

Acentric chromosomes exhibit delayed segregation during mitosis. How these delays affect nuclear envelope reassembly is not fully understood. Lagging acentrics coated with Aurora B induce a highly localized gap in the nuclear envelope to allow acentric entry into daughter nuclei. Gap formation is decreased upon reduction of Aurora B.
Abstract
To determine how chromosome segregation is coordinated with nuclear envelope formation (NEF), we examined the dynamics of NEF in the presence of lagging acentric chromosomes in Drosophila neuroblasts. Acentric chromosomes often exhibit delayed but ultimately successful segregation and incorporation into daughter nuclei. However, it is unknown whether these late-segregating acentric fragments influence NEF to ensure their inclusion in daughter nuclei. Through live analysis, we show that acentric chromosomes induce highly localized delays in the reassembly of the nuclear envelope. These delays result in a gap in the nuclear envelope that facilitates the inclusion of lagging acentrics into telophase daughter nuclei. Localized delays of nuclear envelope reassembly require Aurora B kinase activity. In cells with reduced Aurora B activity, there is a decrease in the frequency of local nuclear envelope reassembly delays, resulting in an increase in the frequency of acentric-bearing, lamin-coated micronuclei. These studies reveal a novel role of Aurora B in maintaining genomic integrity by promoting the formation of a passageway in the nuclear envelope through which late-segregating acentric chromosomes enter the telophase daughter nucleus.
INTRODUCTION
The eukaryotic cell has evolved a number of mechanisms to maintain genome integrity. In response to damaged or improperly replicated DNA, cell cycle checkpoints activate signaling pathways that influence the activity of both positive and negative cell cycle regulators, providing time for repair or elimination of the damaged cell (Abbas et al., 2013). The molecular basis of these signaling pathways has been extensively investigated, with much of the work focused on the action of DNA damage/replication checkpoints during interphase (G1, S, and G2; Rhind and Russell, 2012). Recent studies provide convincing evidence that cells entering metaphase with damaged chromosomes delay anaphase entry, suggesting activation of DNA damage checkpoints during metaphase. Support for this interpretation comes from studies in a number of cell types, demonstrating that these DNA damage–induced metaphase delays depend on the spindle assembly and DNA damage checkpoints (Mikhailov et al., 2002; Fasulo et al., 2012; Cipressa and Cenci, 2013; Zhang and Hunter, 2014). For example, in Drosophila, chk1/grp is required for delaying anaphase entry in response to double-strand breaks (DSBs; Royou et al., 2005).
In spite of these safeguards, cells occasionally exit metaphase with damaged DNA. Unrepaired DSBs are particularly problematic because they produce chromosome fragments lacking either a centromere or a telomere (Kaye et al., 2004). The former, known as acentrics, are incapable of forming normal attachments with the mitotic spindle and are thus expected to exhibit segregation defects. For example, acentrics often fail to segregate properly, resulting in their exclusion from daughter nuclei and inclusion in cytoplasmic micronuclei (Kanda and Wahl, 2000; LaFountain et al., 2001; Fenech et al., 2011). In Drosophila, the behavior of acentrics has been examined by taking advantage of transgenic lines bearing an inducible I-CreI endonuclease (Rong et al., 2002; Royou et al., 2010). I-CreI specifically targets the rDNA locus near the base of the X chromosome (Rong et al., 2002; Maggert and Golic, 2005; Paredes and Maggert, 2009; Golic and Golic, 2011). Surprisingly, I-CreI–induced acentrics segregate in a delayed but otherwise normal manner due to the action of DNA tethers connecting the fragments to their centric partners (Royou et al., 2010; Kotadia et al., 2012). The DNA tether contains histones and is coated with BubR1, Polo, and the chromosome passenger proteins Aurora B and INCENP (Royou et al., 2010). During metaphase, acentrics localize to the outer edge of the metaphase plate, separated from the main mass of chromosomes. On entry into anaphase, they remain in this position while the main mass of sister chromosomes separate. Only during late anaphase do sister acentrics segregate to opposing poles. Polo and BubR1 are not necessary for tether formation but are required for normal poleward movement of the acentrics (Royou et al., 2010).
The extensive delay in acentric segregation raises a number of questions regarding the spatial and temporal control of events during the anaphase–telophase transition, including the timing of nuclear envelope formation (NEF) with respect to chromosome segregation. Drosophila cells undergo “semiclosed” mitosis in which the nuclear envelope only breaks down completely in early anaphase before reassembling again during late anaphase/early telophase (Katsani et al., 2008). Current models of NEF propose that the nuclear envelope forms from mitotic endoplasmic reticulum (ER) extensions, which initiate interactions between chromatin and inner nuclear membrane proteins (Anderson and Hetzer, 2008; Lu et al., 2011). Membrane fusion of nuclear envelope microdomains and nuclear pore complex assembly complete NEF to build a fully functioning nuclear envelope (Baur et al., 2007; Dultz et al., 2008). Chromatin decondensation is concomitant with NEF, and Aurora B has been shown to inhibit both decondensation and NEF on a global scale (Ramadan et al., 2007; Meyer et al., 2010; Afonso et al., 2014).
Understanding the mechanisms by which chromosome segregation and NEF are coordinated is of particular interest. Specifically, how would delayed segregation of acentric chromosomes influence the dynamics of nuclear envelope reformation during telophase? Here we directly address this issue through live and fixed analyses of chromosome segregation and NEF in Drosophila larval neuroblasts bearing normal and lagging acentric chromosomes. Our analysis reveals a highly localized delay of the reassembly of NE components around the lagging acentrics that result in the formation of large gaps in the nascent nuclear envelope. Live analysis reveals that these gaps provide a means for late-segregating acentrics to be included in the newly formed telophase nuclei. Aurora B kinase activity is required for these local nuclear envelope component reassembly delays. If Aurora B levels are reduced, the frequency of cells with delays in the reassembly of NE components upon I-CreI induction is greatly decreased. As a result, reduced Aurora B levels result in a dramatic increase in lamin-coated, acentric-bearing micronuclei. Thus, in addition to its well-established role in the spindle midzone assembly, kinetochore–microtubule attachments, and the abscission checkpoint (Tanaka et al., 2002; Norden et al., 2006; Douglas et al., 2010), we find that Aurora B also plays an important role in maintaining genomic integrity by facilitating the entry of late-segregating acentrics into daughter nuclei through highly localized delays in the reassembly of nuclear envelope.
RESULTS
Acentric chromosomes induce highly localized delays in the reassembly of the nuclear envelope during telophase
To examine the effect of severely delayed acentric chromosome segregation on NEF, we visualized NEF in third-instar Drosophila neuronal stem cells (neuroblasts) in which lagging acentrics were induced by I-CreI. Third-instar neuroblasts divide asymmetrically to produce a large, self-renewing neuroblast and a smaller ganglion mother cell, which divides once more, giving rise to a postmitotic neuron (Doe, 2008). Transgenic Drosophila bearing the heat-inducible I-CreI endonuclease produce DSBs specifically at the rDNA repeats on the X chromosome (Rong et al., 2002; Maggert and Golic, 2005). The resulting acentrics localize to the edge of the metaphase plate and eventually segregate to the poles late in anaphase through the action of a DNA tether attaching each acentric fragment to its centric partner (Royou et al., 2010). In addition to I-CreI, these transgenic lines express the histone 2 variant labeled with red fluorescent protein (H2Av-RFP) and lamin B–green fluorescent protein (GFP), facilitating live analysis of chromosome and nuclear envelope dynamics in neuroblasts (see Materials and Methods).
Live imaging of chromosome segregation and reformation of the nuclear lamina in neuroblasts with undamaged chromosomes (no I-CreI expression) is shown in Figure 1A (top; Supplemental Video S1). In neuroblasts with undamaged chromosomes, NEF occurs rapidly after sister chromatid segregation (Figure 1A, top; Supplemental Video S1). Time 0 marks the onset of anaphase as defined by separation of sister chromosomes. Once NEF is initiated, it proceeds until it is fully completed, as visualized by lamin B-GFP signal completely surrounding the segregated chromatin (compare time points 180–240). On average, initiation and completion of NEF occurred ∼217 ± 102 s (N = 6) and 327 ± 116 s (N = 6), respectively, after anaphase onset. Despite neuroblast divisions being asymmetric with respect to their cell size (Jiang and Reichert, 2013), centrosome morphology (Rebollo et al., 2007), microtubule aster size (Giansanti et al., 2001) and cortical components (Doe, 2008), we found that timing of NEF initiation and completion were synchronous in the two daughter nuclei, which is consistent with previous reports (Katsani et al., 2008).
