

Abstract
The cell surface is the essential interface between a bacterium and its surroundings. Composed primarily of molecules that are not directly genetically encoded, this highly dynamic structure accommodates the basic cellular processes of growth and division as well as the transport of molecules between the cytoplasm and the extracellular milieu. In this review, we describe aspects of bacterial growth, division and secretion that have recently been uncovered by metabolic labeling of the cell envelope. Metabolite derivatives can be used to label a variety of macromolecules, from proteins to non-genetically-encoded glycans and lipids. The embedded metabolite enables precise tracking in time and space, and the versatility of newer chemoselective detection methods offers the ability to execute multiple experiments concurrently. In addition to reviewing the discoveries enabled by metabolic labeling of the bacterial cell envelope, we also discuss the potential of these techniques for translational applications. Finally, we offer some guidelines for implementing this emerging technology.
Keywords: metabolic labeling, click chemistry, bioorthogonal, peptidoglycan, glycolipid, protein secretion
We review aspects of bacterial growth, division and secretion that have recently been uncovered by metabolic labeling of the cell surface.
We review aspects of bacterial growth, division and secretion that have recently been uncovered by metabolic labeling of the cell surface.
INTRODUCTION
The bacterial cell surface is required to resist turgor pressure and maintain cell size and shape. It also protects against non-specific environmental insults as well as the targeted efforts of infected hosts to restrict and ultimately sterilize unwanted invaders. The bacterial cell envelope has been a particularly fruitful target for antibiotic development because it is essential for viability and is composed in part of molecules that are not present in host cells. Both the remarkable biology and medical potential of the cell surface have inspired steady research on the topic for the past half-century.
Here, we focus on aspects of bacterial growth, division and secretion that have recently been uncovered by metabolic labeling of the cell surface. Metabolic labeling is a technique in which a chemically modified precursor, referred to here as a ‘probe’, is incorporated into a macromolecule of interest by the endogenous enzymatic machinery of the cell. Traditionally, this approach involved radioactive or stable isotope probes to study broad classes of biomolecules. Metabolic labeling in this fashion remains useful for many research questions but is not ideal in some important respects. Radiolabeled analytes may not be amenable to detailed chemical analysis unless highly specialized equipment can be dedicated to radioactive techniques. Isotope labeling experiments can provide molecular-level insight but often require collaboration with experts in specialized analytical techniques, such as mass spectrometry or NMR spectroscopy. For both approaches, targeting specific biomolecules in vivo remains technically challenging and to date research has focused primarily on metabolite analysis (Watrous and Dorrestein 2011; Gouzy et al., 2013; Watrous et al., 2013). Lastly, radiolabeling experiments generate expensive, environmentally unfriendly waste streams, and both radio- and isotope-labeled precursors may not be commercially available, or may be prohibitively expensive.
Metabolic labeling used in combination with bioorthogonal chemistry refines and extends traditional metabolic labeling strategies. Instead of containing a radioactive or stable isotope, the “probe” in this approach features a small chemical reporter. The presence of the probe is revealed by chemical reaction with an exogenous ‘label’ which is typically either a fluorophore or affinity tag. Detection in this manner relies on bioorthogonal chemistry (Fig. 1) to form a covalent linkage (Table 1) between the macromolecule of interest and the label (Grammel and Hang 2013; Patterson, Nazarova and Prescher 2014). Metabolic labeling followed by chemoselective ligation can be executed with reagents and equipment that are accessible to biology research groups and indeed the technique has been used extensively for probing the mammalian cell surface (Grammel and Hang 2013). Because the strategy is designed to interfere minimally with the biological system under investigation, it is useful for examining spatial and temporal regulation of cell surface components in their native context. Many of these components, including peptidoglycan and outer membrane glycolipids, are not directly encoded genetically and are therefore not suitable to tagging methods such as fluorescent protein fusions. Metabolic labeling is particularly valuable for studying bacterial species that are refractory to other methods because of environmental niche or difficult genetic manipulation. Finally, the modularity of bioorthogonal chemical detection and ease of appending different labels allow multiple forms of analysis (e.g. imaging and biochemical) in a single experiment.
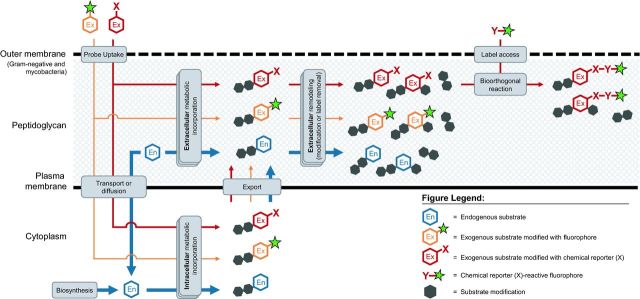
Figure 1.
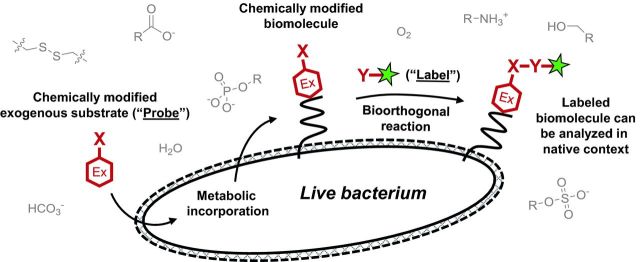
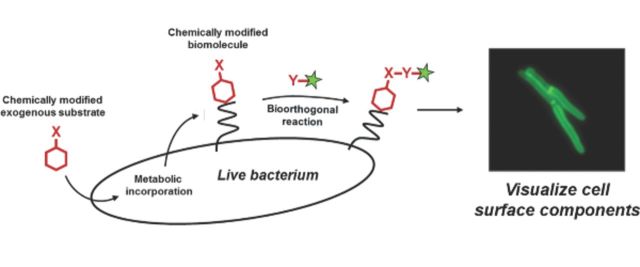
General scheme for metabolic labeling followed by bioorthogonal chemical detection. Chemical or chemoenzymatic synthesis is used to prepare the probe, which is a substrate (red hexagon) modified with a small chemical reporter group X. The endogenous metabolic machinery of the bacterium incorporates the exogenously added probe into the macromolecule of interest. The presence of X is detected by incubating the cell surface of intact (and often live) bacteria with a reaction partner Y containing the label (green star), which is a detectable group such as a fluorophore. X and Y form a covalent bond, thus embedding the label in the macromolecule. Selective, non-toxic reactions that occur in the presence of other cellular biomolecules (gray) are termed ‘bioorthogonal’ and are summarized in Table 1.
Table 1.
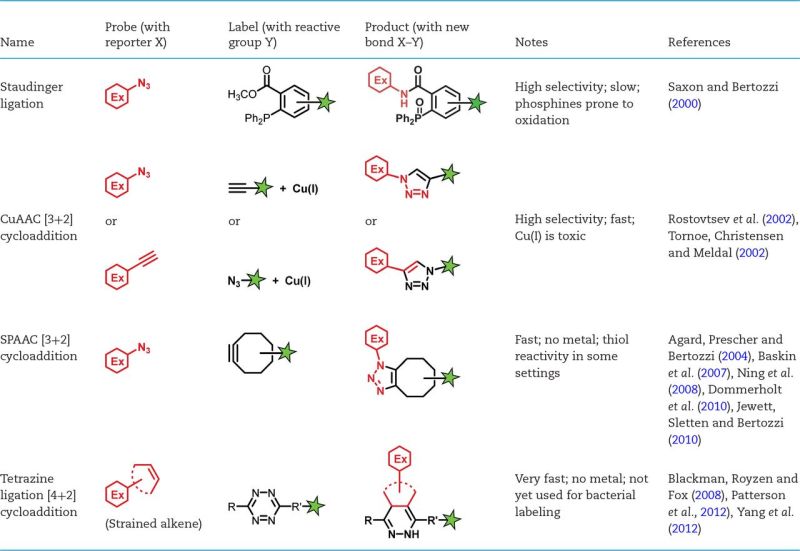
Summary of bioorthogonal reactions that can be used to detect metabolic labeling of the bacterial cell surface. Red hexagon, substrate modified with a chemical reporter. Green star, detection moiety.
 |
Why consider the bacterial cell envelope through the lens of metabolic labeling? We believe that the kinds of data generated by these new technologies have the potential to expand both the breadth and depth of scientific inquiry and to stimulate imaginative new approaches to long-established problems. In this review, we highlight discoveries made by metabolic labeling of the bacterial cell envelope and discuss potential applications of these techniques for both basic and translational research. Although the primary focus of this review is metabolic labeling followed by detection with bioorthogonal chemistry, we also consider one-step methods in which the probe is ‘pre-labeled’, bypassing the chemical ligation step and embedding the detection moiety directly into the macromolecule. For biotechnology applications, we refer readers to (Gautam et al., 2013; Kaewsapsak, Esonu and Dube 2013; Memmel et al., 2013; Fura, Sabulski and Pires 2014; Tra and Dube 2014). Our goal with this review is to narrow the divide between the community of chemists that develop modern metabolic labeling tools and the community of microbiologists that may benefit from using them.
