

This study demonstrates a critical link between hypoxia-inducible factor (HIF) and claudin-1 (CLDN1). HIF1β-deficient intestinal epithelial cells develop abnormal tight junction (TJ) structure and have striking barrier defects. CLDN1 is an HIF target gene, and overexpression of CLDN1 in HIF1β-deficient cells restores TJ structure and function.
Abstract
Intestinal epithelial cells (IECs) are exposed to profound fluctuations in oxygen tension and have evolved adaptive transcriptional responses to a low-oxygen environment. These adaptations are mediated primarily through the hypoxia-inducible factor (HIF) complex. Given the central role of the IEC in barrier function, we sought to determine whether HIF influenced epithelial tight junction (TJ) structure and function. Initial studies revealed that short hairpin RNA–mediated depletion of the HIF1β in T84 cells resulted in profound defects in barrier and nonuniform, undulating TJ morphology. Global HIF1α chromatin immunoprecipitation (ChIP) analysis identified claudin-1 (CLDN1) as a prominent HIF target gene. Analysis of HIF1β-deficient IEC revealed significantly reduced levels of CLDN1. Overexpression of CLDN1 in HIF1β-deficient cells resulted in resolution of morphological abnormalities and restoration of barrier function. ChIP and site-directed mutagenesis revealed prominent hypoxia response elements in the CLDN1 promoter region. Subsequent in vivo analysis revealed the importance of HIF-mediated CLDN1 expression during experimental colitis. These results identify a critical link between HIF and specific tight junction function, providing important insight into mechanisms of HIF-regulated epithelial homeostasis.
INTRODUCTION
The epithelium of the gastrointestinal tract serves as a selective barrier between the host and luminal antigens. Defects in barrier function have been linked to many human diseases, most notably inflammatory bowel diseases such as Crohn's disease and ulcerative colitis (McGuckin et al., 2009). The barrier is maintained by cell–cell interactions known as tight junctions, which serve to seal the epithelial monolayer (Ivanov, 2012). Tight junctions are composed of both cytosolic (i.e., zonula occludins) and integral membrane proteins (i.e., occludin, claudins). Claudins are a large family of tetraspanning integral membrane proteins uniquely responsible for the selective permeability of tight junctions (Van Itallie and Anderson, 2013) and can be categorized as “leaky” or “tight” with regard to their effect on barrier function (Anderson et al., 2004; Anderson and Van Itallie, 2009). Claudin-1 (CLDN1) is an important “tight” claudin and has been shown to be dysregulated in a variety of human diseases, including inflammatory bowel disease (Kucharzik et al., 2001; Weber et al., 2008).
The epithelium of the gut is a unique metabolic environment. Intestinal epithelial cells are exposed to the anoxic lumen at the apical aspect and a highly vascularized submucosa on the basolateral side, resulting in a steep oxygen gradient across the mucosa. Therefore intestinal epithelial cells at baseline reside in a state of much lower oxygen tension than elsewhere in the body, termed “physiologic hypoxia” (Karhausen et al., 2004). This hypoxic microenvironment leads to stabilization of the hypoxia-inducible factor (HIF) complex, a transcription factor comprised of an oxygen-labile α subunit and constitutively expressed β subunit that plays a central role in epithelial cell function and homeostasis. HIF signaling has been linked extensively to barrier function through transcriptional control of intestinal trefoil factor (Furuta et al., 2001), multidrug resistance gene (Comerford et al., 2002), mucin-3 (Louis et al., 2006), and CD73 (Synnestvedt et al., 2002) and has been shown to be protective in murine models of colitis (Karhausen et al., 2004; Cummins et al., 2008; Robinson et al., 2008). The profound barrier-protective effects of HIF signaling have been attributed to regulation of tissue integrity through adenosine signaling, antimicrobial peptides, and modulation of the mucus layer (Kelly et al., 2012, 2013). No direct link has been made between HIF and expression or localization of tight junction components.
In this study, we define a central role for HIF signaling in junctional integrity and homeostasis. Knockdown of HIF1β in vitro resulted in significant defects in barrier function, as well as in gross morphological abnormalities in the architecture of the tight junction. Screening of tight junction proteins by chromatin immunoprecipitation (ChIP)-on-chip highlighted CLDN1 as a potential HIF target. We show significant repression of CLDN1 in HIF1β-deficient intestinal epithelial cells (IECs) by quantitative reverse transcriptase PCR (RT-PCR) and Western blot analysis. Reintroduction of CLDN1 into HIF1β-deficient Caco2 cells resulted in resolution of morphological abnormalities and reversal of defects in paracellular barrier function. Finally, analysis of the CLDN1 promoter revealed multiple hypoxia response element (HRE) consensus sequences, and ChIP and promoter studies confirm CLDN1 as a transcriptional target of HIF. These findings link HIF to barrier function at the level of the tight junction itself and shed further light on the mechanism of the profound barrier-protective effects of HIF signaling.
RESULTS
Depletion of HIF1β in human IECs results in barrier defects
HIF has been integrally linked to barrier function in intestinal epithelial cells through a number of genes important in innate immune defense in vitro and in vivo (Colgan and Taylor, 2010; Glover and Colgan, 2011). However, no direct associations have been made between HIF and epithelial tight junction (TJ) proteins. To determine the relative contribution of HIF to TJ integrity and function, we used short hairpin RNA (shRNA)–mediated knockdown to deplete the common HIF1β subunit in T84 and Caco2 human intestinal epithelial cells. HIF1β knockdown (KD) resulted in significant reduction in HIF1β mRNA (65 ± 9%, p < 0.01) as compared with short-hairpin nontargeting controls (shNTC) as measured by RT-PCR. With this strategy, HIF1β protein was undetectable by Western blot (Figures 1, A and B).
FIGURE 1:

Influence of HIF1β KD on intestinal epithelial cell tight junction structure and function. (A) Lentiviral shRNA–mediated KD of HIF1β in T84 intestinal epithelial cells results in reduced HIF1β mRNA relative to shNTC. (B) HIF1β KD results in loss of HIF1β protein in both normoxia and hypoxia. (C) TER measurements reveal significant transcellular barrier defects in HIF1β KD T84 cells. Data represented as fold of shNTC TER. (D) FITC-dextran flux assay of HIF1β KD T84 cells reveal significant defects in paracellular permeability even with high–molecular weight FITC-dextran. (E) Immunofluorescence for ZO1 in HIF1β KD T84 cells reveals profound morphological abnormalities at the level of the tight junction. (F) Top, representative confocal images showing the distribution of TJ-associated ZO1 in shNTC and HIF1β KD T84 cells, as represented in x-z views (apical to basolateral). Bottom, representative confocal images showing detailed colocalization of ZO1 and F-actin at apical junctions and diminished F-actin labeling of HIF1β KD lateral cell membranes. Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by Student's t test. Scale bars, 10 μm. In all cases, n = 3 independent experiments.
