

The role of the actin-capping protein flightless I in collagen remodeling by mouse fibroblasts is examined. Flightless and nonmuscle myosin IIA cooperate to enable collagen phagocytosis.
Abstract
We examined the role of the actin-capping protein flightless I (FliI) in collagen remodeling by mouse fibroblasts. FliI-overexpressing cells exhibited reduced spreading on collagen but formed elongated protrusions that stained for myosin10 and fascin and penetrated pores of collagen-coated membranes. Inhibition of Cdc42 blocked formation of cell protrusions. In FliI-knockdown cells, transfection with constitutively active Cdc42 did not enable protrusion formation. FliI-overexpressing cells displayed increased uptake and degradation of exogenous collagen and strongly compacted collagen fibrils, which was blocked by blebbistatin. Mass spectrometry analysis of FliI immunoprecipitates showed that FliI associated with nonmuscle myosin IIA (NMMIIA), which was confirmed by immunoprecipitation. GFP-FliI colocalized with NMMIIA at cell protrusions. Purified FliI containing gelsolin-like domains (GLDs) 1–6 capped actin filaments efficiently, whereas FliI GLD 2–6 did not. Binding assays showed strong interaction of purified FliI protein (GLD 1–6) with the rod domain of NMMIIA (kD = 0.146 μM), whereas FliI GLD 2–6 showed lower binding affinity (kD = 0.8584 μM). Cells expressing FliI GLD 2–6 exhibited fewer cell extensions, did not colocalize with NMMIIA, and showed reduced collagen uptake compared with cells expressing FliI GLD 1–6. We conclude that FliI interacts with NMMIIA to promote cell extension formation, which enables collagen remodeling in fibroblasts.
INTRODUCTION
Connective tissue homeostasis is maintained by a dynamic balance involving the coordinated secretion, organization, and degradation of matrix molecules (Everts et al., 1996; Grinnell, 2003). In soft connective tissues, extracellular matrix macromolecules are synthesized and secreted by local fibroblasts, which also contribute to matrix reorganization by compaction and pericellular degradation. The resorptive processes mediated by fibroblasts include the expression and activation of matrix metalloproteinases for protein degradation by the extracellular pathway and the phagocytosis and digestion of matrix molecules in lysosomes that involve the intracellular pathway (Everts et al., 1996; Madsen et al., 2013; Panwar et al., 2013). To enable matrix degradation by phagocytosis, connective tissue cells explore, attach to, and reorganize matrix proteins, processes that require extensive changes in cell shape (Melcher and Chan, 1981).
Some of the structural adaptations that enable matrix remodeling are visible microscopically as cell surface extensions (Bellows et al., 1981); the adhesion of these extensions to underlying matrix substrates is tightly coordinated to facilitate matrix internalization and degradation by phagocytosis (Arora et al., 2011). Because cell adhesions provide structural and functional continuity between extracellular matrix polymers and the actin cytoskeleton, they are potential loci for controlling matrix remodeling (Bouvard et al., 2013; Ciobanasu et al., 2013; Costa et al., 2013; Hoshino et al., 2013). Of note, the organization and remodeling of actin filaments are regulated by a large number of actin-binding proteins, which mediate cycles of actin filament assembly and depolymerization and are enriched in cell adhesions (Wehrle-Haller, 2012; Ciobanasu et al., 2013; Schiller and Fassler, 2013).
Actin filament assembly is regulated by at least six classes of actin-binding proteins, which can sequester actin monomers (e.g., profilin), control filament nucleation (e.g., villin), block the barbed end (e.g., fragmin) or the pointed end of the filament (e.g., α-actinin), sever the filament (e.g., gelsolin) and “nibble” at the filament (e.g., depactin). Further, actin filaments can be cross-linked to form higher-order structures that in turn determine the mechanical stability and shape of the plasma membrane. A large number of actin-filament cross-linking proteins contribute to the formation of several distinct types of actin filament organization, including the tightly packed parallel bundles seen in filopodia, the large arrays of contractile bundles seen in stress fibers, and the branching actin networks of lamellipodia (Mullins et al., 1998). These distinctive actin structures are assembled and disassembled dynamically in concert with actin-binding proteins that respond to various signals to tightly regulate functions such as cell motility, contraction, adhesion (Le Clainche and Carlier, 2008), wound healing (Kopecki and Cowin, 2008), and tissue homeostasis.
Connective tissue homeostasis requires continuous cellular responses to mechanical and chemical signals (Daley et al., 2008). These responses include the generation of propulsive and contractile forces, which are mediated by actin filaments and myosin motor activity (Yamaguchi and Condeelis, 2007). Myosins comprise a superfamily of motor proteins that play important roles in several cellular processes, including force generation, intracellular transport, and cell adhesion (Vicente-Manzanares et al., 2009; Hanein and Horwitz, 2012). Nonmuscle myosin IIA (NMMIIA) and NMMIIB are important regulators of adhesion, polarity, and migration of nonmuscle cells, processes that involve the dynamic remodeling of the actin cytoskeleton and cellular interactions with the extracellular matrix (Alexandrova et al., 2008). Although NMMIIA is dispensable for the assembly and disassembly of nascent cell adhesions, it is required for the subsequent maturation of adhesions (Choi et al., 2008) and may provide a site of convergence for external and cell-generated forces (Galbraith et al., 2002). Of note, the control of adhesion assembly and turnover of cell adhesions can affect the structure, rigidity, and remodeling of extracellular matrix proteins (Block et al., 2008).
Cell–matrix adhesions are dynamic structures that are enriched with actin-binding proteins (Stossel et al., 1985; Vicente-Manzanares and Horwitz, 2011) and in particular with the actin-capping protein flightless I (FliI; Davy et al., 2001; Mohammad et al., 2012). FliI is a conserved member of the gelsolin family (Archer et al., 2004; Ghoshdastider et al., 2013; Nag et al., 2013), which was originally identified in Drosophila melanogaster (Campbell et al., 1993). Mutations in the FliI gene cause defects in Drosophila indirect flight muscles; consequently, they cannot fly. In mammalian cells, FliI regulates cell migration (Cowin et al., 2007) and wound healing (Kopecki and Cowin, 2008); decreased expression of FliI can improve healing in early-gestation fetal wounds (Lin et al., 2011). FliI is also expressed in structures associated with migration, such as neurites, growth cones, and filopodia (Davy et al., 2001; Deng et al., 2007), suggesting a role for FliI in actin filament organization at these sites. In addition to its gelsolin-like domains (GLDs), FliI exhibits a leucine-rich repeat (LRR; Fong and de Couet, 1999), which is not found in other gelsolin family proteins. Protein interactions with the LRR domain may enable intermolecular recognition and structural organization by FliI (Liu and Yin, 1998), but it is not known whether FliI interacts with other cytoskeletal proteins to regulate cell adhesion and matrix remodeling.
We previously showed that FliI promotes cell interactions with the extracellular matrix and enhances cell anchorage (Mohammad et al., 2012). Our new data show that at collagen adhesion sites, FliI interacts with NMMIIA, which upon activation, generates forces required for collagen remodeling. The capping function of FliI regulates the formation of actin-rich filopodial cell extensions independently of other proteins. Collectively our data indicate that FliI interactions with NMMIIA provide a platform for actin filament elongation, cell extension formation, and collagen remodeling.
RESULTS
Characterization of FliI cells
Because previous studies showed that FliI may influence attachment of fibroblasts to collagen, cell migration (Mohammad et al., 2012), and wound repair (Cowin et al., 2007; Kopecki and Cowin, 2008), we examined collagen remodeling in FliI stable cell lines expressing varying levels of FliI. When compared with FliI wild-type (WT) cells, lysates prepared from FliI-overexpressing (OE) cells showed 170% higher FliI expression, whereas FliI-knockdown (KND) cells showed 41% reduced FliI expression (Figure 1A, i and ii). FliI OE, WT, and KND cells stained with Alexa λ488–phalloidin exhibited distinct morphologies after 60 min of spreading on fibrillar collagen. FliI OE cells showed pointed extensions, whereas FliI KND cells exhibited lamellipodia with a smooth-edged morphology (Figure 1Bi). The morphologies of the FliI cell types were similar when plated on monomeric collagen (unpublished data). Because immunostaining protocols involved cell fixation and permeabilization, processes that can disrupt thin cytoplasmic sheets interposed between the cell protrusions, we stained living cells with Alexa 488–wheat germ agglutinin to outline the cell membrane of vital cells. These images are consistent with enhanced development of cell extensions in FliI OE cells (Figure 1Bii).
FIGURE 1:

Characterization and phenotypes of FliI cells. (A) (i, ii) FliI protein expression levels in FliI OE, WT, and KND cells. Compared with FliI WT cells, FliI OE cells show 170% increase of FliI expression, and FliI KND cells show 41% reduction of FliI. Histogram shows quantification of average blot density in Western blots. Experiments were repeated four times with similar results. (B) (i) FITC-phalloidin–stained cells show distinct phenotypes with different cell surface morphologies when plated on fibrillar collagen. FliI KND cells are characterized by a flat, evenly spread, smooth-edged phenotype. FliI OE cells show prominent filopodia with long, actin-containing protrusions. (ii) Cells stained with Alexa 488 wheat germ agglutinin (WGA) to show outline of cell membrane of vital cells. (C) (i) Rescue experiment in which WT FliI-HA was introduced into FliI KND cells. shRNA-resistant construct was transfected into KND cells to block RNA-mediated silencing of FLiI. (ii) Cells transfected with rescue construct exhibit different morphology from FliI KND cells and show longer cell extensions. Experiments were repeated three times. (D) Left, representative images of cells treated with blocking antibody or irrelevant (nebulin antibody). Right, data in histogram show effect of α2β1 blocking antibody on cell attachment. Data are reported as mean ± SD, n = 3, at least 50 cells per group; #p < 0.02 using ANOVA and comparing antibody with control (irrelevant antibody–treated) cells. (E) Projected surface area of spreading FliI KND cells is twofold higher than FliI OE cells on fibrillar or monomeric collagen. Data are reported as mean ± SD, n = 3, with at least 40 cells/group. *p < 0.05 using ANOVA and comparisons between OE with WT or KND cells. (F) (i) Representative images showing round (a), spreading (b), or spread cells (c) as a function of time after plating. Rounded cells have twice the diameter of spreading cells. (ii, iii) FliI OE, WT, and KND cells plated on fibrillar or monomeric collagen. At 60 min, the initial spreading process was largely complete, and there were lower percentages of spread FliI OE cells than WT or FliI KND cells. (G) FLiI OE, WT, and KND cells immunostained for endogenous Myo10 show localized staining at tips of filopodia in FliI OE cells and punctate staining at peripheries of KND cells. (H) Fascin-immunostained cells show localization of fascin to filopodial cell extensions in FliI OE cells. (I) (i) Quantitative analysis of circularity index indicates that KND and WT cells are more circular and are less irregular on their contours than are OE cells (*p < 0.05 by ANOVA; n = 3 experiments/group, with >40 cells analyzed/group). (ii, iii) Histograms show mean ± SD of length and number of cell extensions in OE, WT, and KND cells (n = 3 experiments/group, with at least 40 cells analyzed/group.*p < 0.05 are comparisons of OE with WT and KND cells by ANOVA.