METABOLIC LABELING TECHNIQUES
Metabolic approaches for labeling the bacterial cell surface have been beautifully covered by several recent reviews, notably (van Dam, Olrichs and Breukink 2009; Dube, Champasa and Wang 2011; Foss, Eun and Weibel 2011; Bunschoten et al., 2013; Gautam et al., 2013; Grammel and Hang 2013; Tra and Dube 2014). We will discuss biological insights revealed by the following techniques: labeling of peptidoglycan stem peptide with amino acid analogs; tagging of Gram-negative and mycobacterial outer membrane glycolipids with glycan derivatives; and marking of structural components or effectors of secretion systems with amino acids, glycans and lipids (Fig. 2 and Table 2). First, we highlight some general considerations for using existing tools or designing new ones.
Figure 2.
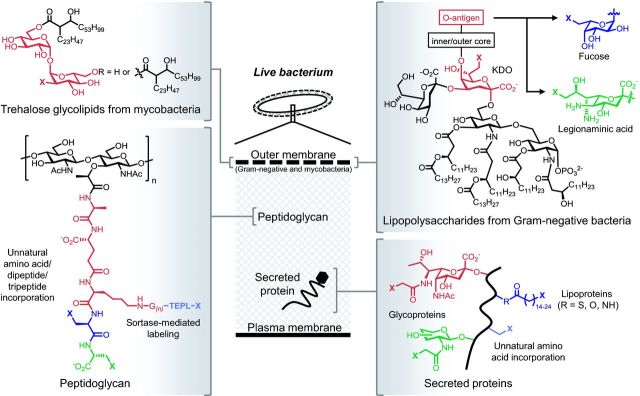
Structure and location of cell envelope macromolecules covered in this review. The embedded metabolic labels (various colors) contain chemical reporters denoted as X. See Table 2 for representative publications and more detailed information regarding metabolic incorporation.
Table 2.
Techniques for metabolic labeling of the bacterial cell surface. Methods are organized by the cell envelope layer containing the target biomolecule and subdivided by the chemical class of the probe. Route indicates whether the probe is incorporated via a cytosolic pathway (internal) or via biosynthetic pathways outside the plasma membrane (external). Target indicates the biomolecule into which the probe is incorporated. Labeling strategy indicates whether the label is attached before or after probe incorporation into the target. Probe source indicates if probe is available commercially (COMM), or must be made by multi-step organic synthesis (MSOS) or solid-phase peptide synthesis (SPPS). Abbreviations: AAA, artificial amino acid; AEK, alanine-glutamate-lysine tripeptide; Alk, alkynyl chemical reporter; Az, azido chemical reporter; FITC, fluorescein fluorophore; FucAl, 6-alkynyl fucose; GlcNAc, N-acetyl glucosamine; KDO, 3-deoxy-D-manno-octulosonic acid; LPS, lipopolysaccharide; NAG, N-acetyl gluosamine; NAM, N-acetyl muramic acid; NBD, nitrobenzofurazan fluorophore; NHS, N-hydroxy succinimide ester; T3SS, type 3 secretion system; TAMRA, carboxy tetramethylrhodamine fluorophore; TMM, trehalose monomycolate; * - likely found in the plasma membrane (Bansal-Mutalik and Nikaido 2014); TDM, trehalose dimycolate; PG, peptidoglycan; PP, peptidoglycan pentapeptide.
| Labeled | Labeling | |||||
|---|---|---|---|---|---|---|
| metabolite | Route | Target | Probe | strategy | Probe source | Selected references |
| Peptidoglycan | ||||||
| Amino acid, chemical handle | External, internal | PG stem peptide | D-cysteine | Post | COMM | de Pedro et al. (1997) |
| External, internal | PG stem peptide | Alk/Az-D-ala | Post | COMM | Kuru et al. (2012), Siegrist et al. (2013) | |
| External, Internal | PG stem peptide | D-ala D-ala with Alk or Az at N or C terminus | Post | COMM, MSOS (3–6 steps) | Liechti et al. (2013) | |
| External | PG stem peptide | Az-LPETG peptide | Post | SPPS | Nelson et al. (2010) | |
| Amino acid, fluorescent | External | PG stem peptide | FITC-LPETG peptide | Pre | SPPS | Nelson et al. (2010) |
| External, Internal | PG stem peptide | NBD-AEK | Pre | SPPS | Olrichs et al. (2011) | |
| External, Internal | PG stem peptide | NBD-D-ala, Coumarin-D-ala, FITC-D-lys, TAMRA-D-lys | Pre | MSOS (1–4 steps) | Kuru et al. (2012) | |
| External | PG stem peptide | FITC-D-lys carboxamide | Pre | MSOS (1–4 steps) | Lebar et al. (2014) | |
| Outer membrane and mycomembrane | ||||||
| Glycan, chemical handle | External, Internal | Fucose-containing bacterial polysaccharides | C6 modified fucoses | Post | MSOS | Yi et al. (2009) |
| Internal | E. coli LPS inner core | 8-azido-8-deoxy-KDO | Post | MSOS | Dumont et al. (2012) | |
| Internal | Fucosylated glycoproteins | Alk-Fuc | Post | MSOS | Besanceney-Webler et al. (2011) | |
| Internal | H. pylori glycoproteins | Ac4GlcNAz | Post | COMM | Koenigs, Richardson and Dube (2009) | |
| Internal | C. jejuni flaggelin | Per-acetylated Az-pseudaminic acid | Post | MSOS | Liu et al. (2009) | |
| External, internal | TMM*, TDM | Az-trehalose | Post | MSOS | Swarts et al. (2012), Urbanek et al. (2014) | |
| Glycan, fluorescent | External | TMM*, TDM | FITC-trehalose | Pre | MSOS | Backus et al. (2011) |
| Artificial amino acid, chemical handle | Internal | Protein, not site-specific | Met replaced by Az-alanine, Az-homoalanine, Az-norvaline, Az-norleucine | Post | COMM, MSOS | Link and Tirrell (2003), Link, Vink and Tirrell (2004) |
| Internal | Protein, site-specific | Tyr replaced by m- or p-acetyl-L-phenylalanine | Post | MSOS | Zhang et al. (2003) | |
| Internal | Protein, site-specific | Met replaced by Az-norleucine AAA | Post | COMM | Link et al. (2006), Tanrikulu et al. (2009) | |
| Internal | Protein, site-specific | Several Alk/Az AAAs as Met surrogates or UAG suppressors | Post | COMM, MSOS | Ngo and Tirrell (2011) | |
| Secretion system components and substrates | ||||||
| Artificial amino acid, cross-linking | Internal | Sec system | SecY–p-benzoyl-phenylalanine AAA | Cross-link | COMM | Mori and Ito (2006) |
| Internal | Translocating polysaccharides | Wza–p-benzoyl-phenylalanine AAA | Cross-link | COMM | Nickerson et al. (2014) | |
| Internal | T3SS secreted proteins | Met replaced by Azidonorleucine AAA | Post (biotin) | COMM | Mahdavi et al. (2014) | |
| Lipid, chemical handle | Internal | Legionella effector proteins | Alkynyl-farnesol-1 | Post | MSOS (3 steps) | Ivanov et al. (2010) |
| Internal | Salmonella T3SS effector proteins | Alkynyl-16 (palmitate analog) | Post | MSOS (6 steps) | Hicks et al. (2011) | |
Experiment design
Any scientific analysis requires attention to possible observer effects. That is, what effect does the act of observation have on the phenomenon under investigation? At a minimum, investigators will want to know whether metabolic labeling grossly impacts bacterial growth, as assessed by optical density measurements, colony-forming units and other viability assays. However, growth inhibition is not necessarily the death knell of a technique as it may still be useful for endpoint analysis. Small molecule inhibitors can also be useful for revealing the essentiality of the pathway under investigation. Moreover it is often possible to find a lower dose at which labeling is achieved but toxicity is in the acceptable range. Certain trehalose analogs, for example, are bacteriostatic in the low millimolar range but have been successfully used to label mycobacterial glycoconjugates at lower concentrations (Backus et al., 2011; Swarts et al., 2012). Even in the absence of overt toxicity, however, care should still be taken that any metabolic perturbations from modified precursor uptake or incorporation are in the acceptable range. For example, D-amino acids modulate peptidoglycan remodeling in some bacterial species (Cava et al., 2011), an effect that may be unacceptable in some experiments but tolerable in others. A good rule of thumb is to titrate the amount of precursor and use the lowest ‘dose’ that yields robust labeling over a no-probe negative control.