TJ integrity was measured by transepithelial resistance (TER) using polarized T84 monolayers grown on membrane-permeable supports. HIF1β KD resulted in a significant reduction (80 ± 4% decreases, p < 0.01) in TER as compared with shNTC (Figure 1C). Furthermore, a comparison of flux rates of various-sized fluorescein isothiocyanate (FITC)–dextrans revealed major deficits in paracellular permeability in cells lacking HIF1β. Significant differences in paracellular permeability were seen over a range of molecular sizes, including 3 kDa (Stokes radius 17 Å, p < 0.05), 10 kDa (Stokes radius 23 Å, p < 0.001), and 40 kDa (Stokes radius 44 Å, p < 0.01; Figure 1D). These data strongly suggest that HIF is a critical determinant in formation and subsequent maintenance of epithelial barrier function.
Loss of HIF signaling results in morphological abnormalities of TJs
In light of the severe functional barrier defects observed in HIF1β KD cells and our recent observation that HIF KD results in defective TJ morphology (Glover et al., 2013), we examined the integrity of TJs in HIF1β KD. Here we visualized the TJ marker zonula occludens 1 (ZO1) by immunofluorescence in confluent shNTC and HIF1β KD T84 monolayers. This analysis revealed the typical chicken-wire patterns of ZO1 staining in shNTC cells, with linear ZO1 contacts between cells and an even distribution of ZO1 throughout cell–cell junctions (Figure 1E). By stark contrast, confluent HIF1β KD monolayers showed dramatic morphological abnormalities characterized by pronounced undulations of ZO1 visualized in the apical lateral membrane. Moreover, ZO1 was unevenly dispersed throughout the junction, giving the junctions a more beaded appearance (Figure 1E). These abnormalities in TJ structure were not unique to T84 cells, as HIF1β KD in Caco2 cells resulted in similar changes to TJ structure. Given the aberrant morphology of HIF1β KD intercellular junctions, we next considered whether HIF1β depletion similarly conferred gross changes in polarized cell height. Phalloidin staining was used to label cytoskeletal F-actin in apical perijunctional regions and lateral membrane cortices. As outlined in Figure 1F, loss of HIF1β resulted in decreased intensity of subapical phalloidin staining, correlating with lateral membrane loss and reduced cell size. Taken together, these results strongly implicated HIF signaling as a critical determinant of TJ architecture, integrity, and membrane assembly.
ChIP-on-chip analysis identifies CLDN1 as an HIF target
To identify the existence of candidate HIF targets within the TJ, a ChIP-on-chip promoter analysis was performed and screened for binding to HIF1α as previously described (Glover et al., 2013; available through the Gene Expression Omnibus database, accession no. GSE43108). Briefly, ChIP was performed on Caco2 IECs with HIF1α-specific polyclonal antibody. ChIP-enriched samples were then hybridized to a custom microarray. Significantly enriched targets included many known HIF targets, including BNIP3L (Sowter et al., 2001; Figure 2A). Of the TJ promoters tiled on the array, only CLDN1 showed significant enrichment for binding to HIF1α (p < 0.001; Figure 2A).
FIGURE 2:

Claudin-1 expression in HIF1β KD T84 cells. (A) HIF ChIP-on-chip highlights CLDN1 as a potential HIF target. Hatched bar represents a positive control (BNIP3L). Bar graph represents log2-fold enrichment of ChIP DNA relative to input chromatin. (B) RT-PCR analysis of HIF1β KD T84s reveals significant repression of CLDN1 mRNA relative to shNTC. (C) Western blotting indicates loss of CLDN1 expression, as quantified by densitometry. (D) T84 shNTC and HIF1β KD cells were fixed and immunostained for CLDN1 (green) and F-actin (phalloidin; red). Representative confocal images were taken of perijunctional actomyosin ring at apical junctions of cells, showing significantly diminished junctional CLDN1 staining. The localization of CLDN1 and F-actin along the z-axis of cells is represented in x-z views (apical to basolateral). Arrowheads indicate enrichment of CLDN1 and F-actin at apical cell–cell contacts. CLDN1 staining and F-actin immunodecoration of apical and cytoskeletal cortices revealed loss of lateral membrane and cellular height.*p < 0.05 and ***p < 0.001 as determined by Student's t test. In all cases, n = 3 independent experiments.
As an extension of these ChIP-on-chip findings, we validated the specific regulation of CLDN1 by HIF in HIF1β KD cells. RT-PCR revealed significantly attenuated expression of CLDN1 mRNA in HIF1β KD T84 cells (59 ± 10% reduction in CLDN1 mRNA, p < 0.001; Figure 2B). No significant differences were observed by RT-PCR in other well-characterized claudins or in barrier-regulating TJ (ZO1, occludin, JAM-A) or adherens junction (AJ; E-cadherin) components (Table 1). Western blotting of cell lysates showed a nearly complete loss of CLDN1 in HIF1β KD cells (80 ± 7% decrease relative to shNTC as measured by densitometry; p < 0.05; Figure 2C).
TABLE 1:
Expression of TJ and AJ components in HIF1β KD T84 cells.
| Name | Fold change | SEM | Significance |
|---|---|---|---|
| CLDN1 | 0.41 | 0.09 | p < 0.001 |
| CLDN2 | 1.53 | 0.75 | ns |
| CLDN3 | 1.12 | 0.22 | ns |
| CLDN4 | 1.56 | 0.39 | ns |
| CLDN5 | 1.5 | 0.92 | ns |
| CLDN6 | 0.83 | 0.08 | ns |
| CLDN7 | 0.77 | 0.09 | ns |
| CLDN10 | 1.55 | 0.62 | ns |
| CLDN15 | 1.01 | 0.31 | ns |
| CLDN18 | 1.32 | 0.71 | ns |
| ZO1 | 0.971 | 0.173 | ns |
| Occludin | 1.44 | 0.29 | ns |
| JAM-A | 1.45 | 0.36 | ns |
| E-cadherin | 1.07 | 0.28 | ns |
Of note, this observation of HIF-dependent CLDN1 expression was not unique to T84 cells. Indeed, HIF1β depletion in Caco2 epithelial cells confirmed this phenotype. It is also remarkable that no differences in CLDN1 expression were noted between normoxic and hypoxic cells by either RT-PCR or Western blot, suggesting a role for basal HIF signaling in the maintenance and expression of CLDN1.
