. 2013;201:709–724. doi: 10.1083/jcb.201301131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann SM, Schramke V, Pedersen RT, Gallina I, Eckert-Boulet N, Oestergaard VH, Lisby M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J Cell Biol. 2014;204:45–59. doi: 10.1083/jcb.201305157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershony O, Pe'er T, Noach-Hirsh M, Elia N, Tzur A. Cytokinetic abscission is an acute G1 event. Cell Cycle. 2014;13:3436–3441. doi: 10.4161/15384101.2014.956486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Goto H, Enomoto M, Tomono Y, Kiyono T, Inagaki M. 14–3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO J. 2010;29:2802–2812. doi: 10.1038/emboj.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaurie-Janvore J, Maiuri P, Wang I, Pinot M, Manneville JB, Betz T, Balland M, Piel M. ESCRT-III assembly and cytokinetic abscission are induced by tension release in the intercellular bridge. Science. 2013;339:1625–1629. doi: 10.1126/science.1233866. [DOI] [PubMed] [Google Scholar]
- Lee DH, Acharya SS, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA, Chowdhury D. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell. 2014;54:512–525. doi: 10.1016/j.molcel.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre C, Fischer B, Kalousi A, Hoffbeck AS, Guirouilh-Barbat J, Shahar OD, Genet D, Goldberg M, Betrand P, Lopez B, et al. The nucleoporin 153, a novel factor in double-strand break repair and DNA damage response. Oncogene. 2012;31:4803–4809. doi: 10.1038/onc.2011.638. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, Lukas J. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- Mackay DR, Makise M, Ullman KS. Defects in nuclear pore assembly lead to activation of an Aurora B-mediated abscission checkpoint. J Cell Biol. 2010a;191:923–931. doi: 10.1083/jcb.201007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DR, Ullman KS, Rodesch CK. Time-lapse imaging of mitosis after siRNA transfection. J Vis Exp. 2010b;6:1878. doi: 10.3791/1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Huttner D, Hickson ID. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013;32:2661–2671. doi: 10.1038/emboj.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Cauvin C, Moch C, Radford SJ, Sampaio P, Perdigoto CN, Schweisguth F, Bardin AJ, Sunkel CE, McKim K, et al. Aurora B and cyclin B have opposite effects on the timing of cytokinesis abscission in Drosophila germ cells and in vertebrate somatic cells. Dev Cell. 2013;26:250–265. doi: 10.1016/j.devcel.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudry P, Lukas C, Macurek L, Neumann B, Heriche JK, Pepperkok R, Ellenberg J, Hodny Z, Lukas J, Bartek J. Nucleoporin NUP153 guards genome integrity by promoting nuclear import of 53BP1. Cell Death Differ. 2012;19:798–807. doi: 10.1038/cdd.2011.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim V, Wilhelm T, Debatisse M, Rosselli F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol. 2013;15:1008–1015. doi: 10.1038/ncb2793. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Zanini IM, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell. 2006;125:85–98. doi: 10.1016/j.cell.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344:189–193. doi: 10.1126/science.1248024. [DOI] [PubMed] [Google Scholar]
- Peddibhotla S, Lam MH, Gonzalez-Rimbau M, Rosen JM. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc Natl Acad Sci USA. 2009;106:5159–5164. doi: 10.1073/pnas.0806671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petsalaki E, Akoumianaki T, Black EJ, Gillespie DA, Zachos G. Phosphorylation at serine 331 is required for Aurora B activation. J Cell Biol. 2011;195:449–466. doi: 10.1083/jcb.201104023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiromizu T, Goto H, Tomono Y, Bartek J, Totsukawa G, Inoko A, Nakanishi M, Matsumura F, Inagaki M. Regulation of mitotic function of Chk1 through phosphorylation at novel sites by cyclin-dependent kinase 1 (Cdk1) Genes Cells. 2006;11:477–485. doi: 10.1111/j.1365-2443.2006.00955.x. [DOI] [PubMed] [Google Scholar]
- Steigemann P, Gerlich DW. An evolutionary conserved checkpoint controls abscission timing. Cell Cycle. 2009;8:1814–1815. [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Tsvetkov L, Xu X, Li J, Stern DF. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J Biol Chem. 2003;278:8468–8475. doi: 10.1074/jbc.M211202200. [DOI] [PubMed] [Google Scholar]
- Wan G, Zhang X, Langley RR, Liu Y, Hu X, Han C, Peng G, Ellis LM, Jones SN, Lu X. DNA-damage-induced nuclear export of precursor microRNAs is regulated by the ATM-AKT pathway. Cell Rep. 2013;3:2100–2112. doi: 10.1016/j.celrep.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, Wong ST, Fu L, Xu B. Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol Cell. 2011;44:597–608. doi: 10.1016/j.molcel.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID. MUS81 promotes common fragile site expression. Nat Cell Biol. 2013;15:1001–1007. doi: 10.1038/ncb2773. [DOI] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.