A second consideration in designing a metabolic labeling experiment is the bacterial species of interest. A precursor molecule that labels a specific cell surface entity in Escherichia coli might not label the same macromolecule in Mycobacterium tuberculosis because of differences in metabolite uptake pathways, enzyme activity and substrate promiscuity, the existence of de novo versus salvage biosynthetic pathways or envelope composition. The same probe used in one species may not label at all in another, may label a different molecule(s) entirely or may label the target molecule by a different route. D-amino acids, for instance, incorporate into peptidoglycan by both extracellular and intracellular routes that are operative to different degrees in different species (Cava et al., 2011). Consequently, some a priori knowledge of the biosynthetic pathways to be targeted is necessary for experimental design and optimization. On the other hand, metabolic labeling techniques have the potential to inform the study of these pathways.
A third consideration is which type of metabolic precursor to use as a probe. While radio- and stable isotope-labeled metabolic precursors are indistinguishable from natural substrates during metabolism, probes modified with chemical reporters are larger than natural substrates and therefore compete with endogenous substrates for the biosynthetic machinery at a kinetic disadvantage. Theoretically, natural substrates should experience more flux through transporters and biosynthetic enzymes than probes with small chemical reporters or pre-labeled probes with reporters already installed (Fig. 3). It follows that a ‘later’ probe, i.e. a probe that enters the target pathway at later point, might lead to better labeling, because the steric penalty of the modified substrate is assessed over fewer steps of transport and enzymatic transformation and because later precursors tend to encounter fewer metabolic branch points, which can divert probes into off-target pathways. However, other considerations often outweigh the theoretical advantages of later probes. For example, late-stage metabolic precursors often lack an efficient transporter to deliver the probe to the correct layer of the cell. Structurally complex probes are rarely commercially available and can be very challenging to synthesize. Finally, in many cases the researcher is interested in the de novo biosynthesis of a cell surface component, a process that is ideally monitored by a probe that incorporates early in the metabolic pathway of a biomolecule.
Figure 3.
Labeling the bacterial cell surface by hijacking endogenous metabolic pathways (blue) with exogenous substrates via one-step (orange) and two-step (red) approaches. Thickness of arrows indicates probable relative processing efficiency of substrates by the cell's metabolic machinery.
Lastly, choices must be made regarding how the label—whether a fluorophore, affinity tag or other functionality—is incorporated into the probe. The one-step or pre-labeled approach involves using a chemical probe with the label already attached before the probe is metabolically incorporated into the biomolecule of interest (Fig. 3, orange pathways). The two-step approach involves initial incorporation of the probe, and then attachment of the label using bioorthogonal chemistry at one or more later time points (Fig. 3, red pathways).
Transport or enzymatic stringency generally necessitate a two-step labeling process in which the precursor contains the smallest possible chemical reporter that can selectively react with the bulkier detection moiety after the precursor is installed in the cell surface. In instances where there is more substrate flexibility, the choice of whether to employ a one- or two-step method is less clear. One-step methods are more straightforward but generally have larger moieties attached to the precursor and label a smaller fraction of the macromolecule pool. Two-step methods initially result in smaller perturbations to the macromolecule structure and a higher proportion of labeling but can be more cumbersome because time is needed for the chemical reaction which attaches the label to the probe, and extra wash steps are required to remove unreacted label from the cell surface. On the other hand, two-step methods facilitate the delivery of a wide variety of labels to a single probe. Since metabolic incorporation of the probe is constant, there is no need to re-evaluate this important parameter for every new label one might wish to append to the biomolecule of interest. Given the multitude of trade-offs, the choice of technique depends on the specific experimental conditions, including but not limited to the target cell surface biomolecule, the species of interest, the doubling time of the organism, the local environment of the organism and the time frame of the experiment. If a two-step method is chosen, a further consideration is the choice of chemical reporter and complementary probe. A brief overview of chemistries used for the two-step labeling technique is given in Table 1. For a comprehensive guide to choosing the chemistry most appropriate for a given metabolic labeling experiment, we refer readers to Patterson, Nazarova and Prescher (2014).
Experiment interpretation
Metabolic labeling depends on the cell's endogenous enzymatic activity and, in some cases, transport machinery to incorporate the probe into the macromolecule of interest (Fig. 3). Assuming no off-target incorporation, apparent labeling is a function of the amount of probe incorporated into the cell surface net of the amount removed from the cell surface by normal remodeling and turnover processes. Interpretation of changes in signal localization or intensity can be complex but a single, well-controlled metabolic labeling experiment also has the potential to test several hypotheses at once. Because of this complexity, however, it is essential to verify that the biomolecule of interest is selectively labeled.
Provided there is no detectable, off-target metabolic incorporation, the first step to deciphering a labeling experiment is to consider the mechanism by which the label might be incorporated (Fig. 3). In some cases, the metabolite is transported into the bacterium, modified and transported back out. In other cases, it is directly incorporated into the cell envelope by enzymes at the surface. For probes that incorporate via an intracellular route, a change in apparent labeling could be the result of the import mechanism, intracellular enzymatic activity and/or substrate concentration, export of modified cell surface precursor, or a combination thereof. For those that incorporate via an extracellular route, the dominant variables are the localization and amount of enzyme and substrate.
Mycobacteria incorporate the trehalose disaccharide into various glycolipids, including trehalose monomycolate (TMM) and trehalose dimycolate (TDM) (Fig. 2 and Table 2). By taking a combined genetic and metabolic labeling approach, we demonstrated that trehalose analogs bearing an azide at the 2-, 4- or 6-position were incorporated into mycobacterial glycolipids by the SugABC-LpqY trehalose recycling pathway but that those modified at the 3-position were incorporated via an extracellular route mediated by the Antigen 85 complex (Swarts et al., 2012; Urbanek et al., 2014). The pathway by which trehalose derivatives incorporate is dependent not only on the position of the modification but also the modification itself. Backus et al. (2011), for example, showed that a keto-trehalose analog functionalized with a fluorophore at the 2-position incorporated into glycolipids by an extracellular, Antigen 85-dependent pathway (Backus et al., 2011). The latter approach is an example of a one-step, pre-labeled probe that succeeded in part because the process under investigation is localized to the cell surface, and because the relevant enzymes are highly promiscuous. The former approach is an example of the two-step strategy that used smaller modifications on the trehalose precursor to distinguish between different metabolic pathways and, serendipitously, to add new knowledge about the substrate specificity of a previously characterized sugar transporter. Together, pre- and post-labeled trehalose analogs constitute a flexible toolset that allows selective interrogation of two distinct, essential pathways in mycobacterial cell envelope biogenesis.