We examined whether the phenotype observed in FliI KND cells was caused by off-target, nonspecific effects of the short hairpin RNA (shRNA). We reintroduced FliI by retroviral introduction of a mutated, shRNA-resistant yet functional FliI into FliI KND cells, and these cells showed extension formation (Figure 1C, i and ii).
We examined the expression of integrins specific for collagen binding by immunoblotting of whole-cell lysates. We found similar expression levels of the α1, α2, α11, α10, and β1 integrins and the fibrillar collagen receptor discoidin domain receptor 1 (DDR1; Supplemental Figure S1A). Surface expression of the β1 integrin (measured by flow cytometry of nonpermeabilized cells with the KMI16 antibody) was higher (p < 0.05) in FliI OE than in WT and KND cells (Supplemental Figure S1B). These results were consistent with our previous data showing that FliI KND cells are less adhesive to collagen and migrate more quickly over collagen than WT cells (Mohammad et al., 2012). The specificity of cell binding through fibrillar collagen-binding integrin receptors was confirmed by incubating FliI OE, WT, and KND cells with a blocking antibody to the α2β1 integrin (Figure 1D; p < 0.02).
Because spreading cells form abundant protrusions (Dubin-Thaler et al., 2004), we compared cell spreading in FliI-stable cell lines. The projected cell surface area of FLiI KND cell line was twofold higher than that of WT or FliI OE cells plated on fibrillar or monomeric collagen (Figure 1E; p < 0.05). We quantified the number of round, spreading, and spread cells as a function of time after plating. We defined a spread cell as one that exhibited a greater than twofold increased radius compared with the mean radius of an initially plated cell (Figure 1Fi). At 30 min, there were lower percentages of FliI OE spreading cells than FliI KND or WT spreading cells (p < 0.05). The initial spreading process was largely complete by 60 min for all cell types, and at 60 min, there were lower percentages of spread FliI OE cells than WT or FliI KND cells (Figure 1F, i and ii). The dynamics of cell spreading for the three different cell types were similar on monomeric collagen and fibrillar collagen.
Spreading on extracellular matrix substrates involves actin filament assembly at the leading edge (Pollard and Borisy, 2003), which can manifest as the generation of membrane extensions such as filopodia. Several proteins, such as the small GTPase Cdc42 (Nobes and Hall, 1995), N-WASP (Pollard and Borisy, 2003), fascin (Zanet et al., 2012), and myosin 10 (Myo10) (Berg and Cheney, 2002), localize and regulate filopodial extensions. Because endogenous and exogenously expressed Myo10 localizes to the tips of actin-rich protrusions in numerous cell types (Bohil et al., 2006), we immunostained FliI cell lines and found that Myo10 localized to the tips of filopodia in FliI OE cells (Figure 1G), whereas Myo10 staining did not localize to tips of WT and FliI KND cells. We also assessed cell surface extensions by immunostaining cells for fascin, which localized to cell surface projections in FliI OE and WT cells (Figure 1H).
Analysis of the circularity index (ImageJ) indicated that FliI KND cells were more rounded than FliI OE cells, which exhibited surface irregularities (Figure 1Ii; p < 0.05). We analyzed and compared the lengths of cell extensions in the FliI cell lines; these analyses showed that at 30 and 60 min after plating, cell extensions were longer and more abundant in FliI OE than in KND and WT cells (Figure 1I, ii and iii; p < 0.05).
Role of Cdc42 in cell extension formation
Small GTPases may regulate the processes by which FliI is involved in actin assembly and cell extension formation (Davy et al., 2001; Higashi et al., 2010). Because spreading cells exhibit various types of cell extensions, including filopodia and lamellipodia, we anticipated that FliI cell lines would exhibit increased Cdc42 and Rac activity during spreading. FliI OE, WT, and KND cells plated on collagen for 30 min showed equivalent levels of Cdc42 activity (Figure 2A, i and ii). Rac activity in FliI KND cells was higher than in FliI WT and FliI OE cells (Figure 2Aiv; p < 0.05). A specific inhibitor of Cdc42 (ML-141) reduced Cdc42 activity (Figure 2Aii; p < 0.01). Cell extension formation in all FliI cell lines was reduced in the presence of the Cdc42 inhibitor (Figure 2B, i and ii; p < 0.01). The number of cell extensions in untreated FliI OE cells was threefold higher than in WT and FliI KND cells (Figure 2Bii; p < 0.01).
FIGURE 2:

Involvement of Cdc42. (A) (i–iv) Lysates from FliI OE, WT, and KND cells plated on collagen for 30 min were incubated with Pak1 protein-binding domain (PBD) beads to estimate active Rac or Cdc42. The three cells types showed similar Cdc42 activity (ii), but Rac activity was higher in FliI KND cells than in WT and FliI OE cells (iv; *p < 0.05 by ANOVA; data from three separate experiments). Cdc42 activity was measured in cells treated with (T) or without (C) the Cdc42 inhibitor ML-141. Note marked inhibition of Cdc42 activation in FliI OE, WT, and KND cells (i, ii) treated with inhibitor but no effect on Rac activation (iii, iv). (ii) Quantitative analysis of blot density in histogram shows effect of Cdc42 inhibitor. Data analyzed by ANOVA, reported as mean ± SD; experiment repeated three times (##p < 0.01, comparisons between inhibitor-treated and respective untreated samples). (B) (i, ii) Phalloidin-stained FliI OE cells show more cell extensions than do WT and FliI KND cells. (ii) Quantitative analysis in the histogram by ANOVA; three separate experiments, 50 cells analyzed from each group; **p < 0.01, comparison of WT and KND cells to OE cells in untreated samples. Cells treated with ML-141 show retarded cell extension formation in all cell types. Histogram shows the analysis on ML-1411–treated and untreated samples by ANOVA; three separate experiments and 50 cells analyzed/group; ##p < 0.01 comparison of treated FliI OE, WT, and FliI KND cells to their respective untreated cells. (C) (i) FLiI KND or WT cells transfected with HA-tagged, constitutively active Cdc42 (G12V). Cells immunostained for HA and double stained with Alexa λ488–phalloidin show lack of extension formation in FliI KND cells transfected with constitutively active Cdc42. (ii) Quantification of cell extensions in FliI KND and WT cells transfected with HA-tagged, constitutively active Cdc42 (G12V; p < 0.05). (iii) Cell lysates of transfected cells were immunoblotted with HA antibody to confirm transfections. (D) Cells treated with cdc-42 inhibitor ML-141 show twofold reduction in collagen internalization by FLiI OE cells. Data are reported in the histogram as mean ± SD, n = 3, analyzed by ANOVA. *p < 0.05, comparison of untreated WT and KND cells to untreated FliI OE cells; #p < 0.05, comparison of treated FliI OE, WT, and FliI KND cells with respective untreated cells.
In experiments to assess the involvement of FLiI in cell extension formation, we found that FliI KND cells transfected with constitutively active, hemagglutinin (HA)-tagged Cdc42 (G12V) did not exhibit cell extension formation. In an experiment of similar design, Cdc42 (G12V)-transfected WT cells showed enhanced cell extension formation. These data indicate that FliI is required for filopodia formation in these cells (Figure 2C, i and ii; p < 0.05). Expression of HA-tagged FliI was confirmed in immunoblots of transfected cells (Figure 2Ciii).
We examined the contribution of Cdc42 to collagen remodeling. Cells were plated on FITC-labeled collagen substrates for 60 min in the presence or absence of ML-141. In untreated controls, the amount of collagen internalized per cell was twofold higher in FLiI OE than in FliI KND cells (Figure 2D; p < 0.05). Treatment with ML-141 inhibited the internalization of labeled collagen by twofold in FliI OE compared with FLiI WT or KND cells (Figure 2D; p < 0.05).
FliI enhances collagen remodeling
Suspended cells rapidly change shape when in contact with solid substrates (Discher et al., 2005). On planar, two-dimensional surfaces, some cells spread uniformly, whereas others form elongated filopodial cell extensions or fan-shaped lamellipodia (Yeung et al., 2005). In three-dimensional matrix cultures, cells often extend protrusions, reflecting their ability to penetrate matrix pores (Martins and Kolega, 2006). We examined the contribution of cell extensions to the invasion of collagen matrices. Cells were plated on collagen-coated porous membranes (3.0-μm pore diameter), and the projection of extensions through the small pores of the membrane in response to a 10% fetal bovine serum gradient was assessed (Nguyen et al., 2000; Wang and Klemke, 2007). By confocal microscopy and serial optical sectioning, we observed that the cell extensions on the ventral surface of cells colocalized with pores in the membrane (Figure 3Ai). A large majority of the FliI OE cells projected extensions through pores on the membrane toward the chemoattractant gradient within 180 min of plating. The extensions were independent of the translocation of cell bodies because 4′,6-diamidino-2-phenylindole–stained cell nuclei were consistently observed on the upper side of the membrane. We assessed filopodial extensions that penetrated membrane pores by confocal microscopy (Figure 3Aii). The number of cell extensions protruding through the pores on the underside of the membrane were fivefold higher in FliI OE than in FliI KND cells (p < 0.001; Figure 3Aiii). We separated mechanically the protrusions that extended through the membrane pores to the underside of the membrane from the cell bodies that were retained on the upper side of the membrane (see Materials and Methods). Immunoblotting of these samples for lamin B confirmed the presence of cell bodies containing cell nuclei on the top surface of the membranes (Figure 3B).