Immunolocalization studies have indicated an association of CLDN1 both with apical TJs and along the lateral plasma membrane in polarized epithelia (Nunbhakdi-Craig et al., 2002; Marchiando et al., 2010). To interrogate whether junctional or lateral CLDN1 populations are selectively depleted in HIF1β-knockdown IECs, we compared CLDN1 localization in shNTC control and HIF1β-knockdown IEC monolayers. CLDN1 staining was prominently decreased both at cell–cell contacts and within lateral membranes of HIF1β-knockdown IECs as compared with controls, with heterogeneous redistribution of CLDN1 to apical clusters in a subset of cells. Of interest, loss of lateral CLDN1 in HIF1β-knockdown IECs correlated with a reduction in lateral membrane length and cell height, as evaluated by F-actin immunostaining (Figure 2D). Further, the lateral distribution of F-actin fluorescence across cell–cell contacts was diminished in HIF1β-knockdown IECs compared with shNTC controls, despite the fact that total cellular levels of actin were not altered.
Dynamics of CLDN1 expression during IEC barrier establishment
We next determined whether HIF contributed to barrier assembly. To explore this, shNTC and HIF1β KD T84 cells were incubated in low-Ca2+ medium overnight to disassemble the junctional complex, then returned to high-Ca2+ medium (i.e., Ca2+ switch assay). TERs were monitored to determine the rate of barrier recovery. HIF1β KD T84 cells had significantly decreased resistances after 24 h relative to shNTC (Figure 3A). No major differences in the rate of TER recovery after Ca2+ switch were observed. Protein was harvested before Ca2+ switch (t = −24 h), upon return to high-Ca2+ medium (t = 0 h), and after recovery (t = 24 h). Western blot analysis revealed a slight increase in CLDN1 in both shNTC and HIF1β KDs at t = 0bh, followed by a decrease to baseline conditions in recovered monolayers (Figure 3B). RT-PCR demonstrated no significant induction of CLDN1 at t = 0 h. These data suggest that whereas HIF1β KD IECs harbor baseline defects in TJ structure and function, the apical junction reassembly response as measured by Ca2+ switch is not significantly affected by the loss of HIF1-dependent CLDN1 expression.
FIGURE 3:
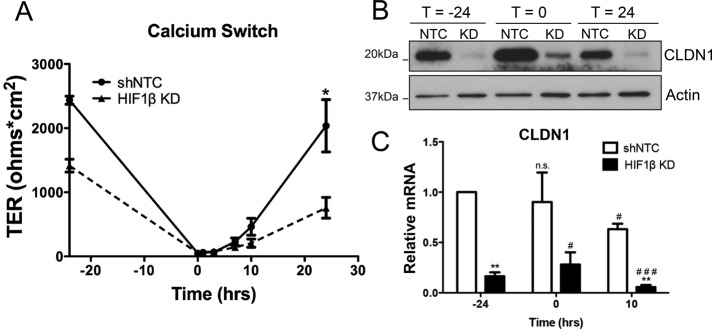
Expression and function of CLDN1 in HIF1β KD epithelia during barrier assembly. (A) shNTC and HIF1β KD T84 cells were subjected to Ca2+ switch, and TER was monitored over time. (B) Western blotting at various time points revealed a slight increase in CLDN1 expression at t = 0 and then repression at t = 24 in both shNTC and HIF1β KD. (C) RT-PCR analysis indicates no increase in transcript at t = 0, with a significant decrease in transcript at t = 10. *Relative to shNTC, #relative to shNTC, t = 0. *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by Student's t test. In all cases, n = 3 independent experiments.
Selective depletion of CLDN1 recapitulates aberrant TJ morphology of HIF1β KD epithelia
We next sought to examine whether selective knockdown of CLDN1 could recapitulate the phenotype of HIF1β-depleted T84 IECs. Stable knockdown of CLDN1 using lentiviral shRNA particles resulted in significant repression of CLDN1 mRNA and protein levels, respectively, relative to shNTC control cells (Figure 4, A and B). Of importance, CLDN1 depletion relative to shNTC controls was found to be similar between CLDN1 KD (74 ± 10%, p < 0.01) and HIF1β KD T84 IECs (65 ± 9%, p < 0.01). In contrast to HIF1β-depleted IECs, no significant changes were detected in TER values (Figure 4C) or in permeability to 3-kDa FITC-dextran in polarized CLDN1 KD IECs compared with shNTC monolayers (Supplemental Figure S1). This finding is consistent with previous work that found little effect of transient siRNA-mediated CLDN1 knockdown on barrier function in model IEC monolayers (Mrsny et al., 2008).
FIGURE 4:

Claudin-1 depletion influences intestinal epithelial cell size and architecture. (A) Stable shRNA-mediated knockdown of CLDN1 in T84 intestinal epithelial cells significantly depletes endogenous CLDN1 mRNA levels relative to shNTC. (B) Western blotting confirms loss of CLDN1 protein expression. (C) TER measurements in shNTC, CLDN1 KD, and HIF1β KD T84 cells. Data are represented as fold change relative to shNTC TER. (D) Confluent shNTC and knockdown T84 monolayers were fixed and immunostained for ZO1. Representative x-y confocal images of apical ZO1 distribution reveal TJ morphological irregularities in CLDN1 KD and HIF1β KD T84 cells. (E) Distribution of epithelial size was quantified by measuring relative cell area in shNTC, CLDN1 KD, and HIF1β KD T84 cell confocal images as represented in D. Results depict reduced uniformity of KD junctional profiles and increased cell size (fourfold) relative to shNTC control IECs. Data are represented as means (400 cells). *p < 0.05 and **p < 0.01 as determined by Student's t test. In all cases, n = 3 independent experiments.
To analyze TJ morphology in CLDN1 KD T84 IECs, we performed ZO1 immunofluorescence staining and confocal microscopy. As outlined in Figure 4, D and E, the morphological characteristics of CLDN1-depleted IECs closely resembled those of HIF1β KD cells, namely undulating apical lateral membranes and increased cell area relative to shNTC control monolayers.
Analysis of myosin motor phosphorylation (pMLC) revealed no changes in KD T84 lysates relative to shNTC cells, suggesting that morphological abnormalities in CLDN1 and HIF1β KD IECS are not manifested through altered signaling at the level of the cortical actomyosin belt.