Exogenous D-amino acids are known to incorporate into the peptidoglycan stem peptide by both intracellular and extracellular routes (Tsuruoka et al., 1984; Caparros, Pisabarro and de Pedro 1992; de Pedro et al., 1997; Cava et al., 2011; Lupoli et al., 2011). The first process is catalyzed by the intracellular D-alanine D-alanine ligase (Ddl) and results in a new D-amino acid at the fifth position of the peptidoglycan stem peptide (Fig. 2). The second process is an L,D- or D,D-transpeptidation reaction that, respectively, results in a new D-amino acid at the fourth or fifth position of the peptidoglycan stem peptide (Fig. 2). Although direct biochemical evidence is still lacking, the positional selectivity of D-amino acid analogs in peptidoglycan labeling and in antibiotic challenge experiments suggests that different bacterial species use diverse pathways of incorporation (Kuru et al., 2012; Siegrist et al., 2013).
A newer variation of the two-step D-amino acid labeling strategy, in which a chemical reporter such as a terminal alkyne is appended to either the N- or C-terminus of a D-alanine D-alanine dipeptide, has the desirable property of controlling chemical reporter incorporation at either the fourth or fifth position, respectively (Liechti et al., 2013). In keeping with the absence of known, extracellular mechanisms by which bacteria incorporate dipeptides, several lines of evidence suggest that the dipeptide derivatives label via the cytoplasmic route (Liechti et al., 2013). The same is true for fluorophore-functionalized peptidoglycan tripeptides (Olrichs et al., 2011) (a one-step labeling approach), whereas sortase-mediated tagging of the biopolymer likely occurs extracellularly (Nelson et al., 2010) (an example of an enzyme-mediated, rather than chemical, two-step labeling approach).
Regardless of incorporation mechanism, however, labeling in some cases is sensitive to subsequent remodeling. For example, several groups have found that deletion of carboxypeptidase-encoding genes dramatically increases peptidoglycan labeling by single D-amino acid analogs (Kuru et al., 2012; Siegrist et al., 2013; Tocheva et al., 2013; Fura, Sabulski and Pires 2014) or by D-alanine D-alanine dipeptides bearing C-terminal reporters (Liechti et al., 2013). In the case of the single D-amino acid derivatives, it is not clear whether this is because carboxypeptidases directly remove labeled pentapeptides or because they remove unlabeled pentapeptides that are the substrate for D-amino acid derivative addition. However, these data suggest that the probes might offer a useful read-out for carboxypeptidase activity in these organisms, provided there are also controls to assess probe incorporation.
As we will emphasize below, metabolic labels as well as the affixed probes can serve as indirect reporters for a variety of cellular pathways. For example, the Gram-negative outer membrane (Shieh et al., 2014) and Gram-positive teichoic acids (Fura, Sabulski and Pires 2014) appear to restrict the access of larger probes to labeled peptidoglycan, while growth and metabolic status (Backus et al., 2011; Kuru et al., 2012; Liechti et al., 2013; Pilhofer et al., 2013; Siegrist et al., 2013; Fura, Sabulski and Pires 2014) likely influence the initial probe incorporation step. However, because multiple factors can impact metabolic labeling, mechanistic insight generally requires that these experiments be paired with additional lines of inquiry (genetic, biochemical, etc.). We now highlight several aspects of cell envelope-related biology that have recently been uncovered at least in part by metabolic incorporation of chemical probes. For reviews of earlier work on the location and timing of cell surface growth that was aided by other labeling methods, please see (Sham et al., 2012; Cava et al., 2013; Kysela et al., 2013; Pinho, Kjos and Veening 2013).
WHAT HAVE WE LEARNED?
Bacterial growth and division
Although fluorescent antibiotic conjugates have been powerful reagents for revealing nascent sites of peptidoglycan synthesis, the size and inhibitory activity of the molecules in most cases have restricted imaging to static snapshots of Gram-positive peptidoglycan (Daniel and Errington 2003; Cabeen and Jacobs-Wagner 2005; Tiyanont et al., 2006). In contrast, optimized labeling by peptidoglycan amino acids does not obviously block cell wall growth and can be used to extract more precise spatial and temporal information. The first version of this technique was a two-step process that utilized D-cysteine as a probe and various thiol-reactive labels for both fluorescence and electron microscopy (de Pedro et al., 1997). Although D-cysteine labeling was instrumental in uncovering a wide range of peptidoglycan-related biology, particularly in Gram-negative species, thiol-reactive chemistries are not suited for live or intact cell imaging. An advantage of detecting D-amino acids with newer, bioorthogonal chemistries (Fig. 1 and Table 1) is the ability to perform multicolor pulse labeling experiments in a variety of contexts, from single species growing in broth culture or within infected host cells (Kuru et al., 2012; Liechti et al., 2013; Pilhofer et al., 2013; Siegrist et al., 2013) to multiple species present in freshwater and saliva samples (Kuru et al., 2012; Cava et al., 2013). These approaches are particularly powerful when combined with loss-of-fluorescence strategies in which bacteria are pulsed to label the entire cell surface, for example with an amine-reactive dye to tag surface-exposed proteins (de Pedro, Grunfelder and Schwarz 2004; Rafelski and Theriot 2006; Aldridge et al., 2012), washed and then allowed to continue growing in the absence of the dye to reveal areas of new cell envelope. Of note, the smaller size of metabolic labeling reagents permits labeling of mycobacterial and Gram-negative species whose outer membranes render them inaccessible or poorly accessible to fluorescent antibiotics.
Growth insights from peptidoglycan labeling
Mycobacteria incubated in the presence of fluorescent vancomycin for one to two generations display spots of staining at the poles and septum (Chauhan et al., 2006; Thanky, Young and Robertson 2007). The lengthy incubation required for robust signal is probably due to the low accessibility of the bulky probe to the mycobacterial peptidoglycan, which is obscured by the highly impermeable mycomembrane. The polar puncta therefore indicate the sites of peptidoglycan synthesis, areas of greater mycomembrane permeability or both. In contrast, mycobacteria labeled with alkyne-D-alanine for much shorter time periods have signal at subpolar regions, along the sidewall and at the septa (Siegrist et al., 2013; Meniche et al., 2014). For M. smegmatis labeled for ∼11–13% of a generation, most bacteria have a larger area of labeling at one end of the cell (Meniche et al., 2014). When the bacteria are pulsed in amine-reactive dye (which covalently tags surface-exposed proteins) and chased with alkyne-D-alanine, the signals are anti-localized, suggesting that the latter reports new cell wall (Meniche et al., 2014). Two studies report similar elongation rates at both poles of genetically engineered M. smegmatis except during a brief period between cytokinesis and physical cell separation (Santi et al., 2013; Wakamoto et al., 2013; Kieser and Rubin 2014). These data differ from earlier work suggesting that the old pole grows more quickly than the new pole throughout the cell cycle (Aldridge et al., 2012; Kieser and Rubin 2014). Our metabolic and amine-reactive dye labeling of wild-type bacteria (Meniche et al., 2014) support an asymmetric model of mycobacterial growth (Aldridge et al., 2012; Joyce et al., 2012; Singh et al., 2013), although further experiments are necessary to address cell cycle stage and population heterogeneity.
Both amine-reactive dye and D-amino acid pulse-chase experiments demonstrate that the Gram-negative bacterium Agrobacterium tumefaciens grows in a unipolar fashion (Brown et al., 2012; Kuru et al., 2012; Cameron et al., 2014). Recently, Cameron et al. (2014) observed that the longer cells of the population tended to have larger areas of alkyne-D-alanine labeling. The increased labeling area may be the result of cell width expansion. Alternatively, these intriguing data may suggest that elongation rate is proportional to pole age, as suggested for mycobacteria (Aldridge et al., 2012; Meniche et al., 2014).