FIGURE 3:

Collagen remodeling and FliI-dependent cell extensions. (A) (i–iii) Cells attached to collagen-coated, 3-μm porous membranes. Extensions protruded through membrane pores toward serum gradient for 4 h. Cell extensions were examined by optical sectioning with a confocal microscope. Note that cell extensions on the ventral surface of cells colocalized with pores in the membrane. (ii) Three-dimensional reconstructions assembled from cell projections through membrane pores. The scale shows the depth (length) of the projections through the membrane pores. (iii) Quantification of cell extensions observed through pores on the underside of the membrane show significantly reduced number of cell extensions in KND and WT cells. Data reported in the histogram are mean ± SD, n = 4, using ANOVA. **p < 0.01, comparison of WT cells to FliI OE cells; ***p < 0.001 comparison of FliI KND cells to FliI OE cells. (B) Protein isolated from cell extensions on the underside of the porous membrane shows absence of the nuclear membrane protein lamin B. (C) (i) FliI OE, WT, and KND cells plated on FITC-collagen show substrate clearance. Dark areas are sites of clearance where fluorescent collagen has been degraded by cells. Note the increased clearance by FliI OE cells at cell extensions compared with WT and FliI KND cells. (ii) Quantification presented in histogram as mean ± SD; n = 3; analysis from 40 cells each using ANOVA. **p < 0.01 comparison of WT and FliI KND cells to FliI OE cells. Treatment with a broad-spectrum MMP inhibitor showed no effect on collagen clearance. (D) Equivalent amount of proteins from the FliI cell lines measured fluorimetrically for estimating internalized FITC-collagen 4 h after plating. Inset, Western blot probed with GAPDH. Data in histogram show increased collagen internalization in FliI OE cells as compared with WT and FliI KND cells, *p < 0.05. Data are reported as mean ± SD, n = 3, at least 50 cells/group analyzed by ANOVA. (E) Quantification of amounts of compacted collagen matrix. Concentric circles radiating from the cell centroid were drawn. The total accumulated intensity of compacted FITC-collagen was measured around each cell. (F) (i, ii) Fluorescence images and quantification show compaction of FITC-collagen around FliI OE, WT, and KND cells. (G) (i, ii) Treatment with blebbistatin blocked collagen accumulation around cells.
We determined whether FliI expression affected remodeling of collagen. In FliI OE cells plated on exogenous fluorescein isothiocyanate (FITC)–labeled collagen for 4 h, there was more collagen clearance at cell peripheries compared with FliI WT or KND cells (p < 0.01; Figure 3C, i and ii). Because collagen substrate clearance could be due to pericellular collagen proteolysis, we treated the cells with the broad-spectrum MMP inhibitor batimastat but found no difference in collagen clearance compared with vehicle controls (Figure 3Cii), indicating that MMPs are probably not involved with collagen clearance.
We evaluated intracellular degraded collagen in cells plated on collagen. There was increased fluorescence detected in fixed and permeabilized FliI OE cells immunostained for three-fourths collagen fragments compared with WT or FliI KND cells (Supplemental Figure S2A, i and ii; p < 0.05; p < 0.001). Fluorescence quantification of cells showed sixfold difference in intracellular degraded collagen between FliI OE and FliI KND cells (Supplemental Figure S2Aii). The differences could be due to contributions by cell surface–degraded substrate collagen, and so we repeated the same experiment in nonpermeabilized cells. There was minimal fluorescence for all cell types, which was significantly different from permeabilized cells (p < 0.01; Supplemental Figure S2Aii). Further, to ensure that the intracellular degraded collagen was indeed substrate collagen, cells were plated on biotinylated collagen for 4 h, removed by trypsinization, and washed. Cell lysates were incubated with high-capacity streptavidin agarose resin (Thermo Scientific) to detect the internalized biotinylated collagen. Boiled samples were run on SDS–PAGE, and blots were probed with antibodies to three-fourths and one-fourth fragments (rabbit antibody to type I collagen one-fourth fragment [IAGQRGGC immunizing peptide; Materials and Methods]). We detected higher amounts of one-fourth and three-fourths degraded collagen in FliI OE cells (p < 0.01; p < 0.01; Supplemental Figure S2B). Fluorimetric measurements of cell lysates with equivalent cell protein (Figure 3D, inset) showed enhanced FITC-collagen internalization by FliI OE compared with FliI WT and KND cells (p < 0.05; Figure 3D).
FliI OE, WT, or KND cells plated on fibrillar collagen aggregated collagen fibrils around their peripheries. The amount of aggregated FITC-collagen around each cell was estimated from consecutively circular measurements radiating from the cell centroid (Figure 3E). FliI OE cells aggregated collagen to a greater extent than did WT cells, whereas FliI KND cells exhibited fourfold less collagen aggregation (p < 0.05; Figure 3F, i and ii).
Because cell-generated contractile forces applied through cell adhesions may compact collagen fibrils (Tamariz and Grinnell, 2002; Petroll and Ma, 2003), we examined collagen compaction after blebbistatin treatment to block myosin II activity. Blebbistatin ablated collagen compaction to the same extent for FliI OE, WT, and KND cells (Figure 3G, i and ii), indicating the involvement of nonmuscle myosin II in this model of collagen remodeling.
Association of FliI with NMMIIA
NMMIIA is involved in force generation through cross-linking and contraction of actin filaments (Cai et al., 2006). Accordingly, we examined expression of nonmuscle myosin IIA and found similar levels in FliI OE, WT, and KND cells (Figure 1Ai). Immunostaining showed abundant NMMIIA at cell adhesions, at the cell periphery, and in cell extensions in FliI OE cells (Figure 4A). FliI (Pearson's r = 0.87) and NMMIIA (Pearson's r = 0.71) colocalized with vinculin at cell adhesions (Figure 4B, i and ii). FliI OE cells transfected with GFP-FliI and immunostained for NMMIIA showed colocalization of exogenous FliI and NMMIIA at adhesion sites (Figure 4Ci). Quantitative analysis (Bolte and Cordelieres, 2006) showed >60% colocalization of transfected FliI and endogenous NMMIIA.
FIGURE 4:

Colocalization of FliI with NMMIIA. (A) Representative images showing localization of nonmuscle myosin IIA in FliI OE, WT, and KND cells. Bar, 10 μm. (B) (i, ii) WT cells show colocalization of endogenous FliI and NMMIIA with vinculin. (C) FliI OE cells show colocalization of exogenous FLiI with NMMIIA at cell extensions. Bar, 10 μm. (D) (i, ii) Immunoblots were probed with an antibody to total myosin light chain (t-MLC) or to phospho-Ser-19 of myosin light chain (p-MLC) and show enhanced phosphorylation of p-MLC in FliI OE cells compared with WT and FliI KND cells. Data are reported as mean ± SD, n = 3, analyzed by ANOVA; **p <0.01 WT and KND cells compared with FliI OE cells as shown in the histogram. (iii) Images show immunostaining for phosphorylated myosin light chain in cells plated on fibrillar collagen (30 min). Bar, 10 μm.
In vitro studies show that NMMIIA filament assembly and motor activity are controlled by myosin light chain phosphorylation on Thr-18/Ser-19 (Ikebe and Hartshorne, 1985). We examined the abundance of NMMIIA phosphorylation in suspended cells and cells plated on collagen. Western blots probed with anti–phosphoserine 19 antibody showed enhanced myosin light chain phosphorylation in FliI OE cells compared with WT and FliI KND cells (p < 0.01), indicating that FliI is functionally associated with NMMIIA activation (Figure 4D, i and ii). These data were confirmed by immunostaining for phospho–myosin light chain (MLC p19 serine), which also showed more abundant staining in FliI OE than FliI WT or FliI KND cells (Figure 4Diii).
We examined the potential association between FliI and NMMIIA. We used mass spectrometry to analyze the potential proteins that may be associated with FliI immunoprecipitates (IPs). We repeated our earlier work (Mohammad et al., 2012), except that in the present study, we plated cells on collagen and used a new FliI antibody (Materials and Methods). FliI immunoprecipitates and immunoprecipitates prepared with an irrelevant antibody (nebulin) were obtained from cells plated on fibrillar collagen; FliI-associated proteins were identified by tandem mass spectrometry. In these analyses, the number of tryptic peptides was assessed (Table 1); 89 peptides matched sequences in mouse NMMIIA, and 31 peptides matched sequences in NMMIIB. These data indicated that NMMIIA in particular associated with FliI; we pursued this analysis using biochemical methods. Cells were plated on monomeric or fibrillar collagen, and NMMIIA immunoprecipitates were immunoblotted for FliI. There was twofold more NMMIIA in FliI immunoprecipitates prepared from cells plated on fibrillar collagen than from cells plated on monomeric collagen (Figure 5A, i, and ii; p < 0.01). This association was confirmed by immunoprecipitation of FliI, which showed that NMMIIA was enriched in FliI immunoprecipitates (Figure 5 B, i, and ii; p < 0.01).
TABLE 1:
Mass spectrometry analysis of FLiI immunoprecipitates.
| Identified protein | Accession number | Molecular weight (kDa) | IP: FliI (number of tryptic peptides) | IP: irrelevant antibody (number of tryptic peptides) |
|---|---|---|---|---|
| Nonmuscle myosin IIA isoform 1 (Mus musculus) | gi|11432644 | 226 | 89 | 1 |
| Actin, cytoplasmic 1 (Homo sapiens) | gi|4501885 | 42 | 17 | 3 |
| Nonmuscle myosin IIB (M. musculus) | gi|33598964 | 229 | 31 | |
| mCG140959, isoform CRA_c (M. musculus) | gi|148692626 | 17 | 8 | |
| Vimentin protein (M. musculus) | gi|202368 | 54 | 17 | 2 |
| Fibronectin 1, isoform CRA_e (M. musculus) | gi|148667852 | 253 | 21 | 2 |
| Myosin regulatory light chain 12B (H. sapiens) | gi|15809016 | 20 | 7 | |
| Hist2h4 protein (M. musculus) | gi|133777677 | 11 | 5 | |
| mCG18413, isoform CRA_b (M. musculus) | gi|148672205 | 51 | 7 | 1 |
| Protein S100-A4 (M. musculus) | gi|33859624 | 12 | 4 | |
| Heat-shock protein hsp84 (M. musculus) | gi|194027 | 83 | 3 | |
| Hist1h3e protein (M. musculus) | gi|119850953 | 15 | 2 | |
| mCG113581 (M. musculus) | gi|148667742 | 13 | 2 | |
| Unnamed protein product | gi|12836870 | 19 | 1 | |
| 40S ribosomal protein | gi|14141193 | 23 | 2 |
FIGURE 5:

FliI interacts with NMMIIA. (A, B) (i, ii) NMMIIA, FliI, or an irrelevant antibody (nebulin) was used to immunoprecipitate proteins on cells plated on fibrillar or monomeric collagen and then immunoblotted for FliI and NMMIIA, respectively. Histograms show quantification of mean blot density of immunoblots. There was twofold reduction of FliI and NMMIIA interaction in cells plated on monomeric collagen. These experiments were repeated four times. Blot density reported as mean ± SD and analyzed by ANOVA. ##p < 0.01, comparison of monomeric collagen to fibrillar collagen in NMMIIA and FliI immunoprecipitates. (C) FliI OE, WT, and KND cells incubated with collagen-coated beads for 0, 30, and 60 min. Beads were isolated, and bead-associated proteins (FLiI and NMMIIA) were examined by immunoblotting. (D) Structural components of FliI and NMMIIA. (E) Pull-down assays show pelleting of NMMIIA rods (1338–1960 aa) with GST-FliI GLD 1–6. (F) Pull-down assays show that NMMIIA rods (1338–1960 aa) do not associate with GST-FliI LRR Sepharose beads. (G) Control pull-down assay shows that GST does not associate with myosin rods. (H) Pull-down assays show limited binding of NMMIIB with FliI GLD 1–6. (I) (i, ii) Analysis of pull-down experiments show quantification of binding of FliI GLD 1–6 to NMMIIA (kD = 0.146 μM) compared with FliI LRR region (kD = 5.074 μM).