Heterologous expression of CLDN1 in HIF1β KD rescues morphological abnormalities
To investigate the influence of CLDN1 in the context of nascent junction integrity, we transfected a human CLDN1 expression plasmid at two concentrations (5 and 10 μg) into subconfluent HIF1β-deficient Caco2 cells. At 24 h posttransfection, the cells were lifted and concentrated onto membrane-permeable supports and localized for TJ structure. Immunofluorescence analyses 48 h posttransfection revealed a CLDN1 dose–dependent recovery of normal TJ morphology, as measured by ZO1 localization. As shown in Figure 5A, increasing concentrations of CLDN1 plasmid resulted in nearly complete normalization of TJ morphology. The abnormal undulations and punctate ZO1 staining were lost in cells transfected with CLDN1 plasmid, and distribution of ZO1 was more uniform than in mock-transfected cells. Quantification of junctional length relative to a straight line between tricellular junctions (TJ length ratio, analysis depicted in Figure 5B) revealed a dose-dependent normalization of the apical lateral membrane in the presence of CLDN1 (Figure 5C, p < 0.001). Western blotting of transfected cells confirmed that CLDN1 levels were increased in a dose-dependent manner (Figure 5D). To determine whether heterologous expression of CLDN1 could rescue the barrier defect of the HIF1β-deficient cells, cells were transfected with 5 or 10 μg of CLDN1 expression plasmid, seeded on permeable inserts, and grown to confluence. FITC-dextran flux revealed a significant decrease in paracellular permeability in transfected monolayers at both plasmid concentrations (p < 0.001; Figure 5E). Western blot analysis confirmed increased levels of CLDN1 in transfected monolayers relative to controls (Figure 5F). These results identify a critical function for CLDN1 in the formation of the nascent TJ in HIF1β-deficient cells. Moreover, these data implicate a central role for HIF-dependent CLDN1 expression in normal TJ structure of IEC.
FIGURE 5:

Tight junction morphology of HIF1β KD after heterologous expression of CLDN1. (A) Immunofluorescence for ZO1 in HIF1β KD Caco2 cells transfected with empty vector reveals characteristic junctional abnormalities. Transfection with 5 or 10 μg of CLDN1 expression plasmid resulted in normalization of ZO1 distribution and loss of membrane undulations as visualized by immunofluorescence. (B) Quantification of undulations was performed by dividing the actual junction length by the distance between tricellular junctions. Examples of abnormal (left) and normal (right) TJ localization by ZO-1 immunofluorescence. (C) TJ length ratio analysis revealed a dose-dependent improvement in junctional length ratio. (D) Western blotting of transfected lysates confirms a dose-dependent increase in CLDN1 expression 48 h posttransfection. (E) FITC-dextran flux assay demonstrated a significant, dose-dependent decrease in paracellular permeability in CLDN1-transfected HIF1β KD Caco2 monolayers. (F) Western blot analysis confirms heterologous CLDN1 expression at day 7 posttransfection. Results are presented as mean ± SEM ratio. *p < 0.05 and ***p < 0.001 as determined by Student's t test. Scale bars, 10 μm.
HIF1 binds and regulates the CLDN1 promoter
To gain insight into the mechanism of HIF-dependent CLDN1 expression, we examined whether HIF regulates the CLDN1 promoter. Analysis of the full-length promoter region of human CLDN1 (Kramer et al., 2000) revealed two HRE consensus sequences located at positions −226 (HRE 1) and −590 (HRE 2) relative to the transcription start site (Figure 6A). HIF1α and HIF2α ChIP was performed in Caco2 cells, and PCR analysis spanning the putative HRE sites demonstrated significant enrichment for both HRE sequences in the CLDN1 promoter relative to an immunoglobulin G (IgG) control (Figure 6, B–E). Of note, CLDN1 promoter enrichment on HIF1α increased in response to 6 h of hypoxia (1% O2), whereas enrichment diminished on HIF2α under hypoxic conditions. Moreover, these results indicate that both HIF1α and HIF2α are actively bound to the promoter of CLDN1 under normoxic and hypoxic conditions.
FIGURE 6:

Binding of HIF1α to the CLDN1 promoter. (A) Promoter analysis of CLDN1 highlights two potential HRE consensus sequences proximal to the transcription start site. ChIP for HIF1α followed by PCR using primers spanning (B) HRE 2 and (C) HRE 1 reveals significant enrichment of the CLDN1 promoter in both normoxia and hypoxia. Error bars represent SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 as determined by Student's t test. Three independent experiments.
To define further the role of HIF and the HRE consensus sequences on CLDN1 expression, we used a CLDN1 promoter—a luciferase reporter plasmid containing 728 base pairs of the CLDN1 promoter proximal to the transcription start site. To define the role of HIF in promoter regulation, we cotransfected CLDN1 reporter plasmids into Caco2 cells with constitutively active HIF1α or HIF2α overexpression plasmids as described previously (Glover et al., 2013). As shown in Figure 7A, overexpression of either HIF1α or HIF2α resulted in a significant induction of CLDN1 promoter activity relative to an empty vector control, (7.6 ± 1.9)-fold (p < 0.05).
FIGURE 7:
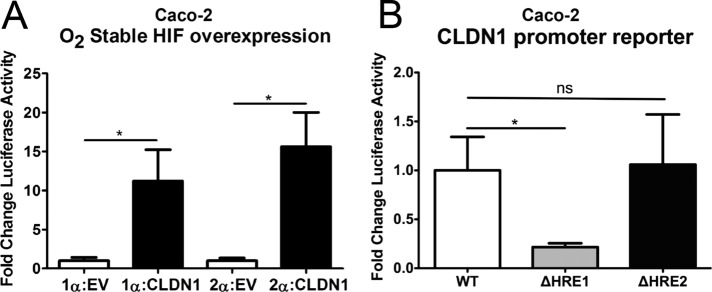
Influence of HIF1α on function the CLDN1 promoter. (A) Cotransfection of CLDN1 luciferase reporter plasmid with HIF1α overexpression plasmid containing a mutated oxygen-dependent degradation domain resulted in significant increase in CLDN1 promoter activity relative to empty vector (EV). (B) Mutagenesis of HRE 2 did not significantly affect basal promoter activity. However, mutagenesis of HRE 1 resulted in a significant decrease in this activity, pointing to its importance in basal regulation of the CLDN1 promoter. Results are presented as mean ± SEM ratio. *p < 0.05 as determined by Student's t test. In all cases, n = 3 independent experiments.