FIGURE 1:

Acentric chromosome fragments enter nascent daughter nuclei through highly localized delays in assembly of the nuclear envelope. (A) Still images from time-lapse movies of mitotic neuroblasts without (top; Supplemental Video S1) and with (bottom; Supplemental Video S2) I-CreI–induced acentrics (red arrows). Acentrics enter daughter nuclei through nuclear envelope gaps (green arrows). The chromosomes are labeled with H2Av-RFP (red) and the nuclear envelope with lamin B-GFP (green). These gaps do not form when acentric fragments are not present. Dashed lines represent cell outlines. Scale bars, 2 μm. (B) Quantification of lamin B-GFP signal across the surface of the nuclear envelope gaps during NEF. Graphs of the lamin B-GFP fluorescence intensity along pole-facing (blue line) and midzone-facing (red line) sections of the newly formed telophase nuclear envelopes with (left) and without acentrics (right). Each graph represents an average of three measurements. Error bars indicate SDs. Green brackets indicate the presence of a nuclear envelope gap. Scale bars, 2 μm. (C) Quantification of lamin B-GFP signal across the midzone face of a newly formed nuclear envelope before and after an acentric enters the daughter nucleus. Graphs of the lamin B-GFP fluorescence intensity along pole-facing (blue line) and midzone-facing (red line) sections of the telophase nuclear envelope before (top) and after (bottom) an acentric has entered the daughter nucleus. Each graph represents an average of three measurements. Error bars indicate SDs. Green brackets indicate the presence of a nuclear envelope gap. Scale bar, 2 μm. (D) Multiplane 3D rendering of an I-CreI–induced acentric entering a nascent daughter nucleus through a nuclear envelope gap (green arrow). Red channel depicts H2Av-RFP. Green channel represents lamin B-GFP.
We next imaged chromosome separation and NEF in neuroblasts bearing I-CreI–induced acentrics (Figure 1A, bottom; Supplemental Video S2). Although initiation of NEF was slightly delayed in divisions with acentrics (298 ± 65 s; N = 32), we observed significant delays in completion of NEF (681 ± 248 s; N = 14; Figure 1A, bottom; compare time points 280–680). The delay in NEF completion, as visualized by a gap in the lamin B-GFP signal, was highly localized, consistently occurring at the site of acentric entry (Figure 1A, green arrows). These gaps in the nuclear envelope serve as passageways for late-segregating acentrics to enter the telophase nuclei. Our analysis revealed that the width of these nuclear envelope gaps did not undergo significant changes in diameter as the acentric passed through to rejoin the main nuclear mass. We found that the localized nuclear envelope gaps have an average width of 0.92 ± 0.27 μm (N = 7). This is approximately sixfold larger than the physical diameter of a nuclear pore (Wente and Rout, 2010). Note that these localized delays in nuclear envelope reassembly only occur in the presence of an acentric and are not present in heat-shocked larvae without I-CreI.
To quantify the presence of localized delays in nuclear envelope reassembly induced by acentric chromosomes, we measured the fluorescence intensity of lamin B-GFP across the surface of the nuclear envelope during telophase (Figure 1B). Compared to regions on the same telophase nucleus without local delays in nuclear envelope reassembly (blue line), fluorescence intensity of lamin B-GFP was markedly reduced across the site where acentrics entered through local delays in nuclear envelope reassembly (Figure 1B, left, red line). We did not observe the same reduced intensity of lamin B-GFP signal on the midzone face in non–I-CreI–expressing control telophase neuroblasts (Figure 1B, right). We found that the reduced intensity of lamin B-GFP signal persisted until after the acentric had passed through the gap, at which point nuclear envelope assembly resumed, as visualized by an increase in lamin B-GFP signal (Figure 1C).
To demonstrate further that these gaps in the nuclear envelope were induced by acentrics, we performed live three-dimensional (3D) rendering of telophase nuclei expressing I-CreI to determine the position of acentrics relative to the nuclear envelope gaps. This analysis revealed that the acentric is positioned within the channel formed by the gap in the nuclear envelope (Figure 1D).
To determine whether these localized gaps in nuclear envelope involve nuclear envelope proteins in addition to lamin B, we examined dividing non–I-CreI–expressing control neuroblasts (Supplemental Video S3) and neuroblasts with I-CreI–induced acentrics (Supplemental Video S4) that coexpressed a GFP-tagged nuclear pore complex protein, Nup107 (Supplemental Figure S1). Embedded in the nuclear membrane, nuclear pore complexes are a highly conversed component of the nuclear envelope (Doucet and Hetzer, 2010). Similar to our live-imaging analysis with lamin B-GFP, we observed localized delays and gaps in the reassembly of the nuclear envelope with GFP-Nup107 (Supplemental Figure S1A, bottom, green arrow; Supplemental Video S4). Similar to our experiments with lamin B-GFP, quantification of GFP-Nup107 fluorescence intensity along the nuclear envelope showed a decrease at the site of acentric entry compared with regions on the same nucleus where nuclear envelope reassembly was not delayed (compare Supplemental Figure S1B to Figure 1B). Staged-matched control and I-CreI–expressing neuroblasts in telophase demonstrate that using GFP-Nup107 to image the nuclear envelope reveals acentric generated gaps in the nuclear envelope (Supplemental Figure S1C). Taken together with the lamin imaging, these studies suggest an absence of the nuclear envelope in the acentric-induced gaps but do not formally exclude the possibility that other components of the nuclear envelope have completely reassembled.
Localized delays and gaps in nuclear envelope formation are determined by the timing of acentric chromosome segregation
To define further the relationship between acentric chromosome segregation and localized gaps in the nuclear envelope, we investigated whether the timing of acentric segregation correlates with gap formation. The initiation of acentric chromosome segregation occurs at varying times after the onset of anaphase segregation of the undamaged autosomes (Royou et al., 2010). In theory, acentrics that segregate soon after the undamaged sister chromatids separate should be more associated with the nascent daughter nuclei than acentrics that segregate much later. Therefore early-segregating acentrics should be able to rejoin the nucleus before NEF is initiated, whereas late-segregating acentrics will remain as discrete entities at this time, necessitating the localized delay in nuclear envelope reformation to allow these late-segregating acentrics to rejoin the nucleus.
To determine whether the timing of acentric segregation is correlated with the presence of localized gaps in the nuclear envelope, we induced I-CreI and imaged live the neuroblast divisions as described. We found that formation of localized gaps is highly correlated with late- segregating acentric chromosomes (Figure 2, A–D). In contrast, early-segregating acentrics were less likely to form localized gaps in the nuclear envelope and rejoined telophase nuclei well before completion of NEF (Figure 2, A–D). To further demonstrate the importance of time and distance of acentric segregation relative to the separation of undamaged chromosomes, we plotted both the time of acentric segregation and the distance of acentrics from the separated sisters (Figure 2F). As expected, this analysis showed that delayed acentric segregation was correlated (r = 0.73) with a greater distance of acentrics from the main mass of separated sisters. For divisions in which acentrics segregate later and thus at a greater distance from the main mass of separated sisters, the formation of a nuclear envelope gap allowed acentric entry into daughter nuclei (Figure 2F, blue diamonds), whereas a lack of gap formation resulted in acentrics forming micronuclei (Figure 2F, green triangles). We interpret these results as evidence that the presence of an acentric fragment separated from the main chromosome mass causes a localized delay in nuclear envelope reassembly, providing a gap for the inclusion of the acentric into the daughter telophase nucleus.