Because FliI and NMMIIA colocalized at cell adhesions, we evaluated their association with collagen adhesions by isolating proteins associated with collagen-coated beads (Arora et al., 2011). Both FliI and NMMIIA were associated with collagen beads 30 min after incubation (Figure 5C), indicating that FliI and NMMIIA are enriched in cell adhesions that bind collagen. Cells incubated with bovine serum albumin–coated beads did not show FliI and NMMIIA enrichment (Figure 5C).
In vitro binding of FliI and NMMIIA
We examined the potential for direct interaction of FliI with NMMIIA in pull-down experiments that assessed the binding of purified NMMIIA rods (residues 1338–1960) to a glutathione S-transferase (GST) fusion protein of the C-terminal of FliI (consisting of FliI GLD 1–6) or to the N-terminal of FliI (consisting of the FliI LRR; Figure 5D). This analysis showed that NMMIIA rods bound tightly to FliI GLD 1–6 but much less tightly to FliI LRR (Figure 5, E and F). Because we used GST-FliI fusion protein for in vitro assays and NMMIIA may bind nonspecifically to GST, we performed an additional control experiment and found that NMMIIA did not bind to GST-Sepharose alone (Figure 5G). In another control experiment, we found that NMMIIB rods do not bind to FliI GLD 1–6 (Figure 5H). Quantitative analysis of these interactions showed tighter binding of FliI GLD 1–6 to NMMIIA (dissociation constant kD = 0.146 μM) than with FliI LRR (kD = 5.074 μM).
NMMIIA is required for collagen compaction and internalization
Cells transfected with NMMIIA small interfering RNA (siRNA) or control siRNA showed 70% reduction of NMMIIA expression compared with nontransfected or control siRNA–transfected samples (Figure 6A). Knockdown of NMMIIA was associated with loss of FliI recruitment to cell extensions (Figure 6B). We found reduced numbers (>50%; p < 0.01) and lengths (<45%; p < 0.05) of cell extensions compared with control siRNA-treated cells (Figure 6C, i and ii). Uptake of labeled FITC-collagen was reduced by 45% in NMMIIA KND cells (p < 0.05; Figure 6C). The amount of compacted collagen was reduced in NMMIIA KND cells compared with cells transfected with control siRNA (p < 0.01; Figure 6E, i and ii). Collectively these data and the interaction of FliI with NMMIIA suggest that these proteins act together in collagen remodeling.
FIGURE 6:

Effect of NMMIIA on cell extension formation. (A) Cells treated with NMMIIA siRNA show reduced levels of NMMIIA compared with nontransfected cells (NT) or cells transfected with control siRNA. (B) Knockdown of NMMIIA reduces formation of cell extensions. (C) (i, ii) FliI OE cells transfected with FliI siRNA show reduced numbers and lengths of filopodial extensions compared with control siRNA–transfected cells. Data in histogram are from three different experiments. Observations recorded on 50 cells in each group presented as mean ± SD using ANOVA. #p < 0.01, comparison of cell extensions; #p < 0.05, comparison of length of cell extensions between NMMIIA siRNA and control siRNA. (D) Knockdown of NMMIIA inhibits collagen uptake. Cells were plated on FITC-labeled collagen, and cell lysates were measured fluorimetrically. Data presented in the histogram show decreased collagen internalization in siRNA-treated cells (#p < 0.05, with respect to control siRNA cells). Data are reported as mean ± SD, n = 5, analyzed by ANOVA. (E) (i, ii) Comparison of compacted collagen in FliI OE cells transfected with NMMIIA siRNA or control siRNA. Cells were plated on FITC-labeled collagen for 4 h. (ii) Quantification shows that knockdown of NMMIIA in FliI OE cells reduces collagen compaction.
Expression of the myofibroblast marker α−smooth muscle actin by fibroblasts is positively associated with collagen remodeling by contraction (Gabbiani, 2003). Accordingly, we examined the expression levels of α−smooth muscle actin in FliI OE, WT, and KND cells (Supplemental Figure S1C) and measured collagen gel contraction (Supplemental Figure S1D). There was no difference in expression levels of α−smooth muscle actin between the different cell types, but collagen gel contraction was reduced in FliI KND cells compared with FliI WT or OE cells.
Function of GLD 1 in FliI
When FliI KND cells were transfected with HA-tagged FliI GLD 2–6 and plated on collagen, this truncated FliI did not colocalize with NMMIIA (Pearson r of FliI-NMMIIA colocalization coefficient = 0.41 for GLD 2-6/NMMIIA and 0.78 for GLD 1-6/NMMIIA; Figure 7A, i and ii; p < 0.01). Further, the truncated FliI GLD 2–6 transfected in FliI KND cells formed fewer cell extensions when compared with WT cells that were transfected with the HA-tagged FliI GLD 1–6 construct (p < 0.01; Figure 7Aiii). We immunoblotted the transfected cells to confirm the expression of the respective constructs in FliI KND and WT cells (Figure 7Aiv). In vitro pull-down binding assays showed reduced binding of GST-tagged FliI 2–6 to purified NMMIIA rods (kD = 0.858 μM; Figure 7B, i and ii).
FIGURE 7:

Role of GLD domain 1 in cell function. (A) (i) FliI KND cells transfected with HA-FliI GLD 2–6 show reduced numbers of cell extensions and greatly diminished colocalization of FliI and NMMIIA. Cells transfected with HA-GLD 1–6 show colocalization of FLiI and NMMIIA at cell attachment sites. (ii) Pearson's correlation was applied to image pairs of FliI and NMMIIA immunostained cells to estimate colocalization of these proteins using ImageJ. Data in histogram show 50% reduction of colocalization of FLiI and NMMIIA in FliI KND cells transfected with FliI GLD 2–6 compared with FliI KND cells transfected with FliI GLD 1–6. Data are reported as mean ± SD and analyzed by ANOVA from observations made on image pairs of 30 cells. **p < 0.01, FliI KND cells transfected with FliI GLD 2–6 compared with WT cells (FLiI GLD 1–6). (iii) Quantification of cell extensions in KND and WT cells transfected with HA-tagged FliI GLD 2–6 and GLD 1–6, respectively (p < 0.01). (iv) Immunoblot shows expression levels of transfected HA-tagged FliI GLD 1–6 and GLD 2–6 in WT and FliI KND cells, respectively. (B) Quantification of FliI binding to NMMIIA shows reduced binding of Sepharose GST FliI GLD 2–6 to purified NMMIIA (kD = 0.8584 μM). (C) (i, ii) Untreated FliI KND cells or FliI KND cells expressing FliI LRR-GLD 2–6 plated on FITC-collagen substrate for 4 h exhibit substantial reduction of substrate clearance (i.e., collagen uptake) compared with FliI OE cells. Quantification of substrate fluorescence is presented in histogram as mean ± SD, n = 3, analyzed by ANOVA; **p < 0.01, comparison of FliI KND and FliI KND cells transfected with LRR-GLD 2–6 cells to FliI OE cells. Batimastat (10 μM) treatment did not alter FITC-collagen substrate clearance. (D) Fluorimetric measurements of cell lysates with equivalent cell protein (inset) show that FliI LRR-GLD 2-6 expression does not enable collagen internalization. KND and FliI LRR-GLD 2–6 cells show reduced collagen internalization compared with FliI OE cells. Experiment repeated three times; data presented in histogram as mean ± SD using ANOVA. *p < 0.05, comparison of KND and FliI LRR-GLD 2–6 to FliI OE cells. (E) (i, ii) Images and quantification show that absence of FliI GLD 1 attenuates collagen compaction.
We examined substrate clearance in stable cell lines expressing LRR-GLD 2–6 by plating cells on FITC-collagen for 4 h and measuring substrate clearance (Arora et al., 2008a). Compared to WT FliI cells, FliI LRR-GLD 2–6 shows twofold reduction in collagen substrate clearance (p < 0.01), and treatment with the MMP inhibitor batimastat did not affect collagen clearance (Figure 7C, i and ii). We also examined collagen internalization by fluorometric measurement of cell lysates with equivalent amounts of cell protein (Figure 7D, inset). There was enhanced FITC-collagen internalization by FliI OE cells compared with FliI KND and FliI LRR-GLD 2–6 (p < 0.05; Figure 7D).
We assessed the contribution of FliI GLD 1 to collagen compaction. The fluorescence intensity and the area of the compacted collagen was 35% less in FliI LRR-GLD 2–6–expressing cells than with cells expressing WT FliI (p < 0.05; Figure 7E, i and ii). Collectively our data suggest that in the absence of GLD 1, FliI does not interact efficiently with NMMIIA and thus fails to colocalize at collagen adhesion sites, which in turn affects collagen remodeling.
Actin filament networks control the shape and internal organization of cells, and one of the first steps in assembly of networks is actin nucleation and capping. On the basis of the actin-filament-capping activity of gelsolin and its amino acid sequence (Sun et al., 1994; Allen et al., 1996), we considered that the actin-capping function of FliI may be associated with GLD 1. We previously showed the capping function of FLiI in vitro (Mohammad et al., 2012), and we used similar experiments to determine the role of FliI GLD 1 in actin capping. Purified FliI GLD 1–6 or GLD 2–6 protein was incubated with pyrene-actin monomers (2 μM) in G-buffer (Materials and Methods) followed by addition of polymerization buffer. In the presence of FliI GLD 1–6, the final pyrene fluorescence was lower than the final fluorescence of 2 μM actin alone (p < 0.001). In contrast, FliI GLD 2–6 did not affect pyrene fluorescence, indicating that FliI GLD 2–6 did not cap actin filaments (Supplemental Figure S1E). These data suggest the importance of GLD 1 in FliI-dependent actin capping or that possibly GLD 1 may regulate the folding or exposure of sites in FliI GLD 2–6 that are required for its activity.