We further extended these studies to define a role for HIF in CLDN1 promoter activity. To do this, we mutated the two putative HRE consensus sequences (from 5′-GCGTG-3′ to 5′-GTTCG-3′) and analyzed them by luciferase assay. As shown in Figure 7B, mutation of the distal HRE 2 site (position −590) did not influence activity relative to wild-type plasmid. By contrast, mutation of the proximal HRE 1 site (position −226) resulted in a significant decrease in luciferase activity (65 ± 11% decrease relative to wild type, p < 0.05), strongly indicating an important role for the HRE 1 site in HIF-dependent regulation of CLDN1 promoter activity.
CLDN1 expression in HIF1β intestinal epithelial–specific knockout mice
Given the profound TJ phenotype observed in HIF1β KD cells in vitro, we sought to determine the relevance of HIF signaling in CLDN1 expression and barrier function in vivo. Here we profiled CLDN1 expression in the proximal and distal intestine using mucosal scrapings from conditional IEC Hif1β-knockout mice. As shown in Figure 8A, this analysis revealed selective repression of CLDN1 mRNA in the duodenum of conditional IEC Hif1β KOs relative to control (53 ± 8%, p < 0.05). No baseline differences in CLDN1 mRNA were observed in colonic mucosa (Figure 8A).
FIGURE 8:

In vivo analysis of HIF regulation of CLDN1. (A) RT-PCR from mucosal scrapings obtained from conditional HIF1β intestinal epithelial KO mice (M. musculus) reveals significant repression of CLDN1 in the duodenum but no difference in the colons of healthy mice. (B) RT-PCR for CLDN1 after induction of TNBS colitis reveals no difference between WT and HIF1β KO mice in vehicle-treated control colon (3–5 mice/genotype). Treatment with TNBS, however, resulted in a significant difference between WT and KO mice (10–15 mice/genotype). (C) Quantitative PCR for the TJ molecule occludin and adherens junction–associated E-cadherin in wild-type and HIF1β KO mice colonic samples. No change in mucosal occludin or E-cadherin was detected in vehicle-treated cohorts. TNBS challenge resulted in repression of occludin in wild-type but not HIF1β KO mice. Results are presented as mean ± SEM; *p < 0.05.
Barrier function in these conditional IEC Hif1β KO animals was previously assessed using FITC-dextran permeability. No differences were observed between control and HIF1β KO animals at baseline (Glover et al., 2013). Induction of experimental colitis using either dextran sodium sulfate or trinitrobenzene sulfonic acid (TNBS), however, resulted in significantly enhanced barrier defects in HIF1β KO animals relative to controls (Glover et al., 2013). Thus we compared CLDN1 expression in these mouse lines after induction of TNBS colitis (Figure 8B). RT-PCR analysis of mucosal scrapings for CLDN1 in tissues obtained from these mice tracked with the observed phenotype. Indeed, vehicle-treated mice showed no difference in CLDN1. Induction of colitis, however, resulted in a significant repression of CLDN1 in HIF1β KO mice relative to controls (p < 0.05; Figure 8B). Previous studies using HIF-overexpressing mice showed that a number of HIF targets are not induced at baseline but are induced during an inflammatory insult (Karhausen et al., 2004). CLDN1 does not appear to be induced in a similar manner but is instead maintained at homeostatic levels during colitis. Similarly, vehicle-treated conditional HIF1β KO mice exhibited no significant change in either mucosal occludin or E-cadherin expression compared with wild-type vehicle controls, strongly arguing against global compensatory changes in these junction components in this model (Figure 8C). Taken together, these results suggest that loss of IEC HIF signaling directly disrupts barrier maintenance and enhances the permeability of the intestinal epithelium during experimental colitis.
DISCUSSION
The intestinal mucosa is composed of a single layer of columnar epithelium that serves as a selective barrier to ever-present luminal antigens. Critical to this barrier function are cell–cell contacts known as TJs, which provide a paracellular seal in epithelial tissues (Hartsock and Nelson, 2008). A key component of these junctional complexes is CLDN1, an integral membrane protein that mediates interactions between neighboring cells and cytosolic tight junction proteins (McCarthy et al., 2000; Cording et al., 2013). A so-called “tight claudin,” CLDN1 plays an integral role in determining barrier function in IECs (Inai et al., 1999) and is a marker of differentiation in the squamous epithelium of the esophagus. Despite its importance, little is known about the transcriptional regulation of CLDN1.
Here we identify HIF as a central transcriptional regulator of CLDN1. Using shRNA-mediated knockdown of HIF1β in T84 intestinal epithelial cells, we observed significant defects in barrier function. Moreover, depletion of HIF1β resulted in profound morphological abnormalities at the level of the tight junction, characterized by uneven distribution of ZO1 and irregular undulations at the level of the TJ. This morphology is reminiscent of our recent work revealing that knockdown of HIF-2α and inhibition of creatine kinase (CK) resulted in junctional defects similar to those observed here with HIF1β (Glover et al., 2013). Such changes in CK expression were mapped to defects in functional energetic coupling between the adherens junction and the actomyosin ring, where CK likely constitutes one terminal of a functional phosphocreatine/creatine kinase energy circuit that supplies energy to myosin motors. Similar structural abnormalities to TJ strand morphology, including the beaded appearance and lateral undulations, were originally observed in RhoA and Rac1 mutants (Jou et al., 1998) and defined important roles for GTPase signaling in spatial organization of TJ proteins. Of interest, RhoA has been implicated at multiple levels in the stabilization of HIF1 (Turcotte et al., 2003, 2004; Pacary et al., 2007; Vogel et al., 2010), suggesting that these original observations of abnormal TJ formation in RhoA mutants could be mediated, at least in part, by aberrant HIF stabilization.
Guided by a HIF ChIP-on-chip promoter array, we identified CLDN1 as a potential HIF target gene. A specific enrichment of CLDN1 within the claudin family of genes provided insight into the junctional defects observed in HIF1β KD IEC. Previous studies with the CLDN1 promoter revealed the relative importance for the promoter SP1 site in butyrate-induced expression of CLDN1 (Wang et al., 2012). Similarly, Lopardo et al. (2008). showed that p63 binds to the CLDN1 promoter and regulates keratinocyte development. Our analysis of the CLDN1 promoter revealed a functional HRE site (position −226) that was relatively more important than another HRE consensus located upstream (position −590). It is noteworthy that CLDN1 was not hypoxia inducible per se; instead, these results suggest that basal levels of stabilized HIF tonally regulate homeostatic levels of CLDN1. This is not unprecedented. For example, we recently demonstrated that IEC human β-defensin-1 expression is regulated through similar mechanisms involving constitutive HIF stabilization under conditions of “physiologic hypoxia” (Kelly et al., 2013). Such observations stem from the steep oxygen gradient that exists from the anaerobic lumen of the intestine across the epithelium to the highly vascularized subepithelium (Colgan and Taylor, 2010). From this perspective, it is perhaps not surprising that the epithelium has evolved a number of features to cope with this metabolic setting. Studies comparing functional responses between epithelial cells from different tissues have revealed that IEC appear to be uniquely resistant to hypoxia (Furuta et al., 2001) and that low levels of oxygenation within the normal intestinal epithelial barrier (i.e., “physiologic hypoxia”) may be a regulatory adaptation that could control some target genes (e.g., CLDN1) through basal HIF stabilization. The extent to which this observation might differ between the colonic mucosa and other more oxygenated tissue beds remains unclear.