FIGURE 2:

Local inhibition of nuclear envelope assembly is determined by acentric segregation timing. (A) Time-lapse images of a larval neuroblast division expressing H2Av-RFP and lamin B-GFP with I-CreI–induced acentrics segregating soon after anaphase initiation. Acentrics (red arrows) lag on the metaphase plate for 40 s after anaphase initiation before they begin their poleward segregation. Nuclear envelope formation begins 340 s after anaphase initiation and is completed at 400 s after anaphase initiation. Nuclear envelope assembly proceeds normally with no nuclear envelope gap present. (B) Time-lapse images of a larval neuroblast division expressing H2Av-RFP and lamin B-GFP with I-CreI–induced acentrics segregating well after anaphase initiation and segregation of the main chromosome complement. Acentrics (red arrows) lag on the metaphase plate for an extended period of 260 s after anaphase initiation and then undergo poleward segregation. Nuclear envelope formation begins 400 s after anaphase initiation and is completed at 1060 s after anaphase initiation. Nuclear envelope assembly is marked by the presence of a nuclear envelope gap (green arrow) through which the acentric fragment enters the daughter nucleus. This gap persists until acentrics have rejoined the nucleus. (C) Schematic depicting the timing and distances of acentric segregation relative to the timing and distance of the main chromosome mass. Time of acentric segregation was measured from sister chromatid separation to separation of the last acentric pair. Distance of acentrics from main nuclear masses was measured from the center of the acentric to the center of the main nuclear mass at the time of last acentric segregation. (D–F) Graphs demonstrating that nuclear envelope gaps are more likely to form in neuroblasts with late-segregating acentrics. (D) Acentrics that do not form gaps (red squares) segregate <100 s after anaphase initiation (50 ± 11, N = 8). Acentrics that form gaps (blue diamonds) segregate at least 100 s after anaphase initiation (220 ± 90, N = 23). p values calculated by independent t test with equal variances not assumed. (E) Acentrics that do not form gaps (red squares) are separated from the main chromosome mass by a shorter distance (3.14 ± 0.8 μm, N = 10) than with acentrics that do form gaps (blue diamonds; 5.38 ± 1.06 μm, N = 30) after anaphase initiation. p values calculated by independent t test with equal variances assumed. (F) Correlation between the timing of acentric segregation after anaphase initiation and the distance between acentrics from the main chromosome mass after anaphase (r = 0.73). Red squares indicated acentrics that rejoin nuclei without gap formation. Blue diamonds represent acentrics that rejoin nuclei through nuclear envelope gaps. Green triangles represent acentrics that do not form gaps rejoin nuclei and form micronuclei. Acentrics that segregate at a shorter time and distance after anaphase initiation are able to reenter daughter nuclei before NEF initiation and do not form gaps (red-shaded area). In contrast, acentrics that segregate later and further from undamaged chromosomes require a nuclear envelope gap to reenter daughter nuclei (blue-shaded area). Solid line represents line of best fit for data (R2 = 0.5). Time and distances are represented as averaged values ± SD. Dotted lines outline the plasma membrane of the cell. Scale bars, 2 μm.
X-irradiation–induced acentrics also produce localized delays and gap formation in nuclear envelope formation
The foregoing results demonstrate that I-CreI–induced late-segregating acentric chromosomes no longer associated with the main mass of segregating chromosomes result in localized delays in nuclear envelope formation. To determine whether acentrics formed by others sources of DSBs also produce localized delays in nuclear envelope formation, we exposed neuroblasts to x-irradiation (Figure 3). X-irradiation produces both single- and double-stranded breaks, resulting in acentric fragments and BubR1-coated DNA tethers (Royou et al., 2005, 2010). In divisions with x-irradiation–induced acentrics, nuclear envelope formation was specifically delayed at the site of acentric entry, resulting in gaps in the nuclear envelope (Figure 3, A and B, green arrow). As seen in an additional irradiated neuroblast, these gaps were strikingly similar to those observed with I-CreI–induced acentrics (compare Figure 3B to Figure 1A, bottom). Of 25 neuroblast divisions examined from irradiated larvae, 15 exhibited chromosome segregation defects (Figure 3C, gray bar). Of these 15 divisions, 8 possessed acentric fragments (Figure 3C, red bar), and gaps in nuclear envelope formation were observed in 4 of these (Figure 3C, blue bar).
FIGURE 3:

X-ray–induced acentric chromosome fragments also enter nascent daughter nuclei through highly localized delays in assembly of the nuclear envelope. (A) Time-lapse images of acentric fragments in the neuroblast division of an x-irradiated larva expressing H2Av-RFP (red) and lamin B-GFP (green). Acentrics resulting from x-irradiation (red arrows) enter the daughter nuclei through a gap in the nuclear envelope (green arrow). Initiation to completion of NEF requires 220 s. (B) A still frame from a time-lapse movie of a nuclear envelope gap forming in a telophase nucleus with irradiation-induced acentrics (different neuroblast than shown in A) expressing H2Av-RFP (red) and lamin B-GFP (green). As seen in A, the irradiation-induced acentric (red arrow) rejoins the main nuclear mass by entering the nucleus through a gap in the nuclear envelope (green arrow). (C) Graphical representation quantifying the percentage of irradiated neuroblasts that had chromosome segregation defects (gray bar), the percentage of neuroblasts with chromosome segregation defects that were acentrics (red bar), and the percentage of neuroblasts with acentrics that formed nuclear envelope gaps (blue bar).
Nuclear envelope gaps are specific to acentric chromosomes and not intact lagging chromosomes
To determine whether nuclear envelope gaps are a result of a physical barrier due to the presence of a lagging chromosome that blocks complete reformation of the nuclear envelope or whether these gaps are specific to acentrics, we visualized lamin reassembly in neuroblasts bearing a long compound second chromosome (C(2)EN). Created by attaching both second chromosome homologues to a common centromere, these artificial chromosomes possess chromosome arms twice the normal length and exhibit extensive lagging during anaphase but are not coated with the tether component BubR1 (Novitski et al., 1981; Gonzalez et al., 1991; Sullivan et al., 1993; Kotadia et al., 2012; Martins et al., 2013). Nuclear envelope formation was examined by immunofluorescence in dividing larval neuroblasts expressing either I-CreI or bearing C(2)EN (Supplemental Figure S2). Supplemental Figure S2A depicts fixed neuroblasts in which I-CreI has been induced. As described in the previous sections, these images clearly show the lagging acentric chromosomes (Supplemental Figure S2A, red arrows) during anaphase and telophase. During telophase, a distinct gap in the nuclear envelope is observed (Supplemental Figure S2A, green arrow).
We used the same fixed protocol to examine whether lagging compound chromosome arms also produce nuclear envelope gaps. Similar to the I-CreI results, fixed and stained neuroblasts bearing the compound chromosome exhibited extensive chromosome lagging during anaphase and telophase (Supplemental Figure S2B, red arrowheads). However, no gaps in the nuclear envelope were observed. Instead, lamin formed around the nucleus and the entire length of the outstretched lagging arm (Supplemental Figure S2B, green arrowhead). Recent studies demonstrated that the compound chromosome undergoes extensive stretching at heterochromatic linker regions (Oliveira et al., 2014). Significantly, the lamin signal could be visualized around these highly stretched regions of the elongated chromosome. We interpret these results as suggesting, but not excluding the possibility, that lagging chromatin by itself is not sufficient to induce nuclear envelope gaps through a physical blockage of nuclear envelope reassembly.
Aurora B kinase is required for highly localized delays in nuclear envelope formation and acentric entry into daughter nuclei
Previous studies demonstrated that injection of chromatin into Xenopus egg extracts resulted in the association of lamins and formation of a nuclear envelope encompassing the outer periphery of the ectopic chromatin mass (Forbes et al., 1983). Given the propensity of lamin to form around chromatin, the highly localized delays in nuclear envelope formation in response to acentric fragments suggest the presence of factors preventing lamin–chromatin association specifically at the site of localized delays and acentric entry. Insight into the mechanism of highly localized delays in nuclear envelope formation comes from the observation that these delays often form in the presence of tethered acentrics. Previous studies demonstrated that Aurora B kinase is an established negative regulator of lamin assembly (Ramadan et al., 2007; Meyer et al., 2010; Afonso et al., 2014). As shown in Figure 4, we observed that Aurora B specifically associated with the tether as previously reported (Royou et al., 2010). Aurora B is a chromosome passenger protein that concentrates at the midzone microtubules once sister chromosome segregation is initiated (Gruneberg et al., 2004). Recent studies demonstrate that a gradient of Aurora B originating from the midzone globally prevents NEF (Afonso et al., 2014). Once the chromosomes have segregated away from the high Aurora B concentrations, NEF is initiated. Here we investigated whether Aurora B associated with the tether during late anaphase/telophase acts locally to prevent lamin assembly and NEF, leading to the formation of highly localized gaps in the reassembly of the nuclear envelope.