DISCUSSION
Previous data showed that FliI regulates mammalian wound healing and cell adhesion (Cowin et al., 2007; Kopecki et al., 2009; Mohammad et al., 2012), but how FliI regulates matrix remodeling is not defined. To obtain a better understanding of the role of FliI in matrix remodeling, we developed stable cell lines that exhibited distinct phenotypes depending on FliI expression levels. We examined the ability of these cells to form extensions and remodel collagen. Our data show that GLD 1 of FliI may be important for FliI function through its binding to NMMIIA, which is in turn important for collagen compaction and remodeling. Immunostaining showed colocalization of NMMIIA with FliI at cell extensions, and in vitro studies with purified proteins indicated a direct interaction of NMMIIA with the GLD 1 of FliI. We proposed earlier that FliI promotes cell interactions with the extracellular matrix and improves anchorage (Mohammad et al., 2012). At collagen adhesion sites, FliI interacts with NMMIIA, where activation may increase the contractile forces required for collagen remodeling. Here we show that FliI may regulate the formation of actin-enriched cell extensions, independent of other proteins, to provide cell-generated contractile forces at cell–substrate adhesions that lead to compaction of collagen (Figure 8).
FIGURE 8:
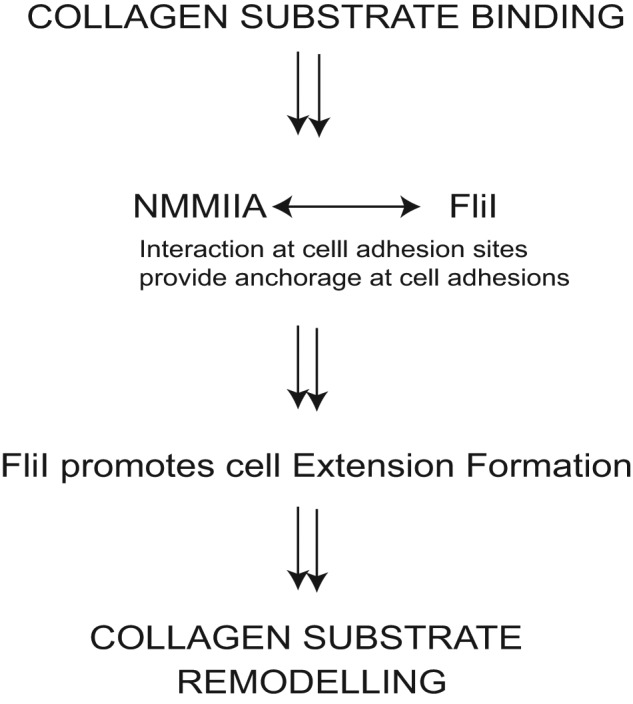
Schematic diagram indicates that at collagen adhesion sites, FliI helps to promote cell interactions with the extracellular matrix and provides anchorage. At cell adhesion sites, FliI interacts with NMMIIA. Phosphorylation of MLC (Ser-19) of NMMIIA is involved in contractile force generation, which results in collagen remodeling. FliI capping function regulates the actin-enriched filopodial cell extensions to provide cell-generated contractile forces at cell–substrate adhesions, which lead to compaction of collagen fibrils.
FliI expression and filopodial extensions
The actin cytoskeleton plays a major role in the ability of cells to form filopodial extensions and cell adhesions and is critically important for the generation of traction forces that are involved in the remodeling of collagen fibrils (Le Clainche and Carlier, 2008). We showed earlier that FliI is an actin-capping protein that localizes to cell adhesions and regulates the speed of cell migration (Mohammad et al., 2012). Capping proteins are important for regulating the availability of free barbed ends to control actin assembly (Edwards et al., 2014), and FliI is believed to act as a mediator for the mammalian formin proteins Daam1 and mDia1 to promote actin assembly (Higashi et al., 2010). Here we show that cells expressing high levels of FliI exhibited actin-rich cell extensions, which may be important for collagen remodeling, since small alterations of the concentrations and functional activity of actin-capping proteins like FliI can profoundly affect the formation of cell protrusions and possibly adhesion to collagen (Kopecki et al., 2007; Mohammad et al., 2012).
FliI belongs to the gelsolin family of actin-binding proteins (Campbell et al., 1993) and, like gelsolin, consists of six domains. Each domain has binding motifs that allow interactions with other proteins for cellular functions. In this context, gelsolin can act as a potent modulator of actin filament restructuring (Kwiatkowski, 1999). The six domains of gelsolin-like proteins act either singularly or in concert with other domains to regulate actin severing, capping, and monomer nucleation. Actin monomer–binding sites have been localized to domains 1 and 4–6; each domain is involved in various aspects of actin capping and actin nucleation (Kwiatkowski et al., 1986, 1989; Way et al., 1989). A third actin-binding site involves domains 2 and 3, which enables binding to the side of actin filaments and is involved with actin filament severing (Pope et al., 1991). Of note, cells expressing FliI that lacked domain 1 phenocopied FliI KND cells, since they did not efficiently form filopodial extensions. In vitro, this truncated protein exhibited reduced actin capping. Actin capping enables the growth of actin filaments, which is required for generation of protrusive structures (Wear and Cooper, 2004). Further, capping proteins are important regulatory proteins involved in controlling stochastic dynamics of cell extensions used by eukaryotic cells to probe the environment (Zhuravlev and Papoian, 2009).
From reconstruction of z-axis confocal microscopy images of three-dimensional collagen matrices, we found that cells expressing high levels of FliI extended finger-like projections through membrane pores coated with collagen. A large number of cell types, including monocytes, endothelial cells, smooth muscle cells, and certain types of cancer cells, develop actin-rich adhesions (podosomes and invadopodia; Linder and Aepfelbacher, 2003) from 0.5 μm to several micrometers in diameter. These structures contain adhesion plaque proteins such as vinculin and talin and are also enriched in integrins, enabling them to form functional bridges between the actin cytoskeleton and the extracellular matrix (Linder and Aepfelbacher, 2003; Buccione et al., 2004; Gimona et al., 2008). Of note, the N-WASP-Arp2/3 complex and cofilin are involved in the formation of invadopodia (Yamaguchi et al., 2005). We found that filopodial extensions expressed in FliI OE cells rapidly cleared collagen from substrates adjacent to cell extensions, and these cells also exhibited increased uptake and pericellular degradation of exogenous collagen. Because the level of FliI expression was positively associated with exogenous collagen uptake, degradation, and compaction, we suggest that FliI is an important mediator required for activation of NMMIIA, which controls collagen remodeling. These data are consistent with previous data showing that FliI levels are increased in wound-healing environments with high levels of matrix degradation (Kopecki and Cowin, 2008; Kopecki et al., 2009).
Despite marked morphological differences, it is believed that cells grown on two-dimensional (2D) substrates exhibit similar cytoskeletal reorganization and shape changes to cells in three-dimensional matrices (Martins and Kolega, 2006). On 2D planar substrates, spreading cells can spontaneously change their shape and cytoskeletal organization between three prototypical forms: round, spiky, or ruffled (Kabaso et al., 2011). In spreading cells, membrane-associated proteins (e.g., WASP, WAVE; Takenawa and Suetsugu, 2007) mediate the generation of filopodial protrusions at the leading edge as a result of actin assembly (Mullins et al., 1998; DeMali and Burridge, 2003). Of note, fascin and Myo10 have been implicated in the clustering of actin filaments into nascent filopodia (Bohil et al., 2006; Gupton and Gertler, 2007). We found that Myo10 is targeted to filopodial tips in cells expressing higher levels of FliI, which is consistent with earlier data showing localization of Myo10 at the tips of actin-rich bundles in spreading cells (Zhang et al., 2004). Myo10 contributes to filopodial dynamics and acts as mobile linker between the actin cytoskeleton and discrete signal transduction pathways (Berg et al., 2000; Berg and Cheney, 2002); it may also serve as a metastatic engine for invasion (Arjonen et al., 2014). Similar to our data on Myo10 localization, we found that fascin was enriched in filopodial cell extensions, which is in line with earlier observations that fascin localizes to the distal tips of filopodia and is also required for promoting assembly and growth of actin filaments (Vignjevic et al., 2006; Zanet et al., 2012). Our immunostaining experiments of Myo10 at the tips of filopodia authenticate the nature of the cell extensions seen in FliI-overexpressing cells.
Treatment of cells with ML-141, an inhibitor of Cdc42 activation (Surviladze et al., 2010), blocked the development of extensions in FliI OE and WT but not FliI KND cells. Further, constitutively active Cdc42 failed to induce filopodial extensions in FliI KND cells. Taken together, these data suggest that FliI is an important regulator of filopodial formation that is downstream of Cdc42. Of note, FliI cooperates with Rho to regulate the actin assembly activity of diaphanous-related formins Daam1 and mDia1 (Higashi et al., 2010), which is particularly important in stress fiber formation but may not play a direct role in cell extension formation and collagen remodeling.
Interaction of FliI with NMMIIA
The association of NMMIIA with FliI was demonstrated by immunoprecipitation and mass spectrometry. Earlier data indicated that NMMIIA, an actin-binding protein, functionally links actin filaments to integrins and is involved in the formation and maturation of cell adhesions (Galbraith et al., 2002; Vicente-Manzanares et al., 2007). Although myosin superfamily motor proteins interact with actin filaments to generate contractile forces (Liu et al., 2014), they may also act as adapter proteins and play critical roles in a variety of cell processes. For example, NMMIIA is necessary not only for E-cadherin–mediated intercellular adhesion and mechanical adhesion, but also for stability of p120 catenin at the intercellular junctions of human embryonic stem cells (Li et al., 2010). In this context, we found that treatment with blebbistatin inhibited collagen compaction and that knockdown of NMMIIA attenuated cell extension formation, which reduced collagen internalization and compaction. Further, knockdown of FliI prevented targeting of NMMIIA to filopodial extensions. Consistent with this observation, in vitro studies showed weaker interactions of FLiI GLD 2–6 with NMMIIA than with FliI GLD 1–6, which may account for the reduced colocalization of FliI GLD 2–6 with NMMIIA in transfected cells.
Previously we showed that FliI helps to promote cell interactions with the extracellular matrix and provides anchorage (Mohammad et al., 2012). Our new data show that at adhesion sites to collagen, FliI interacts with NMMIIA, where phosphorylation of MLC at Ser-19 promotes contractile forces required for collagen remodeling. FliI capping function may regulate the actin-enriched filopodial cell extension formation independently of or in cooperation with other proteins. Collectively our data indicate that FliI interacts with NMMIIA to provide a platform at adhesion sites for actin filament elongation, cell extension formation, and collagen remodeling by compaction and phagocytosis.