The most striking observation from this work was the normalization of TJ morphology and function through heterologous expression of CLDN1 in HIF1β-deficient cells. This finding strongly implies a central role for HIF-regulated CLDN1 expression in normal TJ dynamics. Of interest, selective knockdown of CLDN1 in IECs alters TJ shape and linearity, whereas TER and paracellular flux remain unaffected. As such, the expression of other claudins by T84 IECs likely contributes to TJ barrier function, analogous to observations in other model intestinal epithelia (Mrsny et al., 2008). Given the established role of HIF as a global regulator of IEC barrier function and homeostasis, it is perhaps unsurprising that HIF1β knockdown results in more profound attenuation of barrier properties than individual CLDN1 depletion. It is likely that additional transcriptional targets remain to be identified and that coordinated regulation of such targets confers pleotropic effects on barrier function. Indeed, based on our finding that CLDN1 knockdown recapitulates the morphological aberrations but not the barrier deficits of HIF-depleted cells, it is plausible that compensatory pathways engaged in the setting of CLDN1 deficiency are themselves subject to HIF regulation. Moreover, heterotypic interactions between claudin family members have been identified in epithelial apical junctions (Furuse et al., 1999; Mrsny et al., 2008), suggesting that claudin species other than CLDN1 can readily contribute to TJ function and paracellular flux in intestinal epithelia. Nonetheless, our findings of altered TJ morphology in CLDN1 KD IECs, coupled with restoration of TJ linearity upon heterologous CLDN1 expression in HIF1 β KD IECs, clearly argues for a critical role of HIF signaling in constitutive CLDN1 regulation.
Little is known about the role of CLDN1 in establishing and/or maintaining intestinal epithelial shape and size. Seminal work by Furuse and Tsukita demonstrated that ectopic expression of CLDN1 and CLDN2 in nonepithelial cells could recapitulate TJ-like freeze fracture strands, indicating that these claudins are largely responsible for TJ strand formation (Furuse et al., 1998). Analysis of discrete CLDN1 extracellular loop domains using peptide mimetics revealed homotypic CLDN1 interactions, as well as a close association between CLDN1 and occludin, in the stabilization of TJ structure and function (Mrsny et al., 2008). Changes in CLDN1 expression was found to induce structural and functional changes in colon cancer cell lines, promoting an epithelial-mesenchymal transition shift in cellular phenotype via altered β-catenin/Tcf signaling (Dhawan et al., 2005). Of interest, TJ structural strand abnormalities with selective CLDN1 depletion was described in polarized parotid gland epithelia chronically exposed to the inflammatory cytokine interferon γ (IFNγ; Baker et al., 2008). Previous work from our group reported a role for IFNγ in mediating repression of HIF1β levels and activity, highlighting a potential functional correlation between abrogated HIF signaling and IFNγ-regulated CLDN1 loss (Glover et al., 2011). In our present model of IEC HIF1β depletion, we propose that CLDN1 repression likely abrogates structural architecture of the TJ by altering homeostatic head-to-head interactions between junction-associated claudins and occludin. Given the loss of lateral membrane in HIF1 KD monolayers, CLDN1 loss may further destabilize lateral interactions between claudins within a given epithelial cell. Whether IEC β-catenin signaling might also be influenced by loss of CLDN1 expression is the subject of investigation.
Somewhat surprisingly, we did not observe a difference in epithelial reassembly in HIF1β-deficient cells after Ca2+ switch. Whereas the lower baseline TER observed in these cells was maintained after assembly, these cells were able to recover from Ca2+ switch. It is also noteworthy that CLDN1 expression was induced in shNTC and, to a lesser extent, HIF1β-deficient cells during barrier assembly (Figure 3B). We do not know the mechanism(s) of such induction. It is possible, for example, that transcription factor(s) other than HIF are relatively more important during epithelial barrier establishment. Alternatively, it is possible that the increase in CLDN1 protein observed during barrier reassembly is not dependent on transcription. This last point is supported, at least in part, by the relatively minor induction of CLDN1 mRNA during barrier assembly (Figure 3C). Taken together, our results strongly implicate HIF-regulated CLDN1 as a central orchestrator of TJ homeostasis.
In vivo analysis of CLDN1 expression in intestinal epithelia obtained from WT and conditional HIF1β-null mice revealed no baseline difference between the two groups. However, induction of experimental colitis using TNBS in these mice resulted in a small but nonsignificant increase in CLDN1 in WT mice, with a further repression in the conditional HIF1β-null mice. This correlates with defects in in vivo barrier function noted elsewhere (Glover et al., 2013) and is consistent with the induction of HIF targets seen in other mouse models (Karhausen et al., 2004). These data suggest that HIF-mediated expression of CLDN1 might play an important role in barrier maintenance during an inflammatory insult.
HIF has been extensively linked to beneficial outcomes in murine models of colitis (Karhausen et al., 2004; Cummins et al., 2008; Robinson et al., 2008). Although the endpoints of such signaling vary, significant emphasis has been placed on the barrier-protective aspects of hypoxia and HIF signaling (Colgan and Taylor, 2010; Glover and Colgan, 2011). To date, HIF target genes implicated in such barrier protection are more “nonclassical” in nature, including intestinal trefoil factor (Furuta et al., 2001), multidrug resistance gene (Comerford et al., 2002), mucin-3 (Louis et al., 2006), and CD73 (Synnestvedt et al., 2002). The present results provide the first evidence that HIF affects barrier function of IECs directly through regulation of a TJ component.
MATERIALS AND METHODS
Cell culture
Human T84 and Caco2 intestinal epithelial cells and HeLa cells were cultured in 95% air with 5% CO2 at 37°C. Lentiviral particles encoding shRNA against HIF1β and CLDN1 (MISSION TRC shRNA; Sigma-Aldrich, St. Louis, MO) were transduced into T84 and Caco2 IECs using established protocols. Where indicated, cells were exposed to hypoxia in a humidified cell chamber (Coy Laboratory Products, Grass Lake, MI) using standard conditions of 1% O2, 5% CO2, balance N2.