FIGURE 4:
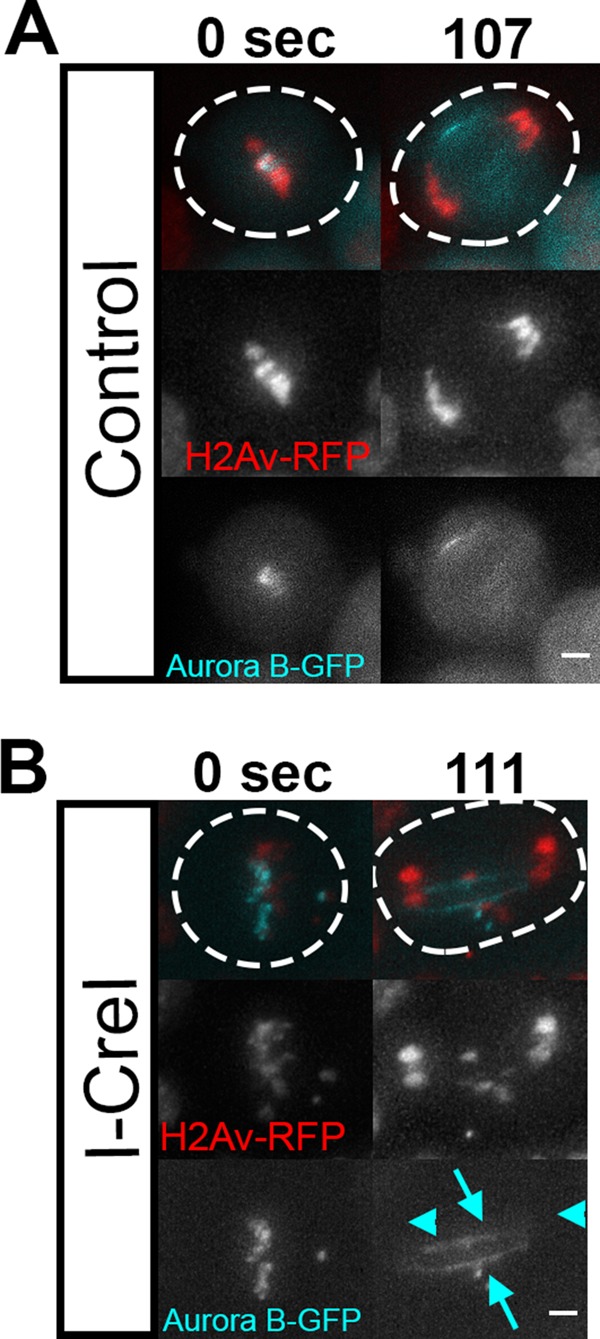
Aurora B is highly associated with acentric fragments and their DNA tethers. (A) Still frames of a control neuroblast (no I-CreI expression) movie show GFP-tagged Aurora B (cyan) on kinetochores during metaphase and at the cleavage furrow during anaphase. Dotted lines outline the plasma membrane of the cell. Scale bars, 2 μm. (B) Still frames from a movie of a dividing neuroblast during the same mitotic stages with I-CreI–induced acentrics decorated with GFP-tagged Aurora B (cyan arrows). Undamaged chromosomes (cyan arrowheads) are not associated with Aurora B (bottom). These results are in accord with Royou et al. (2010). Dotted lines outline the plasma membrane of the cell. Scale bars, 2 μm.
Because Aurora B provides a number of essential functions in the mitotic cell cycle, in order to investigate its role specifically in highly localized nuclear envelope gap formation, we used RNA interference (RNAi) to reduce but not eliminate Aurora B levels in neuroblasts. We achieved conditions in which chromosome morphology, condensation, alignment on the metaphase plate, and segregation proceeded normally. We measured chromosome compaction in I-CreI–expressing control and I-CreI with Aurora B RNAi neuroblasts. Chromosome compaction was determined by measuring the area occupied by the metaphase chromosomes at the time when the last acentric began poleward segregation. This corresponds to the point of maximum chromosome compaction in the cell cycle. We found no difference between the measured areas of the chromosomes in I-CreI–expressing control and I-CreI with Aurora B RNAi neuroblasts (Figure 5A). In addition, Aurora B RNAi–expressing neuroblasts without the I-CreI transgenes revealed no discernible defects in chromosome segregation or any evidence of chromosome bridging. In control and Aurora B RNAi neuroblasts, 0 of 10 and 0 of 13 divisions, respectively, exhibited chromosome bridging.
FIGURE 5:

Aurora B RNAi does not lead to gross changes in chromosome behavior or structure. (A) Measured size of undamaged chromosomes in I-CreI–expressing control (cyan triangles) and I-CreI; Aurora B RNAi neuroblasts (red squares) at the moment of maximal metaphase compaction. (B) Measured size of acentrics generated in I-CreI neuroblasts (cyan triangles) and I-CreI; Aurora B RNAi neuroblasts (red squares) at the beginning of NEF. (C) Distance between acentrics and the main chromosome mass in gap-forming, I-CreI–alone (control) neuroblasts (cyan triangles) and non–gap-forming I-CreI; Aurora B RNAi neuroblasts (red squares) after completion of anaphase. (D) Timing of the initiation of acentric poleward segregation after onset of anaphase in both I-CreI–alone (control) and I-CreI; Aurora B RNAi neuroblasts. The timing of the initiation of acentric poleward segregation was grouped into two categories: 0–4 min (cyan bars) and 4–7 min (red bars). (E) Still images from time-lapse movies of equal (top) and unequal (bottom) segregation of acentrics during anaphase in neuroblasts expressing I-CreI alone (top) or I-CreI plus Aurora B RNAi (bottom). Red arrows show acentrics. Scale bar, 2 μm. Right, graph of the percentage of unequal segregation in both types of neuroblasts. A statistically nonsignificant increase in unequal segregation of acentrics in I-CreI with Aurora B RNAi neuroblasts vs. I-CreI alone (control) neuroblasts was observed. (F) Single-frame pictures of third-instar neuroblasts from wild-type larvae (top) and larvae in which Aurora B is reduced (bottom). Right, graph of the percentage of binucleate neuroblasts in both types of larvae. Cyan arrows point to binucleate cells. Red channel is H2Av-RFP. Gray channel is differential interface contrast (DIC). Scale bar, 6 μm. p values were calculated by independent t test with equal variances not assumed.
To determine whether our Aurora B RNAi conditions caused specific defects in acentric morphology, we measured the area of acentrics of I-CreI–expressing control and I-CreI with Aurora B RNAi neuroblasts at the beginning of NEF (Figure 5B). We found no difference between the measured area of acentrics in I-CreI–expressing control and I-CreI with Aurora B RNAi neuroblasts. We also measured the distance from acentrics to the main nuclear mass at the time of acentric segregation in I-CreI control and I-CreI with Aurora B RNAi neuroblasts. We found no difference in the distance of acentrics from the main chromosome mass in I-CreI control and I-CreI with Aurora B RNAi neuroblasts (Figure 5C). To determine whether induction of Aurora B RNAi influences acentric segregation, we compared the timing and dynamics of acentric poleward segregation in both I-CreI with Aurora B RNAi and I-CreI–expressing control neuroblasts. We found that acentrics began their poleward segregation within similar time frames during anaphase in both I-CreI–expressing control and I-CreI with Aurora B RNAi neuroblasts (Figure 5D). Similarly, there was a small but not statistically significant increase in the frequency of unequal versus equal acentric segregation in I-CreI–expressing neuroblasts with Aurora B RNAi compared with I-CreI alone control neuroblasts (Figure 5E). To ensure that induction of Aurora B RNAi reduced Aurora B levels in the neuroblasts, we assayed the frequency of binucleate cells, a result of failed cytokinesis and a previously described phenotype in Drosophila using RNAi knockdown of Aurora B (Giet and Glover, 2001). We observed a dramatic increase in binucleate neuroblasts (Aurora B RNAi, 22%; N = 18) compared with I-CreI–expressing controls (2.7%; N = 37; Figure 5F). Taken together, these results indicate that reducing Aurora B levels through RNAi does not disrupt chromosome morphology, condensation, or segregation of the normal chromosomes or the acentric fragment.
To determine whether Aurora B is necessary for the highly localized delays in nuclear envelope formation, we compared divisions in I-CreI–expressing control and I-CreI with Aurora B RNAi neuroblasts for the presence of gaps in the nuclear envelope. Through fixed analysis, we discovered that the frequency of gap formation was greatly reduced in neuroblasts bearing an I-CreI–induced acentric with reduced levels of Aurora B (Figure 6A). As seen in Figure 6A, I-CreI–induced acentrics are free of lamins, and a distinct gap is present in the nuclear envelope (green arrow). In contrast, when Aurora B levels are reduced, there is a dramatic increase in the number of acentrics coated with lamins (green arrowheads). Nuclear envelope gaps were formed at frequencies of 79% (N = 13) and 14% (N = 25) in I-CreI–expressing control and I-CreI with Aurora B knockdown neuroblasts, respectively (Figure 6B). In addition, the percentage of telophase cells with lamins ectopically localized to the acentrics increased twofold (from 21% [N = 13] to 41% [N = 25]) in the Aurora B knockdown neuroblasts (Figure 6C).