MATERIALS AND METHODS
Reagents
Rabbit monoclonal antibody to FliI was from Epitomics (Burlington, CA). A blocking antibody to the α2β1 integrin and a rat anti-mouse CD29 (KMI6) antibody were purchased from BD Biosciences (Mississauga, Canada). Protease inhibitor cocktail, the broad-spectrum MMP inhibitor batimastat, nebulin antibody, RIPA buffer, β-actin (clone AC-15), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody, Myo10 antibody, and FITC-conjugated goat anti-mouse antibody and tetra-methyl rhodamine isothiocyanate-phalloidin were obtained from Sigma-Aldrich (Oakville, Canada). Mouse monoclonal anti–influenza A virus HA antibody (MMS-101P) and Cy3-conjugated streptavidin were purchased from Cedarlane (Burlington, Canada). Blebbistatin was obtained from Calbiochem (San Diego, CA). FITC-labeled, bovine type I collagen (5 mg/ml) was purchased from AnaSpec (Fremont, CA). Pyrene-labeled actin, rabbit skeletal muscle actin, and plasma gelsolin were purchased from Cytoskeleton (Denver, CO). MLC p19(ser) was purchased from Cell Signaling (Beverly, MA). Rabbit antibody to type I collagen one-fourth fragment (IAGQRGGC immunizing peptide) was from J. Mort and E. Lee (Shriners Hospital for Children, Montreal, Canada). Affinity-purified rabbit antibody to type I collagen three-fourths cleavage site was from ImmunoGlobe (Himmelstadt, Germany). High-capacity streptavidin agarose resin was purchased from Thermo Scientific, Rockford, IL. Paramagnetic beads (1 μm) were purchased from Bangs Laboratories (Fishers, IN). Sulfo-NHS-Biotin was purchased from Thermo Scientific. Glutathione Sepharose 4B beads were purchased from GE Healthcare, Biosciences, Uppsala, Sweden.
Cell culture
FliI cell lines were maintained in culture with appropriate selection reagents. Cells were cultured at 37°C in complete DMEM as described (Arora et al., 2008b).
Isolation of bead-associated proteins and immunoprecipitation
These methods were described earlier (Arora et al., 2008a). Briefly, collagen bead–associated proteins were isolated with magnetic, collagen-coated beads, and samples were normalized by bead counts. For immunoprecipitation, FliI-overexpressing and WT and KND cells were plated on collagen for 1 h, and washed samples were scraped into RIPA buffer (Sigma-Aldrich). The supernatants of centrifuged samples were incubated with FliI antibody (Epitomics) or NMMIIA antibody, followed by addition of equivalent amounts of protein A–Sepharose beads (Pierce, Thermo Scientific). Immunoblotted samples were probed with appropriate antibodies and secondary antibodies and blots imaged with a Li-Cor imaging system (Mandel, Toronto, Canada).
Mass spectrometry
The method has been described earlier (Mohammad et al., 2012). Briefly, cells were lysed on ice, and supernatant from centrifuged samples was incubated with FliI antibody for 1 h, followed by addition of protein A–Sepharose beads. Proteins associated with beads were eluted with 50 mM glycine buffer (pH, 2.3–2.5). The pH of the eluted proteins was increased to physiological pH by dialysis for 36 h in carbonate buffer (25 mM NH4HCO3, pH 7.5). The dialyzed samples were treated with trypsin (0.1 μg/μl; Roche, Indianapolis, IN) and rotated overnight at 37°C. Subsequently, 0.1% acetic acid was added to the sample, followed by air drying with an evaporator. Lyophilized samples were analyzed by liquid chromatography–tandem mass spectrometry (Mass Spectrometry Facility, Hospital for Sick Children, Toronto, Canada).
Collagen remodeling, clearance, and internalization
Cells were plated on FITC-labeled collagen (1 mg/ml). Fibrillar collagen was prepared by adding the required amount to phosphate-buffered saline (PBS) and raising the pH to 7.5 by addition of NaOH; 1 ml was added to glass coverslips, and the excess was removed after 1 min. The coverslips were dried in sterile conditions before plating cells. Collagen compaction by cells was observed by confocal microscopy. For assessing collagen internalization and clearance, cells were plated on FITC-collagen (1 mg/ml) for 4 h. Collagen clearance was observed by fluorescence microscopy and quantified by ImageJ (National Institutes of Health, Bethesda, MD). To determine the amount of internalized collagen, cells were plated on FITC-collagen for 4 h, followed by trypsinization and washed with PBS. Cell pellets were suspended in lysis buffer, and internalized collagen was quantified by spectrofluorimetry (PhotonTechnology, London, Canada).
Biotinylated collagen and intracellular degraded collagen
Biotinylated collagen was prepared as described (Arora et al., 2000) using rat-tail type I collagen. Briefly, rat-tail type I collagen was diluted in PBS (pH, 8.2) at 4°C to 2 mg/ml solution, which was used as the stock solution for experiments. Sulfo-NHS-Biotin (2 mg/ml) was dissolved in dimethyl sulfoxide) and added to collagen solution in stages with constant stirring at 4°C. For experiments, collagen was diluted to 1 mg/ml. Cell plated on biotinylated collagen were removed by trypsinization and lysed, followed by incubation with high-capacity streptavidin agarose resin (Thermo Scientific). Boiled samples were separated on SDS–PAGE and blots probed with antibodies to three-fourths and one-fourth collagen fragments.
Purification of FliI recombinant proteins
The protocol for purification of FliI recombinant proteins has been described (Mohammad et al., 2012) and was adapted from earlier methods (Frangioni and Neel, 1993; Li et al., 2008).
Pull-down assays
Pull-down assays have been described (Mohammad et al., 2012). Briefly, recombinant FliI proteins bound to Sepharose beads (GST-GLD 1–6, GST-GLD 2–6, or GST-LRR) were incubated with purified myosin rods (from A.B.). The samples were centrifuged (14,000 × g for 5 min); supernatants were removed, and pellets containing Sepharose beads were washed 3× with PBS.
Actin-capping assay
Lyophilized actin (Cytoskeleton, Denver, CO) was resuspended in G-buffer (2 mM Tris, pH 8.0, 0.2 CaCl2, 0.2 ATP, 0.05 mM β-mercaptoethanol). To initiate actin capping, we treated G-actin (2 μM; 15% pyrenyl–G-actin) with polymerization buffer (25 mM Tris, pH 7.0, 50 mM KCl, 2 mM MgCl2, 0.1 mM ATP) and measured the increase in fluorescence in a fluorimeter (PTI, London, Canada; excitation, 365 nm; emission, 386 nm) overnight. To assess actin capping in FliI samples, we included varying concentrations of FliI GLD 1–6 and GLD 2–6 in the polymerization buffer. We used the actin filament–severing/capping protein gelsolin as a positive control.
Isolation of cell extensions
Because cells projected extensions through the pores in the membrane toward the fetal bovine serum gradient, we mechanically separated cell extensions (i.e., protrusions extending through the membrane to its underside) and cell bodies (i.e., cellular component retained on the upper side of the membranes). To avoid cross-contamination of cell body and cell extension fractions, cell bodies on the upper surface were manually removed by scraping, and the cell extensions on the underside were lysed with detergent. Similarly, cell extensions on the lower surface were removed manually, and cell bodies were extracted with detergent (Nguyen et al., 2000).
FliI fusion proteins
A previously described pCDNA3-HA vector (Li et al., 2008), which contains the cDNA fragment encoding GLD (amino acids [aa] 398–1272) of mouse FliI, was generously provided by J. Yuan (Department of Cell Biology, Harvard Medical School, Boston, MA). A primer pair (forward, 5′-GCGCGTCGACAAACCAGCTGAGGCTGGCGGGCGCCTCCCC-3′; reverse, 5′-GCGCGCGGCCGCCTATTAGGCTGGGGCTTGGCGGAACGTG-3′) was designed based on National Center for Biotechnology Information (NCBI) GenBank No. NM_022009.1 to amplify the SalI/NotI sites (underlined) that flanked the FliI GLD cDNA (2.625 kb). The resulting PCR product was digested with SalI/NotI, ligated into the corresponding sites of pGEX-4T-2, and transformed into BL21(DE3) pLysS–competent Escherichia coli cells for protein expression.
A primer pair (forward, 5′-GGTTCCGCGTGGATCCACCAGGATGTACCGTGTAT-3′; reverse, 5′-GATGCGGCCGCTCGAGTCATTAGGCTGGGGCTTG-3′) was designed to amplify GLDs 2–6 of FliI cDNA (1.965 kb, aa 619–1271; pfu DNA Polymerase; Stratagene, Santa Clara, CA). The resulting PCR product was ligated to BamHI/XhoI linearized pGEX-4T-2 using In-fusion HD enzyme premix (Clontech).
GFP FliI
A primer pair (forward, 5′-GCGCCTCGAGAAATGGAGGCCACCGGGGTGCTGCCGTTCG-3′; reverse, 5′-GCGCGGATCCCTATTAGGCTGGGGCTTGGCGGAACGTGCT-3′) was designed based on NCBI GenBank No. NM_022009.1 to create XhoI/BamHI sites (underlined) flanking the FliI full-length cDNA (3.816 kb). The resulting PCR product was digested with XhoI/BamHI, ligated into the corresponding sites of pEGFP-C1, and sequenced (ACGT, Toronto, Canada).
Subcloning FliI-LRR into pCMV-HA
A primer pair (forward, 5′-CGGTCGACCGAGATCTACGCCACCATGGAGGCCACCGGG-3′; reverse, 5′-TGGATCCCCGCGGCCGCCTATTAGGCATCTATGCTTTCCT-3′) was designed based on NCBI GenBank No. NM_022009.1 to amplify the LRR region of FliI cDNA (1.398 kb, aa 1–466; Phusion Hot Start High-Fidelity DNA Polymerase; New England Biolabs, Whitby, ON, Canada), which extends past the end of the LRR repeat at amino acid 380 into part of the linker between the LRR-and the gelsolin-like domains (Liu and Yin, 1998). The resulting PCR product was ligated into BglII/NotI-linearized pCMV-HA (Clontech) using In-fusion HD enzyme premix and sequenced (ACGT).
FliI shRNA–resistant construct
Preparation of FLiI stable knockdown cell lines has been described (Mohammad et al., 2012). To rescue FliI KND cells, the FliI shRNA targeted sequence 5′-GAAGATACACACTATGTTA-3′ was mutated to 5′-GAgGAcACtCACTAcGTcA-3′, which is not destroyed in FliI KND cells. The QuikChange Lightning Site-Directed Mutagenesis Kit (210518; Stratagene) and a modified protocol (Liu and Naismith, 2008) were used to generate the silent mutation in which the HA-tagged mouse FliI in pcDNA (Li et al., 2008) was used as the template. Briefly, a primer pair (forward, 5′-ATACTGTGGAGGACACTCACTACGTCACCAGGATGTACCGTGTATATGGGAAAA-3′; reverse, 5′-CCTGGTGACGTAGTGAGTGTCCTCCACAGTATAGAAGCCACTGGCTGTTC-3′) was designed according to GenBank No. NM_022009.1. The PCR product was digested with 1 μl of DpnI and transformed to the StellarTM-competent cells. The mutations (nucleotides [nt] a1839g, t1842c, a1845t, t1851c, and t1854c) were confirmed by sequencing (ACGT).