Transcriptional analysis
TRIzol reagent (Invitrogen, Carlsbad, CA) was used to isolate RNA from confluent T84 monolayers. cDNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). PCR analysis was performed using SYBR Green (Applied Biosystems, Carlsbad, CA) and the primer sequences described in Supplemental Table S1.
Western blot analysis
The NE-PER extraction kit was used to prepare nuclear lysates from confluent T84 monolayers per manufacturer's instructions (Thermo Scientific, Waltham, MA). Western blotting of these lysates was performed using HIF1β monoclonal mouse antibody (BD Biosciences, San Jose, CA) and TATA binding protein monoclonal mouse antibody (Abcam, Cambridge, United Kingdom). Western blotting on whole-cell lysates was done using rabbit polyclonal anti-CLDN1 (Invitrogen), polyclonal rabbit anti–β-actin (Abcam), and polyclonal antibodies against phosphorylated (Ser-19) and total myosin light chain (Cell Signaling Technology, Danvers, MA).
Immunofluorescence and microscopy
T84 or Caco2 cells were grown to confluence on 0.33-cm2, 0.4-μm permeable polyester inserts (Corning, Corning, NY), fixed with 2% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% normal goat serum (NGS). Rb polyclonal antibody (pAb) against ZO1 (Invitrogen) was used at 1:100 dilution in 5% NGS. Secondary antibody was Alexa Fluor 568 goat anti-rabbit (Invitrogen), used at 1:500 dilution in 5% NGS. Immunolabeling was visualized with an AxioCam MR c5 attached to an AxioImager A1 microscope (Zeiss, Oberkochen, Germany). Where indicated, series of confocal fluorescence images were obtained using a Zeiss Axiovert 200M laser-scanning confocal/multiphoton-excitation fluorescence microscope with a Meta spectral detection system (Zeiss NLO 510 with META; Zeiss, Thornwood, NY) as previously described (Dobrinskikh et al., 2013). Quantification of cell area using relative pixel values was performed using ImageJ (National Institutes of Health, Bethesda, MD).
Calcium switch and permeability assays
T84 cells were grown to confluence on 0.33-cm2, 0.4-μm permeable polyester inserts (Corning). Calcium switch was performed by incubating cells in Ca2+-free MEM for suspension culture (S-MEM; Sigma Aldrich) with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 14 mM NaHCO3, and 5% dialyzed fetal bovine serum for 16 h. Cells were then returned to normal Ca2+ medium. TER was measured using the EVOM2 voltohmmeter (World Precision Instruments, Sarasota, FL). Paracellular permeability was assayed using a FITC-flux assay described previously (Furuta et al., 2001). Briefly, confluent T84 monolayers on 0.33-cm2, 0.4-μm permeable polyester inserts were washed and equilibrated in Hank's balanced salt solution (Sigma-Aldrich). FITC-dextran (Sigma Aldrich), 1 mg/ml, was added to the apical compartment, and samples were taken from the basolateral compartment every 30 min for 2 h. Fluorescence was determined using a Glomax Multi fluorescent plate reader (Promega, Madison, WI) and represented as the change in fluorescence over time.
Chromatin immunoprecipitation and promoter experiments
ChIP was performed on confluent Caco2 IECs as previously described (Clambey et al., 2012; Glover et al., 2013). Briefly, cells were exposed to either normoxia or hypoxia for 6 h and fixed in 1% formaldehyde for 10 min at 4°C. Cells were sonicated to shear genomic DNA and incubated overnight with 5 μg of either control Rb IgG or Rb pAb against HIF1α (NB100-134; Novus Biologicals, Littleton, CO). The resulting complexes were precipitated using Fastflow G-Sepharose beads (GE Healthcare, Little Chalfont, United Kingdom), eluted, purified, and analyzed by PCR using HRE spanning primers (Supplemental Table S1).
CLDN1 promoter luciferase reporter plasmid (Switchgear Genomics, Carlsbad, CA) was mutated using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) with primers against both HRE sites. Using Lipofectamine LTX (Invitrogen), 100 ng of plasmid was transfected into subconfluent HeLa cells and luciferase activity determined at 24 h using LightSwitch luciferase reagent (Switchgear Genomics) and luminescence determined using the Glomax Multi plate reader (Promega).
CLDN1 overexpression
CLDN1 overexpression plasmid (Thermo Scientific) was transfected into subconfluent Caco2 HIF1β KD cells with 5 or 10 μg of plasmid on 10-cm dishes using Lipofectamine LTX and Plus reagent per manufacturer's instructions (Invitrogen). Controls were mock transfected. Cells were incubated overnight, trypsinized, and plated on 0.33-cm2, 0.4-μm polyester inserts (Corning). Monolayers were harvested for immunofluorescence at 48 h postplating. Quantification of junctional length (A) relative to a straight line between tricellular junctions (B) was calculated as a TJ length ratio (equal to A/B) from individual image tracings (80 measurements for control, 91 for 5 μg of plasmid, and 62 for 10 μg of plasmid transfection) using ImageJ (Schneider et al., 2012).
Animals
HIF1β conditional KO mice (Mus musculus) were generated as previously described (Glover et al., 2013). RNA was isolated from IEC HIF1β KO (n = 5) or Cre-negative littermate control (n = 5) whole colonic tissue or mucosal scrapings, reverse transcribed, and assayed using PCR. TNBS experimental colitis was induced as previously described (Karhausen et al., 2004). Briefly, mice were sensitized by epicutaneous administration of 1% TNBS (Sigma Aldrich) in 80% EtOH on day 0, followed by intrarectal application of 2.5% TNBS in 50% ethanol on day +7. RNA was isolated from whole colonic tissue or mucosal scrapings of duodenum, ileum, cecum, and colon, reverse transcribed, and assayed using PCR. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Colorado.
Statistical analysis
GraphPad Prism 5 (GraphPad Software, La Jolla, CA) was used to generate figures and perform statistical analyses, including paired and unpaired t tests. Probability values of p < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants DK50189, HL60569, and DK095491 and the Crohn's and Colitis Foundation of America.