FIGURE 6:

Local inhibition of nuclear envelope assembly is mediated by Aurora B kinase. (A) Fixed images of telophase nuclei from I-CreI–expressing control and I-CreI with Aurora B depleted–neuroblasts bearing I-CreI–induced acentrics stained with anti-lamin (green) and DAPI (red). Acentrics induced in I-CreI–expressing controls show little to no association with lamin B and form a nuclear envelope gap (green arrow). In contrast, acentrics in neuroblasts with reduced Aurora B result in both the absence of a nuclear envelope gap and clear associations with lamins on acentrics (green arrowheads). (B) I-CreI–induced acentrics in control neuroblasts resulted in nuclear envelope gaps in 79% (N = 13) of telophase nuclei. In contrast, acentrics in neuroblasts depleted of Aurora B resulted in a significant reduction (14%; p < 0.05; N = 25) in the formation of nuclear envelope gaps in telophase nuclei. (C) Acentrics in I-CreI–expressing control neuroblasts were coated with lamin B in 21% (N = 13) of telophase nuclei. In contrast, telophase nuclei of neuroblasts depleted of Aurora B resulted in an increase (41%; N = 25) of acentrics coated with lamins (not significant). (D) Time-lapse images of dividing neuroblasts from I-CreI–expressing controls (top) and I-CreI with Aurora B RNAi (bottom; Supplemental Video S5) expressing lamin B-GFP (green) and H2Av-RFP (red). I-CreI–induced acentrics are present in both. In I-CreI–expressing controls, acentrics (red arrows) enter telophase nuclei through nuclear envelope gaps (green arrow). In contrast, neuroblasts bearing acentrics with reduced Aurora B show no gaps. Consequently, acentrics (red arrows) in Aurora B–depleted neuroblasts remain outside of the nucleus and form micronuclei coated with lamin B (green arrowheads). (E) Bar graphs of micronuclei frequencies in I-CreI–expressing control, I-CreI with Aurora B RNAi, and Aurora B RNAi without I-CreI control neuroblasts. (F) Box plots showing the time elapsed from the onset of anaphase to the completion of NEF in neuroblasts expressing I-CreI and I-CreI with Aurora B RNAi. Early-segregating I-CreI–induced acentrics do not form gaps, whereas late-segregating I-CreI–induced acentrics do form gaps. Late-segregating acentrics in neuroblasts with reduced Aurora B expression fail to form gaps and complete NEF in a similar time frame to early-segregating wild-type acentrics. These results indicate that Aurora B mediates the local inhibition of NEF in neuroblasts. Asterisks indicate statistical significance. (G) Time-lapse images of dividing neuroblasts from I-CreI–expressing, dimethyl sulfoxide (DMSO)–treated controls (left) and I-CreI–expressing Binucleine-2–treated neuroblasts (right) expressing lamin B-GFP (green) and H2Av-RFP (red). In DMSO-treated controls, acentrics (red arrows) enter telophase nuclei through nuclear envelope gaps (green arrows). In contrast, Binucleine-2–treated neuroblasts show no gaps. Consequently, acentrics (red arrows) in Aurora B–depleted neuroblasts remain outside of the nucleus and form micronuclei coated with lamin B (green arrowhead).
In accord with these fixed results, live imaging of acentrics in I-CreI–expressing control neuroblasts formed nuclear envelope gaps (Figure 6D, green arrow) through which the acentrics passed to join daughter nuclei. Strikingly, acentric fragments in divisions with reduced Aurora B were associated with ectopic lamin (Figure 6D, green arrowheads; Supplemental Video S5). Presumably, because of this absence of highly localized delays in nuclear envelope formation, acentric fragments in I-CreI with Aurora B RNAi–expressing neuroblasts were not able to rejoin daughter nuclei and therefore formed micronuclei at a higher rate than with controls (see later discussion; Figure 6E).
Increased failure of nuclear envelope gap formation when Aurora B levels are reduced indicates that gap formation is dependent on Aurora B. Because our Aurora B RNAi conditions did not affect whole or acentric chromosome segregation or geometry, our results suggest that nuclear envelope gap formation is likely mediated by Aurora B through prevention of local nuclear envelope reassembly specifically at the site of acentric entry into daughter nuclei as opposed to altering chromosome segregation or compaction. In addition, the ectopic assembly of lamins on acentric fragments in reduced Aurora B conditions shows that the formation of nuclear envelope gaps is not due to an inability of the acentrics to recruit nuclear envelope components.
Failure to form highly localized delays in nuclear envelope formation results in increased rates of micronucleus formation
As described previously, when I-CreI induction results in late segregation of acentrics, there is a local delay in completion of NEF. In contrast, when Aurora B levels are reduced, these delays are eliminated (Figure 6F). In neuroblasts expressing I-CreI and Aurora B RNAi, acentrics form micronuclei at a higher rate (67%; N = 9) than acentrics in I-CreI–expressing controls (15%; N = 33). Therefore formation of micronuclei appears to be a direct consequence of the failure to form highly localized delays in nuclear envelope formation when Aurora B levels are reduced.
A small-molecule inhibitor of Aurora B prevents nuclear envelope gap formation
To demonstrate further that Aurora B is responsible for gap formation, we treated neuroblasts with the Aurora B small-molecule inhibitor Binucleine-2 (Afonso et al., 2014). Similar to our results with Aurora B RNAi, the inhibition of Aurora B by Binucleine-2 decreased nuclear envelope gap formation and increased the rate of lamin-coated, acentric-bearing micronuclei (Figure 6G). To test whether Aurora B inhibition diminished tether function, we examined the localization of the tether component BubR1 (Royou et al., 2010) to I-CreI–Induced DNA tethers. We found that Binucleine-2 inhibition of Aurora B did not disrupt tether integrity, as evidenced by the ectopic localization of BubR1 on the acentric and tether in control (Supplemental Figure S3A) and Binucleine-2–treated (Supplemental Figure S3B) neuroblasts. Taken together, these results suggest that a key function of Aurora B is to locally prevent lamin assembly, resulting in a nuclear envelope gap through which late-segregating chromatin separated from the main chromosome mass can enter the telophase nucleus (Figure 7).
FIGURE 7:

A model of Aurora B–dependent formation of nuclear envelope gaps. (A) NEF is completed within 349 s after the metaphase-to-anaphase transition in non–I-CreI–expressing control neuroblasts with undamaged chromosomes. NEF initiates 60 s after cessation of poleward movement of the chromosomes and is quickly completed to form an intact nuclear envelope. In neuroblasts with acentrics (B, C), the initiation of NEF is slightly delayed relative to neuroblasts with undamaged chromosomes (A). In the event of early acentric segregation (B), acentrics have sufficient time to rejoin daughter nuclei before the initiation of NEF; thus localized delays in NEF are not induced, and no nuclear envelope gap is observed. In contrast, late-segregating acentrics (C) have less time with respect to the initiation of NEF to rejoin daughter nuclei and are more likely to exist as distinct entities when NEF initiates. This results in localized delays in NEF at the site where Aurora B–decorated acentrics facilitate entry into daughter telophase nuclei through the formation of nuclear envelope gaps.
DISCUSSION
Much remains unknown concerning the cellular response to defects and delays in chromosome segregation during anaphase and telophase. The anaphase–telophase transition is a complex process requiring the coordination of chromosome movements with cytoskeletal and membrane dynamics. On completion of chromosome segregation during anaphase, extensive chromosome reorganization and decondensation occurs while reassembly of the nuclear envelope is initiated (Güttinger et al., 2009; Katsani et al., 2008). Spindle disassembly is completed, and the centrosome reassociates with the newly formed nuclear envelope (Robinson et al., 1999). Directly after the chromosomes have cleared the plate during anaphase, cytokinesis is initiated. Because of the complexity and speed of these events, compensating for delays and defects in chromosome segregation is particularly challenging. A number of studies demonstrate that some cells respond to delays in chromosome segregation by delaying initiation of cytokinesis (Norden et al., 2006) or alternatively elongating the spindle and daughter cells to accommodate increased arm length (Kotadia et al., 2012).
In the studies presented here, we focus on the effects of lagging acentric chromosomes on NEF. Recent work shows that delayed chromosome segregation induces a delay in NEF initiation (Afonso et al., 2014). Here we specifically investigated whether severely delayed acentric segregation results in corresponding delays in NEF completion as well. We reasoned that severely delayed acentric fragments must be able to enter daughter nuclei by a mechanism distinct from delayed NEF initiation, as acentric segregation occurs around the same time as the nuclear envelope starts to reform. By using a subset of nuclear envelope components (lamin B and Nup107) to visualize nuclear envelope reassembly, we found that severely lagging acentrics induce highly localized delays in NEF completion specifically at the site where lagging acentrics rejoin the main chromosome mass.