Stable cell line overexpressing FliI
A primer pair (forward, 5′-GCGCGCGGCCGCCGCCACCATGGAGGCCACCGGGGTGCTG-3′, reverse, 5′-GCGCGTCGACCTATTAAGCATAGTCGGGCACATCGTACGG-3′) was designed according to GenBank No. NM_022009.1 and CY103298.1 to create and amplify the NotI/SalI sites (underlined), HA-tagged, full-length FliI cDNA, using pCDNA3-FliI-HA as template. The resulting PCR product was ligated into a retroviral vector pLNCX2-N1 (Clontech) and digested with NotI/SalI. The construct was cotransfected with pVSV-G (Clontech) into GP-293 cells (Clontech; provided by H. Sarantis and S. D. Gray-Owen, University of Toronto, Toronto, ON, Canada) for retrovirus production.
Stable cell lines expressing HA-tagged FliI LRR-GLD 2–6 or GLD 2–6
Forward (5′-CTCGGAGTTCGCCAGTGGCTTCTATACTGTGGAAGATACACACT-3′) and reverse (5′-ACTGGCGAACTCCGAGTAGTCAAGGCGTGGCTTCTCCAGGCCCT-3′) primers were designed based on NCBI GenBank No. NM_022009.1 to delete (Phusion high-fidelity DNA polymerase; New England Biolabs) FliI GLD 1 (nt 1471–1815, aa 491–605) in pcDNA3-FliI-HA. The resulting HA-tagged FliI LRR-GLD 2–6 was subcloned into HpaI/EcoRI linearized pMSCVpuro using In-fusion HD enzyme premix (Clontech) and sequenced (ACGT).
To obtain HA-tagged FliI GLD 2–6 (2.011 kb, including ATG transcription start codon, aa 619–1271-HA), a primer pair (forward, 5′-AGATCTCTCGAGGTTAACCGCCACCATGACCAGGATGTACCGTG-3′; reverse, 5′-CTACCCGGTAGAATTCTCATTAAGCATAGTCGGGC-3′) was designed based on NCBI GenBank No. NM_022009.1 to perform PCR (Phusion high-fidelity DNA polymerase) by using pcDNA3-FliI-HA as template. The resulting PCR product was ligated into HpaI/EcoRI linearized pMSCVpuro using In-fusion HD enzyme premix and sequenced (ACGT).
Retroviruses prepared using the foregoing two constructs were used to infect FliI KND cells, respectively. Puromycin-resistant (10 μg/ml) cells were selected.
Preparation of myosin rods
Myosin rods were provided by A.B. as frozen samples stored in buffer consisting of 20 mM Tris, pH 7.5, 0.6 M NaCl, 1 mM dithiothreitol (DTT), and 0.02% NaN3. The proteins were thawed and centrifuged at 200,000 × g for 30 min, and the supernatant was dialyzed against assembly buffer containing 5 mM 1,4-piperazinediethanesulfonic acid, pH 6.5, 20 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 0.02% NaN3 overnight. Filamentous rods were pelleted in a microcentrifuge at 6000 × g for 10 min before binding assays. Protein concentrations were confirmed by bicinchoninic acid assay and checking OD280 of dialyzed samples after addition of 0.6 M NaCl.
Statistical analysis
For all continuous variable data, means, SDs, and SEMs were computed. When appropriate, comparisons between two samples were made by Student's t test with statistical significance set at p < 0.05. For multiple comparisons, analysis of variance (ANOVA) was used, followed by Tukey's test for assessment of individual differences. All experiments were performed at least three times in triplicate. In figures, statistical analyses are illustrated and show comparisons of cells with either WT or FliI KND cells contrasted with FliI-overexpressing cells, as these comparisons enabled the most clear-cut demonstration of the effect of FliI on various cell measures.
Supplementary Material
Acknowledgments
This work was supported by a Canadian Institute of Health Research operating grant to C.A.M. (MOP-36332). C.A.M. is supported by a Canada Research Chair (Tier1). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations used:
- DDR1
discoidin domain receptor
- DTT
dithiothreitol
- FITC
fluorescein isothiocyanate
- FliI
flightless I
- GLD
gelsolin-like domain
- HA
hemagglutin
- KND
knockdown
- LRR
leucine-rich region
- NMMIIA
nonmuscle myosin IIA
- OE
overexpressing
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-11-1536) on April 15, 2015.
REFERENCES
- Alexandrova AY, Arnold K, Schaub S, Vasiliev JM, Meister JJ, Bershadsky AD, Verkhovsky AB. Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS One. 2008;3:e3234. doi: 10.1371/journal.pone.0003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen PG, Laham LE, Way M, Janmey PA. Binding of phosphate, aluminum fluoride, or beryllium fluoride to F-actin inhibits severing by gelsolin. J Biol Chem. 1996;271:4665–4670. doi: 10.1074/jbc.271.9.4665. [DOI] [PubMed] [Google Scholar]
- Archer SK, Behm CA, Claudianos C, Campbell HD. The flightless I protein and the gelsolin family in nuclear hormone receptor-mediated signalling. Biochem Soc Trans. 2004;32:940–942. doi: 10.1042/BST0320940. [DOI] [PubMed] [Google Scholar]
- Arjonen A, Kaukonen R, Mattila E, Rouhi P, Hognas G, Sihto H, Miller BW, Morton JP, Bucher E, Taimen P, et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J Clin Invest. 2014;124:1069–1082. doi: 10.1172/JCI67280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Conti MA, Ravid S, Sacks DB, Kapus A, Adelstein RS, Bresnick AR, McCulloch CA. Rap1 activation in collagen phagocytosis is dependent on nonmuscle myosin II-A. Mol Biol Cell. 2008a;19:5032–5046. doi: 10.1091/mbc.E08-04-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Manolson MF, Downey GP, Sodek J, McCulloch CA. A novel model system for characterization of phagosomal maturation, acidification, and intracellular collagen degradation in fibroblasts. J Biol Chem. 2000;275:35432–35441. doi: 10.1074/jbc.M003221200. [DOI] [PubMed] [Google Scholar]
- Arora PD, Marignani PA, McCulloch CA. Collagen phagocytosis is regulated by the guanine nucleotide exchange factor Vav2. Am J Physiol Cell Physiol. 2008b;295:C130–137. doi: 10.1152/ajpcell.00168.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Wang Y, Janmey PA, Bresnick A, Yin HL, McCulloch CA. Gelsolin and non-muscle myosin IIA interact to mediate calcium-regulated collagen phagocytosis. J Biol Chem. 2011;286:34184–34198. doi: 10.1074/jbc.M111.247783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows CG, Melcher AH, Aubin JE. Contraction and organization of collagen gels by cells cultured from periodontal ligament, gingiva and bone suggest functional differences between cell types. J Cell Sci. 1981;50:299–314. doi: 10.1242/jcs.50.1.299. [DOI] [PubMed] [Google Scholar]
- Berg JS, Cheney RE. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat Cell Biol. 2002;4:246–250. doi: 10.1038/ncb762. [DOI] [PubMed] [Google Scholar]
- Berg JS, Derfler BH, Pennisi CM, Corey DP, Cheney RE. Myosin-X, a novel myosin with pleckstrin homology domains, associates with regions of dynamic actin. J Cell Sci. 2000;113:3439–3451. doi: 10.1242/jcs.113.19.3439. [DOI] [PubMed] [Google Scholar]
- Block MR, Badowski C, Millon-Fremillon A, Bouvard D, Bouin AP, Faurobert E, Gerber-Scokaert D, Planus E, Albiges-Rizo C. Podosome-type adhesions and focal adhesions, so alike yet so different. Eur J Cell Biol. 2008;87:491–506. doi: 10.1016/j.ejcb.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Bohil AB, Robertson BW, Cheney RE. Myosin-X is a molecular motor that functions in filopodia formation. Proc Natl Acad Sci USA. 2006;103:12411–12416. doi: 10.1073/pnas.0602443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Pouwels J, De Franceschi N, Ivaska J. Integrin inactivators: balancing cellular functions in vitro and in vivo. Nat Rev Mol Cell Biol. 2013;14:430–442. doi: 10.1038/nrm3599. [DOI] [PubMed] [Google Scholar]
- Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- Cai Y, Biais N, Giannone G, Tanase M, Jiang G, Hofman JM, Wiggins CH, Silberzan P, Buguin A, Ladoux B, Sheetz MP. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J. 2006;91:3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell HD, Schimansky T, Claudianos C, Ozsarac N, Kasprzak AB, Cotsell JN, Young IG, de Couet HG, Miklos GL. The Drosophila melanogaster flightless-I gene involved in gastrulation and muscle degeneration encodes gelsolin-like and leucine-rich repeat domains and is conserved in Caenorhabditis elegans and humans. Proc Natl Acad Sci USA. 1993;90:11386–11390. doi: 10.1073/pnas.90.23.11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciobanasu C, Faivre B, Le Clainche C. Integrating actin dynamics, mechanotransduction and integrin activation: The multiple functions of actin binding proteins in focal adhesions. Eur J Cell Biol. 2013;92:339–348. doi: 10.1016/j.ejcb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Costa P, Scales TM, Ivaska J, Parsons M. Integrin-specific control of focal adhesion kinase and RhoA regulates membrane protrusion and invasion. PLoS One. 2013;8:e74659. doi: 10.1371/journal.pone.0074659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowin AJ, Adams DH, Strudwick XL, Chan H, Hooper JA, Sander GR, Rayner TE, Matthaei KI, Powell BC, Campbell HD. Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. J Pathol. 2007;211:572–581. doi: 10.1002/path.2143. [DOI] [PubMed] [Google Scholar]
- Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J Cell Sci. 2008;121:255–264. doi: 10.1242/jcs.006064. [DOI] [PubMed] [Google Scholar]
- Davy DA, Campbell HD, Fountain S, de Jong D, Crouch MF. The flightless I protein colocalizes with actin-and microtubule-based structures in motile Swiss 3T3 fibroblasts: evidence for the involvement of PI 3-kinase and Ras-related small GTPases. J Cell Sci. 2001;114:549–562. doi: 10.1242/jcs.114.3.549. [DOI] [PubMed] [Google Scholar]
- DeMali KA, Burridge K. Coupling membrane protrusion and cell adhesion. J Cell Sci. 2003;116:2389–2397. doi: 10.1242/jcs.00605. [DOI] [PubMed] [Google Scholar]
- Deng H, Xia D, Fang B, Zhang H. The Flightless I homolog, fli-1, regulates anterior/posterior polarity, asymmetric cell division and ovulation during Caenorhabditis elegans development. Genetics. 2007;177:847–860. doi: 10.1534/genetics.107.078964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- Dubin-Thaler BJ, Giannone G, Dobereiner HG, Sheetz MP. Nanometer analysis of cell spreading on matrix-coated surfaces reveals two distinct cell states and STEPs. Biophys J. 2004;86:1794–1806. doi: 10.1016/S0006-3495(04)74246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, Cooper JA. Capping protein regulators fine-tune actin assembly dynamics. Nat Rev Mol Cell Biol. 2014;15:677–689. doi: 10.1038/nrm3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts V, van der Zee E, Creemers L, Beertsen W. Phagocytosis and intracellular digestion of collagen, its role in turnover and remodelling. Histochem J. 1996;28:229–245. doi: 10.1007/BF02409011. [DOI] [PubMed] [Google Scholar]