In addition to the ability to penetrate bacteria with outer membranes, small D-amino acids are able to label intracellular structures e.g. developing spores. Sporulation in the model organism Bacillus subtilis begins with asymmetric cell division, which is followed by engulfment of the nascent spore, formation of the thick cell wall cortex and coat and finally lysis of the mother cell. Although cell wall synthesis during engulfment had been suggested by earlier work (Meyer et al., 2010), it was not clear whether the peptidoglycan was degraded and re-synthesized or present for the entire process. Tocheva et al. (2013) initially used electron cryotomography to demonstrate the continuous existence of a peptidoglycan layer during B. subtilis sporulation and germination. They confirmed that cell wall surrounds the prespore throughout engulfment by labeling sporulating B. subtilis with fluorescent D-amino acids. Taken together, their data suggest that this Gram-positive organism is able to thin its peptidoglycan during sporulation and then to re-thicken it to form the cortex. The capacity to control the number of peptidoglycan layers and hence thickness of the biopolymer is consistent with the notion that Gram-positive and Gram-negative bacteria share similar, circumferential peptidoglycan architecture.
D-amino acid labeling has facilitated super-resolution imaging of nascent peptidoglycan from a variety of bacterial species (Kuru et al., 2012; Meniche et al., 2014). The increased precision of this imaging modality was instrumental in building a new model for mycobacterial elongation. Despite the medical importance of these bacteria, the site and geometry of polar extension as well as the principles that coordinate the addition of different cell envelope layers remain largely unclear. Using fluorescent protein fusions, Meniche and colleagues first showed that the terminal synthetic enzymes for peptidoglycan, arabinogalactan and mycolic acids colocalize at the septum and at a previously uncharacterized, subpolar elongation zone (Meniche et al., 2014). The intermediate filament-like protein DivIVA (Wag31) binds these enzymes but high-resolution imaging of amine-reactive dye and alkyne-D-alanine labeling showed that DivIVA is physically separated from the site of new peptidoglycan synthesis/remodeling (Meniche et al., 2014). In contrast to the proposed role of DivIVA in other species, these data suggest a model whereby the protein creates a zone of polar exclusion in the mycobacterial cell, which then forces cell wall deposition to occur at a subpolar location.
The flexibility of two-step cell surface labeling and ease of amino acid analog synthesis for one-step methods offer the potential for a wide range of peptidoglycan detection strategies beyond conventional fluorescence microscopy. For example, we have developed fluorogenic (activated by chemical reaction) azide probes in the near-infrared region with the long-term goal of detecting peptidoglycan in whole animals (Shieh et al., 2014). Nelson et al. (2010) used a three-step approach to perform immunocryoelectron microscopy on isolated Staphylococcus aureus peptidoglycan. A non-exhaustive list of other, theoretical detection approaches for metabolic labeling includes biochemical analysis by affinity tags, positron emission tomography by radioisotope tracers, mass spectrometry imaging by mass tags and Raman spectroscopy using an embedded alkyne or isonitrile group (Crawford et al., 2012; Wei et al., 2014). The versatility of metabolic labeling offers many exciting opportunities for studying bacterial physiology and pathogenesis.
Growth insights from outer membrane and mycomembrane labeling
Although the relationship between bacterial growth and peptidoglycan synthesis is well studied, comparatively little is known about the spatial and temporal regulation of other layers of the cell envelope. Several groups have assessed growth and/or outer membrane fluidity using detection moieties that attach directly to surface components (Muhlradt et al., 1973; Schindler, Osborn and Koppel 1980; de Pedro, Grunfelder and Schwarz 2004; Gibbs et al., 2004; Ghosh and Young 2005; Rafelski and Theriot 2006; Winther et al., 2009; Aldridge et al., 2012; Ursell et al., 2012). In most cases, agents that bind covalently or non-covalently to the cell envelope will result in indiscriminate labeling with respect to the age of the particular component to which it binds. Thus, these studies have inferred growth and fluidity dynamics from pulse-chase and fluorescence recovery after photobleaching (FRAP) experiments.
The ability to track new surface components directly can simplify experimental detection and interpretation. One advantage of metabolic labeling is that it often affords control over the age of the cell envelope biomolecule that is marked. Given sufficient evidence that a labeled metabolite accurately reports nascent cell surface material and does not appreciably diffuse prior to detection, varying the length of bacterial incubation as well as the reporter on the metabolite can permit a researcher to perform ‘virtual time-lapse microscopy’, in which information on cell wall synthesis and dynamics can be obtained without live cell imaging (Kuru et al., 2012). Tagging of non-peptidoglycan constituents of the bacterial envelope has been achieved by metabolic labeling with glycans (Koenigs, Richardson and Dube 2009; Liu et al., 2009; Yi et al., 2009; Backus et al., 2011; Besanceney-Webler et al., 2011; Dumont et al., 2012; Swarts et al., 2012; Memmel et al., 2013; Mas Pons et al., 2014), amino acids (Link and Tirrell 2003; Zhang et al., 2003, Zhang et al., 2011; Okuda and Tokuda 2009; Frobel, Rose and Muller 2011; Ieva et al., 2011; Lin et al., 2011; Abe et al., 2012; Okuda, Freinkman and Kahne 2012; Zoufaly et al., 2012; Pavlova, Ieva and Bernstein 2013; Nickerson et al., 2014) and lipids (Rangan et al., 2010) conjugated to chemical reporters or directly to detection agents such as fluorophores or photo-cross-linkers. However, these studies have generally not reported spatial or temporal details at the subcellular level.
One exception is the observation that TMM and TDM (and possibly other metabolites) labeled with particular trehalose derivatives accumulate along the sides and at the poles of mycobacterial cells (Backus et al., 2011; Swarts et al., 2012). The analogs that generate the apparent polar localization—3-azido-trehalose and a 2-fluorescein-modified keto-trehalose—incorporate into the glycolipids via an extracellular reaction by the Antigen 85 complex. The Antigen 85 enzymes transfer mycolates from TMM to either arabinogalactan or to another molecule of TMM to generate TDM (Sathyamoorthy and Takayama 1987; Belisle et al., 1997). Both reactions result in the concomitant loss of free trehalose. The enzymes can also catalyze the reverse reaction from TDM to TMM, which is thought to be the source of extracellular labeling by trehalose analogs (Backus et al., 2011; Swarts et al., 2012). However, these experiments involved a relatively long (one or more generations) incubation prior to labeling analysis. Whereas an initial pulse might result in incorporation of 3-azido-trehalose and 2-fluorescein-trehalose into TMM alone, and would be a specific read-out for Antigen 85 activity, subsequent incubation likely labels both TMM and TDM, and would reflect a combination of enzyme activity, cell growth and diffusion of the glycolipids in the inner and outer membranes, respectively (Bansal-Mutalik and Nikaido 2014). More experiments are necessary to determine when and where the glycolipids are added to the mycobacterial cell surface. However, the polar accumulation of trehalose-labeled metabolites in mycobacteria is intriguing given that mycobacteria grow from their tips (Hett and Rubin 2008; Kieser and Rubin 2014). Our model for mycobacterial growth predicts that precursors of arabinogalactan and mycolic acids are transported out of the cell from the same cellular location as those of peptidoglycan (Meniche et al., 2014). Further support for this model comes from a recent study in which fluorescently tagged proteins involved in mycolic acid synthesis and transport machinery localize to poles; however, the microscopy was not of sufficient resolution to determine true polar versus subpolar localization (Carel et al., 2014). These observations are further supported by data showing that antibiotics targeting different layers of the cell envelope, including peptidoglycan, arabinogalactan and mycolic acids, cause polar lysis (Bardou et al., 1996; Makarov et al., 2009; Aldridge et al., 2012; Kumar et al., 2012). The ability to localize both the proteins and the metabolites of different cell wall and membrane synthetic pathways may help researchers unravel the spatial and temporal sequence of mycobacterial envelope assembly.