As previously reported, delays in the time interval between anaphase initiation (sister chromosome segregation) and initiation of NEF occur in acentric-bearing cells compared with controls (Afonso et al., 2014). Although we observed delays in NEF initiation in the presence of lagging chromosomes, we also observed significant highly localized delays in the completion of NEF that are required for acentric entry into daughter nuclei.
Previous studies revealed that acentric chromosomes segregate to daughter nuclei using a DNA tether–like structure connecting, and perhaps pulling, an acentric fragment to its centric partner (Royou et al., 2010). Segregation of these acentric fragments is severely delayed. The tethers are coated with the spindle checkpoint components BubR1 and Polo and the chromosome passenger proteins INCENP and Aurora B (Royou et al., 2010). Of these, the Aurora B kinase is particularly interesting because it is responsible for maintaining the chromosomes in a condensed state and preventing nuclear envelope assembly until after the chromosomes have completed segregation (Ramadan et al., 2007; Afonso et al., 2014).
To investigate whether Aurora B plays a role in the formation of the acentric-induced gaps in nuclear envelope formation, we reduced Aurora B in neuroblasts by RNAi. We found that the frequency of acentric-induced nuclear envelope gaps were reduced in neuroblasts depleted of Aurora B. In addition, we found that acentrics in neuroblasts depleted of Aurora B were coated with lamins and left outside of the main nuclei as micronuclei. Increased failure of nuclear envelope gap formation when Aurora B levels are reduced indicates that gap formation is dependent on Aurora B. Because our partial knockdowns of Aurora B did not affect normal or acentric chromosome segregation or compaction, our results suggest that nuclear envelope gap formation is mediated by Aurora B through prevention of nuclear envelope reassembly specifically at the site of acentric entry into daughter nuclei as opposed to altering chromosome segregation or compaction. In addition, the ectopic assembly of lamins on acentric fragments in reduced Aurora B conditions shows that the formation of nuclear envelope gaps is not due to an inability of the acentrics to recruit nuclear envelope components. Furthermore, lamin association along the outstretched arms of compound chromosomes suggests but does not exclude that gaps in the nuclear envelope are not simply due to a physical blockage of nuclear envelope formation created by the tether.
Recent studies show that the nuclear envelope reforms around separated sister chromosomes once they have segregated outside of the highly concentrated Aurora B gradient in the mitotic midzone (Afonso et al., 2014). In these studies, lagging acentrics remained free of nuclear envelope components during anaphase despite the initiation of nuclear envelope reformation around the intact chromosomes at the poles. That is, the nuclear envelope cannot reform around lagging acentrics that remain in the highly concentrated Aurora B gradient in the midzone. Although we have not specifically ruled out the role of the midzone gradient of Aurora B in nuclear envelope gap formation, we believe that the localized formation of gaps in the nuclear envelope is the specific result of acentrics and tethers coated with ectopic Aurora B. The rationale for this view is that nuclear envelope gaps do not occur in neuroblasts with undamaged chromosomes, despite the presence of an Aurora B midzone gradient. That is, nuclear envelope gaps are only observed with acentrics associated with highly localized and concentrated levels of Aurora B at the site where a gap is needed to include acentrics in daughter nuclei. Our studies complement the work showing an Aurora B gradient in regulating nuclear envelope formation on a global scale (Afonso et al., 2014) by suggesting that an increased local concentration of Aurora B can induce a corresponding local delay in nuclear envelope reformation to ensure the inclusion of acentrics in daughter nuclei.
Because Aurora B phosphorylation of histone H3 inhibits HP1 recruitment to the chromatin (Hirota et al., 2005) and HP1 is known to interact with the nuclear lamina (Ye and Worman, 1996), it would be interesting to test whether this Aurora B–mediated prevention of lamin–tether interaction is the effect of disrupted HP1 recruitment to histone H3. Alternatively, the Aurora B–mediated regulation of the state of chromosome condensation at telophase may influence local lamin–chromatin interactions and gap formation (Ramadan et al., 2007).
One of the most striking consequences of reducing Aurora B levels in the presence of I-CreI–induced double-stranded breaks is the formation of large numbers of micronuclei. This appears to be a direct consequence of the failure to form a nuclear envelope gap, blocking the acentric chromosome from entering the daughter telophase nucleus. Historically, micronuclei have been used as biomarkers for genomic instability, and their increased presence of micronuclei in human cells is typically associated with cancer (Bonassi et al., 2011; Santos et al., 2010; Celik et al., 2013). As such, there has been much interest in understanding the cellular response to micronucleus formation and subsequent micronucleus fate (Fenech et al., 2011; Huang et al., 2011; Kirsch-Volders et al., 2011; Hatch et al., 2013; Ji et al., 2013). Previous studies showed a direct correlation between Aurora B levels and micronucleus frequency (Tatsuka et al., 1998). Here we find that reducing Aurora B levels can also lead to an increased frequency of micronuclei in the presence of double-stranded breaks. Taken together, these results suggest multiple distinct mechanisms by which micronuclei can form. Furthermore, we provide an easy method for generating large amounts of micronuclei, which can then be used for further studies into micronucleus contribution toward genomic instability.
Taken together, our results suggest that Aurora B–mediated highly localized delays in the final stages of NEF result in a portal through which the tethered acentrics pass to rejoin the daughter telophase nuclei, preserving genomic integrity. Whether this portal is a novel structure of the nuclear envelope remains to be determined.
MATERIALS AND METHODS
Fly stocks
All stocks were raised on standard Drosophila media (Sullivan et al., 2000) at room temperature and then incubated at 29°C for at least 24 h before imaging. The following Gal4 drivers were used: elav-Gal4 (Lin and Goodman, 1994), Wor-Gal4 (Cabernard and Doe, 2009), and actin-Gal4 (stock #25708; Bloomington Drosophila Stock Center [BDSC], Bloomington, IN; Ito et al., 1997). Using these drivers, we constructed the following larvae: elav-Gal4; H2Av-RFP/upstream activation sequence (UAS)–lamin-GFP; I-CreI, Sb (Figures 1, A–D, 2, A and B, 5E, and 6, D and G), Wor-Gal4; I-CreI, Sb (Supplemental Figure S2A), Actin-Gal4; I-CreI, Sb (Figure 6A), elav-Gal4; H2Av-RFP/UAS-GFP-Nup107; I-CreI, Sb (Supplemental Figure S1A-C), and elav-Gal4; H2Av-RFP (Figure 5F). For Aurora B RNAi experiments, ial RNAi (#28691, BDCS) was introduced to create the following larvae: actin-Gal4; I-CreI/ial (Figure 6A), elav-Gal4; H2Av-RFP/UAS-lamin-GFP; I-CreI/ial (Figures 5E and 6D), and elav-Gal4l H2Av-RFP; ial (Figure 5F). The lamin-GFP (#7376) and GFP-Nup107 (#35514) were obtained from BDSC. The Aurora B-GFP lines, kindly provided by the Lipsick lab (Stanford University, Stanford, CA), were used to create the following larvae: elav-Gal4; H2Av-RFP/UAS-AuroraB-GFP (Figure 4A) and elav-Gal4; H2Av-RFP/UAS-AuroraB-GFP; I-CreI, Sb (Figure 4B). For the experiments with the compound chromosome (Supplemental Figure S2B), a yw;C(2)EN,bw,sp (Novitski et al., 1981) stock was obtained from DBSC (#2974). GFP-BubR1 lines previously characterized by Buffin et al. (2005) were used to create elav-Gal4; H2Av-RFP/GFP-BubR1; I-CreI, Sb larvae (Supplemental Figure S3).
I-CreI– and x-irradiation–induced chromosome breaks
I-CreI expression was induced in elav-Gal4; H2Av-RFP/lamin-GFP; I-Cre, Sb larvae with I-CreI driven by a heat shock 70 promoter. Female third-instar larvae were subjected to a 1 h of 37°C heat shock, followed by a 1-h recovery period at room temperature. The larval brains were then dissected and imaged as described next. To induce more-generalized DNA damage, female third-instar larvae were exposed to x-irradiation using a Torrex x-ray generator. This was followed by a 1- to 6-h recovery period (higher doses required a longer recovery period). The larval brains were then dissected and imaged as described next.
Live and fixed neuroblast cytology
For live imaging, female Drosophila third-instar larval brains were dissected in phosphate-buffered saline (PBS; pH 7.4) and placed between a slide and coverslip. The resulting capillary forces resulted in a gentle squashing of the preparation (Buffin et al., 2005). The preparation was imaged immediately and for a maximum period of 60 min. Best imaging was achieved using neuroblasts along the periphery of the squashed brain.