Abbreviations used:
- ChIP
chromatin immunoprecipitation
- CLDN1
claudin-1
- FITC
fluorescein isothiocyanate
- HIF
hypoxia-inducible factor
- HRE
hypoxia response element
- IEC
intestinal epithelial cell
- JAM-A
junctional adhesion molecule-A
- KD
knockdown
- RT-PCR
reverse transcriptase PCR
- shRNA
short hairpin RNA
- TER
transepithelial resistance
- TJ
tight junction
- TNBS
trinitrobenzene sulfonic acid
- ZO1
zonula occludens 1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1194) on April 22, 2015.
*These authors contributed equally and should be regarded as joint first authors.
†These authors should be regarded as joint senior authors.
The authors declare no financial interests in any of the work presented here.
REFERENCES
- Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM, Fanning AS. Setting up a selective barrier at the apical junction complex. Curr Opin Cell Biol. 2004;16:140–145. doi: 10.1016/j.ceb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Baker OJ, Camden JM, Redman RS, Jones JE, Seye CI, Erb L, Weisman GA. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am J Physiol Cell Physiol. 2008;295:C1191–C1201. doi: 10.1152/ajpcell.00144.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. 2012;109:E2784–E2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–3394. [PubMed] [Google Scholar]
- Cording J, Berg J, Kading N, Bellmann C, Tscheik C, Westphal JK, Milatz S, Gunzel D, Wolburg H, Piontek J, et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J Cell Sci. 2013;126:554–564. doi: 10.1242/jcs.114306. [DOI] [PubMed] [Google Scholar]
- Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, Neff J, Washington MK, Beauchamp RD. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J Clin Invest. 2005;115:1765–1776. doi: 10.1172/JCI24543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrinskikh E, Lanzano L, Rachelson J, Cranston D, Moldovan R, Lei T, Gratton E, Doctor RB. Shank2 contributes to the apical retention and intracellular redistribution of NaPiIIa in OK cells. Am J Physiol Cell Physiol. 2013;304:C561–C573. doi: 10.1152/ajpcell.00189.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143:391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147:891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, Dobrinskikh E, Kendrick AA, Kelly CJ, Burgess A, et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc Natl Acad Sci USA. 2013;110:19820–19825. doi: 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover LE, Colgan SP. Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology. 2011;140:1748–1755. doi: 10.1053/j.gastro.2011.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover LE, Irizarry K, Scully M, Campbell EL, Bowers BE, Aherne CM, Kominsky DJ, MacManus CF, Colgan SP. IFN-γ attenuates hypoxia-inducible factor (HIF) activity in intestinal epithelial cells through transcriptional repression of HIF-1β. J Immunol. 2011;186:1790–1798. doi: 10.4049/jimmunol.1001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inai T, Kobayashi J, Shibata Y. Claudin-1 contributes to the epithelial barrier function in MDCK cells. Eur J Cell Biol. 1999;78:849–855. doi: 10.1016/S0171-9335(99)80086-7. [DOI] [PubMed] [Google Scholar]
- Ivanov AI. Structure and regulation of intestinal epithelial tight junctions: current concepts and unanswered questions. Adv Exp Med Biol. 2012;763:132–148. doi: 10.1007/978-1-4614-4711-5_6. [DOI] [PubMed] [Google Scholar]
- Jou TS, Schneeberger EE, Nelson WJ. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J Cell Biol. 1998;142:101–115. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Investigation. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Colgan SP, Frank DN. Of microbes and meals: the health consequences of dietary endotoxemia. Nutr Clin Pract. 2012;27:215–225. doi: 10.1177/0884533611434934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Glover LE, Campbell EL, Kominsky DJ, Ehrentraut SF, Bowers BE, Bayless AJ, Saeedi BJ, Colgan SP. Fundamental role for HIF-1alpha in constitutive expression of human beta defensin-1. Mucosal Immunol. 2013;6:1110–1118. doi: 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer F, White K, Kubbies M, Swisshelm K, Weber BH. Genomic organization of claudin-1 and its assessment in hereditary and sporadic breast cancer. Hum Genet. 2000;107:249–256. doi: 10.1007/s004390000375. [DOI] [PubMed] [Google Scholar]
- Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopardo T, Lo Iacono N, Marinari B, Giustizieri ML, Cyr DG, Merlo G, Crosti F, Costanzo A, Guerrini L. Claudin-1 is a p63 target gene with a crucial role in epithelial development. PLoS One. 2008;3:e2715. doi: 10.1371/journal.pone.0002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–1627. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Skare IB, Lynch RD, Schneeberger EE. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J Cell Sci. 2000;113:3387–3398. doi: 10.1242/jcs.113.19.3387. [DOI] [PubMed] [Google Scholar]
- McGuckin MA, Eri R, Simms LA, Florin TH, Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15:100–113. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- Mrsny RJ, Brown GT, Gerner-Smidt K, Buret AG, Meddings JB, Quan C, Koval M, Nusrat A. A key claudin extracellular loop domain is critical for epithelial barrier integrity. Am J Pathol. 2008;172:905–915. doi: 10.2353/ajpath.2008.070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, 3rd, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–978. doi: 10.1083/jcb.200206114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacary E, Tixier E, Coulet F, Roussel S, Petit E, Bernaudin M. Crosstalk between HIF-1 and ROCK pathways in neuronal differentiation of mesenchymal stem cells, neurospheres and in PC12 neurite outgrowth. Mol Cell Neurosci. 2007;35:409–423. doi: 10.1016/j.mcn.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte S, Desrosiers RR, Beliveau R. HIF-1alpha mRNA and protein upregulation involves Rho GTPase expression during hypoxia in renal cell carcinoma. J Cell Sci. 2003;116:2247–2260. doi: 10.1242/jcs.00427. [DOI] [PubMed] [Google Scholar]
- Turcotte S, Desrosiers RR, Beliveau R. Hypoxia upregulates von Hippel-Lindau tumor-suppressor protein through RhoA-dependent activity in renal cell carcinoma. Am J Physiol Renal Physiol. 2004;286:F338–F348. doi: 10.1152/ajprenal.00254.2003. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. Claudin interactions in and out of the tight junction. Tissue Barriers. 2013;1:e25247. doi: 10.4161/tisb.25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel S, Wottawa M, Farhat K, Zieseniss A, Schnelle M, Le-Huu S, von Ahlen M, Malz C, Camenisch G, Katschinski DM. Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/rho-associated kinase-dependent cofilin phosphorylation. J Biol Chem. 2010;285:33756–33763. doi: 10.1074/jbc.M110.132985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Wang PY, Wang X, Wan YL, Liu YC. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Digestive Dis Sci. 2012;57:3126–3135. doi: 10.1007/s10620-012-2259-4. [DOI] [PubMed] [Google Scholar]
- Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88:1110–1120. doi: 10.1038/labinvest.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.