In addition to their utility in basic bacterial cell biology, some metabolic labeling methods have exciting potential for diagnostic development because they afford genus or even species-specific labeling without the need for genetic engineering. A fluorescent trehalose derivative, for example, is selective for mycobacterial glycolipids (Backus et al., 2011) (although it would likely also label related organisms such as Corynebacterium and Nocardia) while an azido analog of 5-N-acetimidoyl-7-N-acetyllegionaminic acid (Leg) is selective for an O-antigen structure that comprises the lipopolysaccharide (LPS) of Legionella pneumophila but not other Legionella species (Mas Pons et al., 2014). Because of differences in endogenous fucose metabolism, an alkynyl analog incorporates into surface glycoproteins of Parabacteroides distasonis much better than those of Bacteroides fragilis, a property that could be exploited for differential labeling of Bacteroidales in the mammalian gut (Besanceney-Webler et al., 2011). Other metabolic labeling methods offer more coarse-grained selectivity: sortase-mediated tagging (Nelson et al., 2010) labels peptidoglycan of a variety of Gram-positive species that express the sortase enzyme, while an azido derivative of 3-deoxy-d-mannooctulosonic acid (KDO) incorporates into an essential component of the LPS inner core found in nearly all Gram-negative species (Dumont et al., 2012). Recently, Lebar et al. (2014) demonstrated that E. coli incorporates fluorescent D-amino acids but not carboxamides into peptidoglycan while B. subtilis labels weakly with fluorescent D-amino acids but strongly with carboxamides. The ability to incorporate D-amino carboxamides may depend on the presence of transpeptidases with specificity for meso-DAP-containing muropeptides, suggesting that such compounds might differentiate species based on enzymatic substrate preference. We have also found that in live bacteria, the Gram-negative outer membrane (Shieh et al., 2014), and to a lesser extent, the M. tuberculosis mycomembrane (unpublished), appear to exclude certain fluorophores, presumably because their size, geometry, hydrophobicity or charge make them unsuitable for passive diffusion or transit through outer membrane porins. Therefore, judicious combination and timing of different fluorophore reaction partners have the potential to impart Gram stain-like discrimination to D-amino acid labeling.
Cell division
Bacterial division is often observed by electron microscopy, to assess the developing septum in the context of other cellular components, or by fluorescence microscopy, to assess the presence or absence of protein markers for different stages of the division process. While such studies have been illuminating, electron microscopy is laborious and relatively low throughput while fluorescent protein fusions can, in some cases, alter the behavior of the protein of interest [for examples, see Swulius and Jensen (2012) and Meniche et al. (2014)] and are not possible in organisms that are not genetically tractable. An alternative way to mark the bacterial division site is by cell envelope labeling. Bacterial division proceeds in several steps: (1) formation of the cytoplasmic division apparatus; (2) synthesis of plasma membrane and cell wall; (3) septal peptidoglycan hydrolysis and daughter cell separation. Therefore, mid-division can be marked by metabolic labeling of plasma membrane lipids, peptidoglycan and components of the outer membrane. For non-diffusible envelope components, this strategy also has the potential to mark cell division in progress as well as division events that have already occurred, for example, from label that is retained at the daughter cell poles.
Metabolic labeling of the bacterial division site is particularly powerful when combined with other division markers. Eun et al. (2013) identified a small molecule, termed Divin, which inhibits cell division by disrupting the assembly of late division proteins. The authors first used phase contrast microscopy and FRAP to demonstrate that Divin treatment blocks cytokinesis in Caulobacter crescentus. After showing that the molecule causes late division proteins to disassemble from the division site, they then employed a coumarin-labeled D-amino acid (HADA) (Kuru et al., 2012) to show that peptidoglycan remodeling continues following departure of FtsI from the septum but not after disassembly of other cell division proteins FtsQ, FtsL, FtsW and FtsB.
Chlamydia research has traditionally been limited by the obligate intracellular lifestyle of the organism as well as its resistance to standard genetic techniques. By taking a metabolic labeling approach, Liechti, Kuru and colleagues were able to generate and test new hypotheses related to the role of peptidoglycan in the Chlamydia trachomatis division process. They observed that fluorescence from incorporated D-alanine D-alanine dipeptide probe was concentrated in a ring-like structure at the putative division site of RBs despite an incubation time of several generations (Liechti et al., 2013). These data suggest that either the septum is the primary site of probe incorporation in C. trachomatis or that non-septal cell wall (including areas that were derived from division of a labeled bacterium) is remodeled in such a way that there is rapid loss of the dipeptide label. Either of these possibilities may explain the difficulty of detecting peptidoglycan by standard biochemical techniques (Desmarais et al., 2013). The idea that C. trachomatis may synthesize new peptidoglycan for division but not growth is consistent with earlier work (Mohammadi and Breukink 2014) and particularly intriguing given the lack of an FtsZ sequence homolog in chlamydiae genomes. Most bacterial species organize cell wall synthesis at the division site via the tubulin homolog FtsZ, which acts both as a contractile ring to drive cytokinesis as well as a platform to recruit other cell division proteins. Jacquier et al. (2014) recently demonstrated that chlamydiae may instead depend on RodZ and the actin homolog MreB to direct cell division. Recruitment of RodZ to the Waddlia chondrophila septum is blocked by phosphomycin and augmented by penicillin, antibiotics that respectively inhibit early-stage peptidoglycan synthesis and late-stage peptidoglycan remodeling in other organisms. These experiments further support the idea that peptidoglycan, either the mature structure or precursors thereof, help coordinate chlamydial division.
In Gram-negative organisms like E. coli, the outer membrane and peptidoglycan constrict together during cell division (Egan and Vollmer 2013). In mycobacteria, the inner layers of the cell envelope contribute to septum formation while the outer layers remain intact and bridge the developing daughter cells (Hett and Rubin 2008). Septal hydrolysis precedes the uneven rupture of the outer layers and the characteristic ‘V-snap’ shape. Because this model is based on electron microscopy (Takade et al., 1983; Dahl 2004), however, the precise coordination of synthesis and degradation of the different layers is not clear. Although we readily observe D-alanine labeling of septal peptidoglycan in asynchronous mycobacterial cultures (Meniche et al., 2014), obvious mid-cell labeling is not apparent with trehalose analogs alone (Backus et al., 2011; Swarts et al., 2012; Urbanek et al., 2014). These data are consistent with the hypothesis that trehalose-based cell surface components do not participate in septum formation, although shorter time course experiments and more complete characterization of the labeled ‘trehalome’ are clearly warranted.
As discussed earlier, the growth dynamics of non-peptidoglycan portions of the bacterial cell envelope have been extrapolated from pulse chase and FRAP using reagents that bind non-selectively to the molecules of interest (Muhlradt et al., 1973; Schindler, Osborn and Koppel 1980; de Pedro, Grunfelder and Schwarz 2004; Gibbs et al., 2004; Ghosh and Young 2005; Rafelski and Theriot 2006; Winther et al., 2009; Aldridge et al., 2012; Ursell et al., 2012). These techniques are less conducive to the study of the smaller surface area of the septum as well as the shorter time scales of the division process (elongation in these studies is often extended by treating the bacteria with division inhibitors). The ability to label newly synthesized, non-peptidoglycan constituents with metabolic reporters that are suitable for correlative light and electron microscopy (Sjollema et al., 2012) may provide unique insights into the mechanisms of bacterial cell division, particularly for organisms with unusual cell envelopes.
Coordination of growth and division
In the rod-shaped bacterial models E. coli, B. subtilis and C. crescentus, elongation and division are respectively controlled by the cytoskeletal proteins MreB and FtsZ. Fenton and Gerdes used bacterial two hybrid analysis and cross-linking to confirm earlier suggestions of putative interactions between the two proteins in E. coli (Butland et al., 2005; Hu et al., 2009; Tan, Awano and Inouye 2011; Fenton and Gerdes 2013). Although wild-type MreB was recruited to the FtsZ ring in dividing cells, a point mutant that was physically unable to bind FtsZ did not. Expression of the mutant MreB allele caused the bacteria to elongate excessively, suggesting that the cells were no longer competent to divide. FtsZ formed rings at the proper locations in these cells but failed to recruit PBP1B or PBP2, enzymes that are critical for septum synthesis. Indeed, whereas wild-type cells labeled with fluorescent D-amino acids along the periphery and at FtsZ rings, E. coli expressing the mutant MreB allele stained only along periphery. Thus, by marrying classical genetics with biochemistry and chemical probes, the authors demonstrated that bacterial division and elongation are coupled by a direct interaction between FtsZ and MreB.