Fixed images were obtained by dissecting female third-instar larval brains in 0.7% NaCl and then soaking them for 5 min in 0.5% Na citrate to induce brain cell swelling. Brains were squashed in fixative (1.85% formaldehyde, 45% acetic acid), and then slides were frozen in liquid nitrogen. After washing in PBS, samples were immersed in a 5% dried milk/0.2% Triton X-100 solution for 1 h before being incubated overnight at 4°C with anti-lamin antibody (1:1000 dilution in the milk solution). Samples were then washed three times in PBST (PBS + 0.2% Triton X-100) and incubated at room temperature with Alexa 546–conjugated anti-rabbit secondary antibody (1:300 dilution). The samples were washed three more times in PBS before addition of 4′,6-diamidino-2-phenylindole (DAPI) in Vectashield. This fixing and staining procedure was adapted from Cenci et al. (2003).
Microscopy and image acquisition
Figure 1D was acquired with a 60×/1.4 numerical aperture (NA) objective on an inverted spinning disk microscope (ImproVision, Waltham, MA) equipped with a Hamamatsu (San Jose, CA) C9100-50 electron-multiplying charge-coupled device (EM CCD) camera. Three-dimensional reconstruction and brightness adjustments were performed with Volocity Image Analysis software (ImproVision). Figures 1C and 3B were acquired with a Leica DN5500B wide-field upright microscope equipped with a Leica DFC360 FX camera using a 63× objective with an NA of 1.4 (Leica, Buffalo Grove, IL). All other images were acquired with a Leica DMI6000B wide-field inverted microscope equipped with a Hamamatsu EM CCD camera (ORCA C9100-02) with a binning of 1 and obtained using a 100× Plan-Apochromat objective with NA 1.4. Time-lapse movies were composed of images taken at 20-s intervals unless otherwise indicated. All movie images were then deconvolved with Leica AF software using six iterations of a blind deconvolution algorithm with a 1.5 refractive index, except for Figures 1C and 3B, which were deconvolved with Autoquant X3, and Figures S3, A and B, and 6G, which were deconvolved with Autoquant X2 (Media Cybernetics, Rockville, MD). Both movies and fixed images were projected two-dimensional images (maximum intensity). All images were assembled using ImageJ software (National Institutes of Health, Bethesda, MD), Adobe Photoshop (Adobe, San Jose, CA; Figures 1, 2, 3, 5, and 6 and Supplemental Figures S1–S3), and Adobe Illustrator (Figures 2C, 4, and 7). Selected stills (both experimental and control) were processed with Adobe Photoshop to increase brightness.
Measurements
Anaphase initiation was determined in time-lapse movies as the last frame before sister chromatid separation. Initiation of poleward movement by the final acentric fragment was measured as the last frame before the final two acentric chromosomes segregated. The measured distance from the center of the acentrics to the center of the main nuclear masses at this time was used as the distance of the acentrics from the main nuclear mass. The start of nuclear envelope formation was measured as the first frame in which a nuclear envelope reappeared around segregating sister chromatids. Complete nuclear envelope formation was scored as the first frame in which quantification of GFP signal intensities along lines tangent to polar and opposing faces of daughter nuclei were of similar shape and magnitude. These measurements, as well as those appearing in Figure 1, B and C, and Supplemental Figure 1B were obtained in ImageJ by plotting the profile of signal intensity along regions of interest tangent to polar and opposing faces of daughter nuclei.
The diameter of the nuclear envelope gap was measured as the distance of the width of the gap in the lamin B-GFP signal using frames from time-lapse movies in which the acentric is passing through the gap. Pooled data calculated from fixed images required the presence of two nuclei in the same decondensation state on the same Z-plane with an acentric fragment between them that was also on the same Z-plane. Presence or absence of a nuclear envelope gap or lamin association with the acentric was then scored. The frequency of micronucleus formation was determined in time-lapse movies in which both the segregating chromosomes and nuclear envelope were clearly visible throughout the length of the movie.
Measurements for I-CreI values from Figures 2, D–F, 5, A–E, and 6, E and F, were taken from the same pool of data. Measurements for I-CreI; Aurora B RNAi values from Figures 5, A–E, and 6, E and F, were taken from the same pool of data.
All measurements were made using ImageJ software. Statistical analysis was calculated using SPSS software (International Business Machines Corporation, Armonk, NY). Bonferroni one-way analysis of variance tests were used to calculate p values for Figure 6F. Independent sample t tests were used to calculate p values for Figures 2, D and E, 5, A–C, E, and F, and 6, B, C, and E. Final graphic representations were created in SPSS (Figure 6F) and Excel (Figures 2, C–E, 5, A–F, and 6, B, C, and E, and Supplemental Figure 1B). For box plots, boxes represent data within the 25th to 75th percentile range. Black bar is data median, and whiskers represent the 95th percentile range. Circles in box plot are outliers.
Supplementary Material
Acknowledgments
We thank Paul Fisher at Stony Brook University, School of Medicine (Stony Brook, NY), for providing the anti-lamin antibody. We thank the Tamkun, O'Farrell, and Karpen labs for their insightful comments and Bill Saxton and Susan Strome for use of their microscope. In addition, we thank Ben Abrams. W.S. was supported by National Institutes of Health Grant GM046409-19 and a University of California Cancer Research Coordinating Committee grant.
Abbreviations used:
- DSB
double-stranded break
- H2Av-RFP
histone 2Av–red fluorescent protein
- NEF
nuclear envelope formation
- UAS
upstream activation sequence.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-01-0026) on April 15, 2015.
*These authors contributed equally to this work.
REFERENCES
- Abbas T, Keaton MA, Dutta A. Genomic instability in cancer. Cold Spring Harb Perspect Biol. 2013;5:a012914. doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonso O, Matos I, Pereira AJ, Aguiar P, Lampson MA, Maiato H. Feedback control of chromosome separation by a midzone Aurora B gradient. Science. 2014;345:332–336. doi: 10.1126/science.1251121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, Hetzer MW. Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J Cell Biol. 2008;182:911–924. doi: 10.1083/jcb.200805140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur T, Ramadan K, Schlundt A, Kartenbeck J, Meyer HH. NSF- and SNARE-mediated membrane fusion is required for nuclear envelope formation and completion of nuclear pore complex assembly in Xenopus laevis egg extracts. J Cell Sci. 2007;120:2895–2903. doi: 10.1242/jcs.010181. [DOI] [PubMed] [Google Scholar]
- Bonassi S, El-Zein R, Bolognesi C, Fenech M. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: evidence from human studies. Mutagenesis. 2011;26:93–100. doi: 10.1093/mutage/geq075. [DOI] [PubMed] [Google Scholar]
- Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE. Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr Biol. 2005;15:856–861. doi: 10.1016/j.cub.2005.03.052. [DOI] [PubMed] [Google Scholar]
- Cabernard C, Doe CQ. Apical/basal spindle orientation is required for neuroblast homeostasis and neuronal differentiation in Drosophila. Dev Cell. 2009;17:134–141. doi: 10.1016/j.devcel.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Celik DA, Kos¸ar PA, Ozçelik N, Erog˘lu E. Cytogenetic finding of breast cancer cases and their first-degree relatives. J Breast Cancer. 2013;13:285–290. doi: 10.4048/jbc.2013.16.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci G, Siriaco G, Raffa GD, Kellum R, Gatti M. The Drosophila HOAP protein is required for telomere capping. Nat Cell Biol. 2003;5:82–84. doi: 10.1038/ncb902. [DOI] [PubMed] [Google Scholar]
- Cipressa F, Cenci G. DNA damage response, checkpoint activation and dysfunctional telomeres: face to face between mammalian cells and Drosophila. Tsitologiia. 2013;55:211–217. [PubMed] [Google Scholar]
- Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- Doucet CM, Hetzer MW. Nuclear pore biogenesis into an intact nuclear envelope. Chromosoma. 2010;119:469–477. doi: 10.1007/s00412-010-0289-2. [DOI] [PubMed] [Google Scholar]
- Douglas ME, Davies T, Joseph N, Mishima M. Aurora B and 14-3-3 coordinately regulate clustering of centralspindlin during cytokinesis. Curr Biol. 2010;20:927–933. doi: 10.1016/j.cub.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dultz E, Zanin E, Wurzenberger C, Braun M, Rabut G, Sironi L, Ellenberg J. Systematic kinetic analysis of mitotic dis- and reassembly of the nuclear pore in living cells. J Cell Biol. 2008;180:857–865. doi: 10.1083/jcb.200707026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo B, Koyama C, Yu KR, Homola EM, Hsieh TS, Campbell SD, Sullivan W. Chk1 and Wee1 kinases coordinate DNA replication, chromosome condensation, and anaphase entry. Mol Biol Cell. 2012;23:1047–1057. doi: 10.1091/mbc.E11-10-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M, Kirsch-Volders M, Natarajan AT, Sarralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26:125–132. doi: 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- Forbes DJ, Kirschner MW, Newport JW. Spontaneous formation of nucleus-like structures around bacteriophage DNA microinjected into Xenopus eggs. Cell. 1983;34:13–23. doi: 10.1016/0092-8674(83)90132-0. [DOI] [PubMed] [Google Scholar]