- Fong KS, de Couet HG. Novel proteins interacting with the leucine-rich repeat domain of human flightless-I identified by the yeast two-hybrid system. Genomics. 1999;58:146–157. doi: 10.1006/geno.1999.5817. [DOI] [PubMed] [Google Scholar]
- Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshdastider U, Popp D, Burtnick LD, Robinson RC. The expanding superfamily of gelsolin homology domain proteins. Cytoskeleton (Hoboken) 2013;70:775–795. doi: 10.1002/cm.21149. [DOI] [PubMed] [Google Scholar]
- Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 2003;13:264–269. doi: 10.1016/s0962-8924(03)00057-6. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB. Filopodia: the fingers that do the walking. Sci STKE. 2007;2007:re5. doi: 10.1126/stke.4002007re5. [DOI] [PubMed] [Google Scholar]
- Hanein D, Horwitz AR. The structure of cell-matrix adhesions: the new frontier. Curr Opin Cell Biol. 2012;24:134–140. doi: 10.1016/j.ceb.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Ikeda T, Murakami T, Shirakawa R, Kawato M, Okawa K, Furuse M, Kimura T, Kita T, Horiuchi H. Flightless-I (Fli-I) regulates the actin assembly activity of diaphanous-related formins (DRFs) Daam1 and mDia1 in cooperation with active Rho GTPase. J Biol Chem. 2010;285:16231–16238. doi: 10.1074/jbc.M109.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Sci. 2013;126:2979–2989. doi: 10.1242/jcs.079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260:10027–10031. [PubMed] [Google Scholar]
- Kabaso D, Shlomovitz R, Schloen K, Stradal T, Gov NS. Theoretical model for cellular shapes driven by protrusive and adhesive forces. PLoS Comput Biol. 2011;7:e1001127. doi: 10.1371/journal.pcbi.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecki Z, Arkell R, Powell BC, Cowin AJ. Flightless I regulates hemidesmosome formation and integrin-mediated cellular adhesion and migration during wound repair. J Invest Dermatol. 2009;129:2031–2045. doi: 10.1038/jid.2008.461. [DOI] [PubMed] [Google Scholar]
- Kopecki Z, Cowin AJ. Flightless I: an actin-remodelling protein and an important negative regulator of wound repair. Int J Biochem Cell Biol. 2008;40:1415–1419. doi: 10.1016/j.biocel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Kopecki Z, Luchetti MM, Adams DH, Strudwick X, Mantamadiotis T, Stoppacciaro A, Gabrielli A, Ramsay RG, Cowin AJ. Collagen loss and impaired wound healing is associated with c-Myb deficiency. J Pathol. 2007;211:351–361. doi: 10.1002/path.2113. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ. Functions of gelsolin: motility, signaling, apoptosis, cancer. Curr Opin Cell Biol. 1999;11:103–108. doi: 10.1016/s0955-0674(99)80012-x. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Janmey PA, Yin HL. Identification of critical functional and regulatory domains in gelsolin. J Cell Biol. 1989;108:1717–1726. doi: 10.1083/jcb.108.5.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Stossel TP, Orkin SH, Mole JE, Colten HR, Yin HL. Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature. 1986;323:455–458. doi: 10.1038/323455a0. [DOI] [PubMed] [Google Scholar]
- Le Clainche C, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- Li J, Yin HL, Yuan J. Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J Cell Biol. 2008;181:321–333. doi: 10.1083/jcb.200711082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhou J, Wang L, Shin ME, Su P, Lei X, Kuang H, Guo W, Yang H, Cheng L, et al. Integrated biochemical and mechanical signals regulate multifaceted human embryonic stem cell functions. J Cell Biol. 2010;191:631–644. doi: 10.1083/jcb.201006094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Waters JM, Powell BC, Arkell RM, Cowin AJ. Decreased expression of Flightless I, a gelsolin family member and developmental regulator, in early-gestation fetal wounds improves healing. Mamm Genome. 2011;22:341–352. doi: 10.1007/s00335-011-9320-z. [DOI] [PubMed] [Google Scholar]
- Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YT, Yin HL. Identification of the binding partners for flightless I, a novel protein bridging the leucine-rich repeat and the gelsolin superfamilies. J Biol Chem. 1998;273:7920–7927. doi: 10.1074/jbc.273.14.7920. [DOI] [PubMed] [Google Scholar]
- Liu Z, Ho CH, Grinnell F. The different roles of myosin IIA and myosin IIB in contraction of 3D collagen matrices by human fibroblasts. Exp Cell Res. 2014;326:295–306. doi: 10.1016/j.yexcr.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen DH, Leonard D, Masedunskas A, Moyer A, Jurgensen HJ, Peters DE, Amornphimoltham P, Selvaraj A, Yamada SS, Brenner DA, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. 2013;202:951–966. doi: 10.1083/jcb.201301081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins GG, Kolega J. Endothelial cell protrusion and migration in three-dimensional collagen matrices. Cell Motil Cytoskeleton. 2006;63:101–115. doi: 10.1002/cm.20104. [DOI] [PubMed] [Google Scholar]
- Melcher AH, Chan J. Phagocytosis and digestion of collagen by gingival fibroblasts in vivo: a study of serial sections. J Ultrastruct Res. 1981;77:1–36. doi: 10.1016/s0022-5320(81)80064-0. [DOI] [PubMed] [Google Scholar]
- Mohammad I, Arora PD, Naghibzadeh Y, Wang Y, Li J, Mascarenhas W, Janmey PA, Dawson JF, McCulloch CA. Flightless I is a focal adhesion-associated actin-capping protein that regulates cell migration. FASEB J. 2012;26:3260–3272. doi: 10.1096/fj.11-202051. [DOI] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Larsson M, Robinson RC, Burtnick LD. Gelsolin: the tail of a molecular gymnast. Cytoskeleton (Hoboken) 2013;70:360–384. doi: 10.1002/cm.21117. [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Wang HJ, Zalzal S, Nanci A, Nabi IR. Purification and characterization of beta-actin-rich tumor cell pseudopodia: role of glycolysis. Exp Cell Res. 2000;258:171–183. doi: 10.1006/excr.2000.4929. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Panwar P, Du X, Sharma V, Lamour G, Castro M, Li H, Bromme D. Effects of cysteine proteases on the structural and mechanical properties of collagen fibers. J Biol Chem. 2013;288:5940–5950. doi: 10.1074/jbc.M112.419689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroll WM, Ma L. Direct, dynamic assessment of cell-matrix interactions inside fibrillar collagen lattices. Cell Motil Cytoskeleton. 2003;55:254–264. doi: 10.1002/cm.10126. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Pope B, Way M, Weeds AG. Two of the three actin-binding domains of gelsolin bind to the same subdomain of actin. Implications of capping and severing mechanisms. FEBS Lett. 1991;280:70–74. doi: 10.1016/0014-5793(91)80206-i. [DOI] [PubMed] [Google Scholar]
- Schiller HB, Fassler R. Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 2013;14:509–519. doi: 10.1038/embor.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stossel TP, Chaponnier C, Ezzell RM, Hartwig JH, Janmey PA, Kwiatkowski DJ, Lind SE, Smith DB, Southwick FS, Yin HL, et al. Nonmuscle actin-binding proteins. Annu Rev Cell Biol. 1985;1:353–402. doi: 10.1146/annurev.cb.01.110185.002033. [DOI] [PubMed] [Google Scholar]
- Sun HQ, Wooten DC, Janmey PA, Yin HL. The actin side-binding domain of gelsolin also caps actin filaments. Implications for actin filament severing. J Biol Chem. 1994;269:9473–9479. [PubMed] [Google Scholar]
- Surviladze Z, Waller A, Strouse JJ, Bologa C, Ursu O, Salas V, Parkinson JF, Phillips GK, Romero E, Wandinger-Ness A, et al. Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information; 2010. A potent and selective inhibitor of Cdc42 GTPase. Available at http://www.ncbi.nlm.nih.gov/books/NBK51965/ (accessed November 2014) [PubMed] [Google Scholar]
- Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- Tamariz E, Grinnell F. Modulation of fibroblast morphology and adhesion during collagen matrix remodeling. Mol Biol Cell. 2002;13:3915–3929. doi: 10.1091/mbc.E02-05-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Horwitz AR. Adhesion dynamics at a glance. J Cell Sci. 2011;124:3923–3927. doi: 10.1242/jcs.095653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignjevic D, Kojima S, Aratyn Y, Danciu O, Svitkina T, Borisy GG. Role of fascin in filopodial protrusion. J Cell Biol. 2006;174:863–875. doi: 10.1083/jcb.200603013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Klemke RL. Biochemical purification of pseudopodia from migratory cells. Methods Mol Biol. 2007;370:55–66. doi: 10.1007/978-1-59745-353-0_5. [DOI] [PubMed] [Google Scholar]
- Way M, Gooch J, Pope B, Weeds AG. Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J Cell Biol. 1989;109:593–605. doi: 10.1083/jcb.109.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wear MA, Cooper JA. Capping protein: new insights into mechanism and regulation. Trends Biochem Sci. 2004;29:418–428. doi: 10.1016/j.tibs.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Wehrle-Haller B. Structure and function of focal adhesions. Curr Opin Cell Biol. 2012;24:116–124. doi: 10.1016/j.ceb.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- Zanet J, Jayo A, Plaza S, Millard T, Parsons M, Stramer B. Fascin promotes filopodia formation independent of its role in actin bundling. J Cell Biol. 2012;197:477–486. doi: 10.1083/jcb.201110135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Berg JS, Li Z, Wang Y, Lang P, Sousa AD, Bhaskar A, Cheney RE, Stromblad S. Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat Cell Biol. 2004;6:523–531. doi: 10.1038/ncb1136. [DOI] [PubMed] [Google Scholar]
- Zhuravlev PI, Papoian GA. Molecular noise of capping protein binding induces macroscopic instability in filopodial dynamics. Proc Natl Acad Sci USA. 2009;106:11570–11575. doi: 10.1073/pnas.0812746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.