While elongation and division are well characterized in rod-shaped, model organisms like E. coli, they remain largely uncharacterized in species of different shapes or that lack MreB or FtsZ homologs. Fleurie et al. (2014) found that the oval-shaped pathogen Streptococcus pneumonia utilizes StkP, DivIVA and GpsB to coordinate peripheral and septal cell wall synthesis. By tracking fluorescent protein fusions and peptidoglycan D-amino acids in the presence or absence of these proteins, the authors provide evidence for a model in which the different modes of cell wall synthesis are tuned by a single machinery to create an ellipsoid shape.
Life outside the test tube
Construction and remodeling of the cell wall are well described for bacteria growing in vitro under idealized laboratory conditions (Typas et al., 2012). However, very little is known about how these processes occur in more physiologically relevant habitats. For pathogenic and commensal species, the relative importance of many cell wall-acting enzymes changes with host environment (Boneca 2005; Goodman et al., 2009; Frirdich and Gaynor 2013), suggesting that physical and chemical alterations in vivo and the presence of host biomolecules place unique demands on the cell wall. However, mechanisms of cell wall adaptation to environmental fluctuation are unlikely to be uncovered solely by transcriptional or proteomic profiling or biochemical analysis. For example, in the oligotrophic environmental bacterium C. crescentus, the localization of several peptidoglycan-acting proteins varies according to osmolality (Hocking et al., 2012). These changes occur independently of gene expression. Defining the impact of the external milieu on the bacterial cell wall is not straightforward but is, in our opinion, an exciting opportunity for new, microscopy-based methods.
Host environment
We initially developed a metabolic labeling approach to probe bacterial peptidoglycan during cellular infection (Siegrist et al., 2013). Given that the growth of a L. monocytogenes D-alanine auxotroph in cultured mammalian cells is rescued when D-alanine is added to the tissue culture medium (Thompson et al., 1998), we reasoned that host cells would be able to take up D-alanine and perhaps other D-amino acids. Moreover, because eukaryotic cells do not generally produce D-amino acids, we hypothesized that D-alanine analogs might selectively label bacteria inside of host cells. Time course and two-color pulse-chase labeling experiments with D-alanine derivatives revealed that the general spatial and temporal distributions of fluorescence were conserved for L. monocytogenes growing in vitro or during macrophage infection (Siegrist et al., 2013).
More recently, similar techniques have been used to shed light on the so-called ‘chlamydial anomaly’. The Chlamydiae comprise a diverse group of obligate intracellular bacteria. The presence or absence of peptidoglycan in these organisms has been controversial (Moulder 1993; Chopra et al., 1998; Ghuysen and Goffin 1999; McCoy and Maurelli 2006). Although they are sensitive to cell wall-acting antibiotics and their genomes encode functional homologues of peptidoglycan synthesis enzymes, biochemical proof for the existence of chlamydial peptidoglycan has been elusive.
Using a combination of electron cryotomography, mass spectrometry and fluorescent D-amino acid labeling, Pilhofer et al. (2013) demonstrated the presence of the biopolymer in Protochlamydia amoebophila but not in Simkania negevensis, two examples of environmental chlamydiae. Soon after, Liechti et al. (2013) successfully labeled the pathogen C. trachomatis growing in L2 mouse fibroblast cells using D-alanine D-alanine modified with an alkyne at its N-terminus. Dipeptides modified at the C-terminus as well as single D-amino acid analogs did not label the bacteria. The authors found that modified single D-alanine, in contrast to dipeptide derivatives, failed to rescue bacterial plaquing in the presence of D-cycloserine, an antibiotic that inhibits the alanine racemase and Ddl. The authors concluded that the lack of alkyne-D-alanine labeling was likely because the single D-amino acid analogs alone are poor substrates for the chlamydial Ddl, whereas the lack of labeling by C-terminally modified D-alanine D-alanine was the result of post-incorporation removal by transpeptidation, carboxypeptidation or both. In support of the latter, inhibiting these enzymatic processes by ampicillin treatment facilitated labeling by the C-terminally modified dipeptide. Purified Ddl has high substrate specificity for the N-terminal D-alanine (Barreteau et al., 2008) so an alternative interpretation is that the single D-alanine probes can be ligated to D-alanine only in one orientation (and not to each other) (Cava et al., 2011). In this way, they would not rescue growth of D-cycloserine-treated bacteria but, in the presence of endogenous D-alanine from untreated bacteria, would incorporate and label peptidoglycan very similarly to the C-terminally modified dipeptides. Presumably, the incorporated D-alanine probes would also be subject to the same transpeptidation/carboxypeptidation that removes the label from dipeptide incorporated into peptidoglycan.
Chlamydiae are dimorphic, alternating between the infectious form (elementary body, EB), which is metabolically inactive and extracellular, and the non-infectious form (reticulate body, RB), which is metabolically active and replicates intracellularly (McCoy and Maurelli 2006). Peptidoglycan can be biochemically detected in P. amoebophila purified from infected amoeba but stained with fluorescent D-amino acids only when the organism is replicating intracellularly (Pilhofer et al., 2013), consistent with the hypothesis that new cell wall is produced only when the bacterium is metabolically active and growing. Likewise, signal from dipeptide-labeled C. trachomatis was not observed until 8 h post-infection, a time point that corresponds with the early stages of EB-to-RB transition (Liechti et al., 2013).
A strict correlation between cell wall labeling and growth status could allow researchers to use the former as a read-out for the physiology of other bacterial species that alternate between growth and quiescence. This approach is similar in concept to work by Hatzenpichler et al. (2014), who used incorporation of L-azidohomoalanine, a surrogate for L-methionine, to visualize translationally active cells in samples from diverse environments, including oral biofilm, freshwater and anoxic sediments. We have found that L. monocytogenes labels poorly with D-alanine analogs when growth is limited in vitro (Siegrist et al., 2013) or during macrophage infection (unpublished). On the other hand, Fura, Sabulski and Pires (2014) have demonstrated that signal from D-amino acid derivatives increases when B. subtilis is incubated during stationary phase compared to exponential phase. These data are consistent with earlier work showing that a variety of bacterial phyla produce and incorporate D-amino acids at elevated levels during entry into stationary phase (Caparros, Pisabarro and de Pedro 1992; Lam et al., 2009; Cava et al., 2011). The discrepancy between the results in B. subtilis and our findings in L. monocytogenes may be because of species-specific differences in incorporation routes and/or stationary phase remodeling of peptidoglycan by transpeptidases. Combining transpeptidase-dependent and -independent methods of peptidoglycan labeling may help discriminate the growth status of different bacterial species in a variety of environmental contexts.
One application for such a strategy would be during infection with pathogenic mycobacteria. There is mounting evidence that these organisms adopt a range of metabolic states and replication rates to adapt to different host environments during both active and chronic infection (Bouley et al., 2001; Solomon, Leung and Isberg 2003; Barry et al., 2009; Gill et al., 2009). This model has broad implications for tuberculosis treatment but is difficult to evaluate by standard, bulk population measurements of disease burden (Crimmins and Isberg 2012; Helaine and Holden 2013; Manina and McKinney 2013). Like D-amino acids, trehalose is a non-mammalian component of the mycobacterial cell envelope. In contrast to D-amino acid reporters, which were designed to label a feature common to nearly all bacteria, trehalose analogs target surface glycolipids that are conserved in a narrow range of species. Backus et al. (2011) found that M. tuberculosis growing in vitro or in murine macrophage-like J774 cells or primary bone marrow-derived macrophages were labeled with a fluorophore-modified trehalose analog. After approximately one generation of in vitro growth, M. tuberculosis exhibited uniform labeling around the cell with some accumulation at the poles. By contrast, intracellular M. tuberculosis and M. bovis BCG exhibited more heterogeneous labeling, with some bacteria staining like their in vitro-grown counterparts and others remaining completely unlabeled. Even mycobacteria within a single macrophage exhibited significant variability in fluorescence intensity. By examining markers for endosomes, phagosomes and lysosomes, the authors showed that trehalose-labeled bacteria tended to be less associated with markers of mature, degradative compartments than the general population following immune activation by interferon-γ. Although the effect was modest, these data suggest either that mycobacteria in less hospitable phagosomes grow poorly and therefore do not synthesize as much new cell envelope or that the glycolipids labeled by trehalose derivatives inhibit phagosome maturation (Spargo et al., 1991; Indrigo, Hunter and Actor 2003). Discriminating between these hypotheses may allow the probes to be used not only for asking basic questions about bacterial pathogenesis but also in a more diagnostic capacity to monitor infection during therapy.