- Giansanti MG, Gatti M, Bonaccorsi S. The role of centrosomes and astral microtubules during asymmetric division of Drosophila neuroblasts. Development. 2001;128:1137–1145. doi: 10.1242/dev.128.7.1137. [DOI] [PubMed] [Google Scholar]
- Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol. 2001;152:669–682. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golic MM, Golic KG. A simple and rapid method for constructing ring-X chromosomes in Drosophila melongaster. Chromosoma. 2011;120:159–164. doi: 10.1007/s00412-010-0297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Casal Jimenez J, Ripoli P, Sunkel CE. The spindle is required for the process of sister chromatid separation in Drosophila neuroblasts. Exp Cell Res. 1991;192:10–15. doi: 10.1016/0014-4827(91)90150-s. [DOI] [PubMed] [Google Scholar]
- Gruneberg U, Neef R, Honda R, Nigg EA, Barr FA. Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2. J Cell Biol. 2004;166:167–172. doi: 10.1083/jcb.200403084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol. 2009;10:178–191. doi: 10.1038/nrm2641. [DOI] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47–60. doi: 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hou H, Yi Q, Zhang Y, Chen D, Jiang E, Xia Y, Fenech M, Shi Q. The fate of micronucleated cells post X-irradiation detected by live cell imaging. DNA Repair (Amst) 2011;10:629–638. doi: 10.1016/j.dnarep.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurons and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]
- Ji W, Bian Z, Yu Y, Yuan C, Liu Y, Li C, Zhu J, Jia X, Guan R, Zhang C, et al. Expulsion of micronuclei containing amplified genes contributes to a decrease in double minute chromosomes from malignant tumor cells. Int J Cancer. 2013;134:1279–1288. doi: 10.1002/ijc.28467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Reichert H. Analysis of neural stem cell self-renewal and differentiation by transgenic RNAi in Drosophila. Arch Biochem Biophys. 2013;534:38–43. doi: 10.1016/j.abb.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Kanda T, Wahl GM. The dynamics of acentric chromosomes in cancer cells revealed by GFP-based chromosome labeling strategies. J Cell Biochem Suppl. 2000;35:107–114. doi: 10.1002/1097-4644(2000)79:35+<107::aid-jcb1133>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Katsani KR, Karess RE, Dostatni N, Doye V. In vivo dynamics of Drosophila nuclear envelope components. Mol Biol Cell. 2008;19:3652–3666. doi: 10.1091/mbc.E07-11-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol. 2004;14:2096–2106. doi: 10.1016/j.cub.2004.10.051. [DOI] [PubMed] [Google Scholar]
- Kirsch-Volders M, Plas G, Elhajouli A, Lukamowicz M, Gonzalez L, Vande Loock K, Decordier I. The in vitro MN assay in 2011: origin and fate, biological significance, protocols, high throughput methodologies, and toxicological relevance. Arch Toxicol. 2011;85:873–899. doi: 10.1007/s00204-011-0691-4. [DOI] [PubMed] [Google Scholar]
- Kotadia S, Montembault E, Sullivan W, Royou A. Cell elongation is an adaptive response for clearing long chromatid arms from the cleavage plane. J Cell Biol. 2012;199:745–753. doi: 10.1083/jcb.201208041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFountain JR, Jr, Oldenbourg R, Cole RW, Rieder CL. Microtubule flux mediates poleward motion of acentric chromosome fragments during meiosis in insect spermatocytes. Mol Biol Cell. 2001;12:4045–4065. doi: 10.1091/mbc.12.12.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motorneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Lu L, Ladinsky MS, Kirchhausen T. Formation of the postmitotic nuclear envelope from extended ER cisternae precedes nuclear pore assembly. J Cell Biol. 2011;194:425–440. doi: 10.1083/jcb.201012063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggert KA, Golic KG. Highly efficient sex chromosome interchanges produced by I-CreI expression in Drosophila. Genetics. 2005;171:1103–1114. doi: 10.1534/genetics.104.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins T, Kotadia S, Malmanche N, Sunkel CE, Sullivan W. Strategies for outcrossing and genetic manipulation of Drosophila compound autosome stocks. G3 (Bethesda) 2013;3:1–4. doi: 10.1534/g3.112.004481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Drozdowska A, Dobrynin G. A role for Cdc48/p97 and Aurora B in controlling chromatin condensation during exit from mitosis. Biochem Cell Biol. 2010;88:23–28. doi: 10.1139/o09-119. [DOI] [PubMed] [Google Scholar]
- Mikhailov A, Cole RW, Rieder CL. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr Biol. 2002;12:1797–1806. doi: 10.1016/s0960-9822(02)01226-5. [DOI] [PubMed] [Google Scholar]
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosomes breakage. Cell. 2006;125:85–98. doi: 10.1016/j.cell.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Novitski E, Grace D, Strommen C. The entire compound autosomes of Drosophila melanogaster. Genetics. 1981;98:257–273. doi: 10.1093/genetics/98.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RA, Kotadia S, Tavares A, Mirkovic M, Bowlin K, Eichinger CS, Nasmyth K, Sullivan W. Centromere-independent accumulation of cohesion at ectopic heterochromatin sites induced chromosome stretching during anaphase. PLoS Biol. 2014;12:e1001962. doi: 10.1371/journal.pbio.1001962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, Maggert KA. Expression of I-CreI endonuclease generates deletions within the rDNA of Drosophila. Genetics. 2009;181:1661–1671. doi: 10.1534/genetics.108.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan K, Bruderer R, Spiga FM, Poppo O, Baur T, Gotta M, Meyer HH. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature. 2007;450:1258–1262. doi: 10.1038/nature06388. [DOI] [PubMed] [Google Scholar]
- Rebollo E, Sampaio P, Januschke J, Llamazares S, Varmark H, González C. Functionally unequal centrosomes drive spindle orientation in asymmetrically dividing Drosophila neural stem cells. Dev Cell. 2007;12:467–474. doi: 10.1016/j.devcel.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Rhind N, Russell P. Signaling pathways that regulate cell division. Cold Spring Harb Perspect Biol. 2012;4:a005942. doi: 10.1101/cshperspect.a005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Wojcik EJ, Sanders MA, McGrail M, Hays TS. Cytoplasmic dynein is required for the nuclear attachment and migration of centrosomes during mitosis in Drosophila. J Cell Biol. 1999;146:597–608. doi: 10.1083/jcb.146.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YS, Titen SW, Xie HB, Golic MM, Bastiani M, Bandyopadhyay P, Olivera BM, Brodsky M, Rubin GM, Golic KG. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 2002;16:1568–1581. doi: 10.1101/gad.986602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royou A, Gagou ME, Karess R, Sullivan W. BubR1- and Polo-coated DNA tethers facilitate poleward segregation of acentric chromatids. Cell. 2010;140:235–245. doi: 10.1016/j.cell.2009.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royou A, Macias H, Sullivan W. The Drosophila Grp/Chk1 DNA damage checkpoint controls entry into anaphase. Curr Biol. 2005;15:334–339. doi: 10.1016/j.cub.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Santos RA, Teixeira AC, Mayorano MB, Carrara HHA, Andrade JM, Takahashi CS. Basal levels of DNA damage detected by micronuclei and comet assays in untreated breast cancer patients and healthy women. Clin Exp Med. 2010;10:87–92. doi: 10.1007/s10238-009-0079-4. [DOI] [PubMed] [Google Scholar]
- Sullivan W, Ashburner A, Hawley RS. Drosophila Protocols. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Sullivan W, Daily DR, Fogarty P, Yook KJ, Pimpinelli S. Delays in anaphase initiation occur in individual nuclei of the syncytial Drosophila embryo. Mol Biol Cell. 1993;4:885–896. doi: 10.1091/mbc.4.9.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJ, Nasmyth K. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosomal bi-orientation by altering kinetochore-spindle pole connections. Cell. 2002;108:317–329. doi: 10.1016/s0092-8674(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Tatsuka M, Katayama H, Ota T, Tanak T, Odashima S, Suzuki F, Tereda Y. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998;58:4811–4816. [PubMed] [Google Scholar]
- Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol. 2010;2:1–19. doi: 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271:14653–14656. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134:1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.