An exciting opportunity presented by cell surface engineering is the ability to attach environment-sensitive probes directly to the surface of a bacterium (Ali et al., 2012). Metabolic labeling offers a complementary approach to genetically encoded sensors. Yang and coworkers combined the two techniques in a proof-of-concept work by incorporating an azido amino acid derivative at various sites of the pH-responsive, E. coli chaperone HdeA and subsequently attaching an azide-reactive dye that is sensitive to surrounding hydrophobicity but not to pH (Yang et al., 2012). In labeled HdeA, conformational changes under low pH altered the local environment of the dye and resulted in brighter fluorescence. Because the fluorescence read-out is dependent on protein conformation, and thus reversible, this system could in principle allow investigators to track the pH experienced by enteric bacteria as they pass though the stomach and enter the gut. By contrast, reaction-based small molecule pH probes (Chan, Dodani and Chang 2012) are often irreversible and independent of the conformation of the biomolecule to which they are attached. Labeling different layers of the bacterial cell envelope by this approach might allow subcellular resolution of pH and other properties of the biochemical milieu.
Bacterial growth varies qualitatively as well as quantitatively. Indeed, changing shape is a widespread strategy for surviving stressful environments (Justice et al., 2008; Frirdich and Gaynor 2013). The processes by which this occurs are still emerging but likely require both cytoskeletal proteins and peptidoglycan-acting enzymes. Importantly, this stress response can alter antibiotic susceptibility and may itself be a promising drug target (Justice et al., 2008). Ranjit and Young investigated what could potentially be a new category of morphological plasticity during infection, that is, shape regeneration after peptidoglycan digestion by the innate immunity protein lysozyme. Bacteria that have lost their peptidoglycan can, in some cases, resynthesize the biopolymer and regain their wild-type shape (Zinder and Arndt 1956; Lederberg and St Clair 1958; Birdsell and Cota-Robles 1967). Because an external template is not required, it was not clear whether recovery of rod morphology would rely on the same pathways that maintain shape in the presence of peptidoglycan. After inducing spheroplast formation by lysozyme treatment, Ranjit and Young found that E. coli relied on the Rcs stress response as well as several envelope proteins to regenerate their normal shape (Ranjit and Young 2013). The investigators pre-labeled live cells with fluorescent D-amino acid prior to spheroplasting to confirm the lack of correlation between residual peptidoglycan left in the spheroplasts and the ability of a mutant strain to recover its shape. Strikingly, the mechanisms required for de novo production of cell shape are dispensable for normal laboratory growth. Because E. coli and other intestinal flora are likely subject to cell wall-damaging lysozyme pressure in their natural habitat, the authors speculate that the pathways required for de novo shape generation may be especially important for commensals and pathogens to survive the in vivo environment.
Aquatic environment
Another example of bacterial morphologic adaptation is the stalk. Stalks are appendage-like extensions of the cell wall common to bacteria that inhabit nutrient-poor, aquatic environments. These prosthecae allow the bacterial cell to increase its surface area available for nutrient absorption without changing its surface-to-volume ratio (Young 2006). Jiang et al. (2014) investigated the mechanisms underlying the natural variation in stalk position along the bacterial cell body. Using a combination of amine-reactive dye and fluorescent D-amino acid labeling, the authors found that stalks were synthesized by peptidoglycan insertion at the base, regardless of stalk positioning or species of origin.
Cell surface enzyme activity
With the proper controls for the variables that might impact labeling detection, metabolic incorporation in some cases can be used as a read-out for cell envelope enzyme activity. This concept is related to that of activity-based protein profiling, which uses chemical probes that bind covalently to the active sites of mechanistically related enzymes (Li, Overkleeft and Florea 2012). Similarly, extracellular incorporation and removal of labeled metabolites into and out of the larger biomolecule of interest depends critically on the action of the mechanistically related enzymes (Alvarez et al., 2014). We have already discussed the potential for peptidoglycan amino acids (single D-amino acids and carboxamides as well as dipeptide probes) to report carboxypeptidation and transpeptidation and for labeling by certain trehalose derivatives to serve as a read-out for Antigen 85 activity. In this section, we will consider additional examples.
As exemplified by the detection of dipeptide analog labeling in Chlamydia (Liechti et al., 2013), metabolic labeling can provide evidence for the existence of enzymatic activity that is predicted by genomic analysis but difficult to evaluate by standard biochemical techniques. There are 22 putative glycosyltransferases encoded in the Helicobacter pylori strain 26695 genome, half of which lack predicted functions (Champasa et al., 2013). The first hint that the activity of these enzymes might extend to cell surface substrates other than flagellin (Schirm et al., 2003) came from metabolic labeling experiments (Koenigs, Richardson and Dube 2009). Incubation of the bacteria in azido-GlcNAc did not result in measurable labeling, possibly because the reporter group is removed by the NagA-catalyzed deacetylation step of the GlcNAc recycling pathway (Park 2001) or because azido-GlcNAc inhibits NagA and is not processed into the UDP-GlcNAc donor required for the glycosylation reaction. However, peracetylated azido-GlcNAc permitted metabolic labeling that was detectable by Staudinger ligation (Table 1) of the FLAG epitope tag. Subsequent fractionation studies showed that different subcellular compartments, including the plasma membrane and periplasm, had varied distributions of FLAG-tagged glycoproteins (Champasa et al., 2013). In mammalian cells, peracetylation of glycan analogs is a well-established technique for facilitating cell entry by passive diffusion. Non-specific esterases remove the acetyl groups and allow the glycan derivatives to access the endogenous metabolic pathways for incorporation into biomolecules (Luchansky et al., 2003). However, this non-specific esterase activity has been shown to exist at very low levels in E. coli (Antonczak, Simova and Tippmann 2009), which had significant implications for the interpretation of prior metabolic labeling studies by peracetylated glycosylated amino acid derivatives in this organism (Xu et al., 2004; Zhang et al., 2004; Check Hayden 2009). Therefore, to test whether labeled H. pylori biomolecules were bona fide glycoproteins, Koenigs, Richardson and Dube (2009) showed that azide-dependent FLAG-tag signal in cell lysates was susceptible to β-elimination with sodium hydroxide and digestion by PNGase F, suggesting the presence of both O- and N-linked glycans, respectively. In a follow-up study from the same group, FLAG-tagged lysates were enriched by affinity chromatography and subjected to mass spectrometry analysis. Fragmentation analysis revealed that at least a subset of these proteins contained a Staudinger ligation-glycan adduct that was released upon β-elimination (Champasa et al., 2013). Glycosylation of the surface-associated virulence protein urease was confirmed by pull-down and mass spectrometry experiments. These data support the notion that some fraction of the peracetylated GlcNAc may be deacetylated and incorporated into O-linked glycoproteins in H. pylori. In other work, the presence of multiple, glycosylated H. pylori proteins has been confirmed by hydrazide labeling and mass spectrometry analysis (Hopf et al., 2011).
D,D-transpeptidases join the penultimate D-alanine and meso-diaminopimelic acid (DAP) or L-lysine of the peptidoglycan stem peptide to form 4–3 cross-links. L,D-transpeptidases join two meso-DAP residues to form 3–3 cross-links. In most organisms for which peptidoglycan has been analyzed, the 4–3 cross-links predominate. However, in Actinobacteria, the Rhizobiales and clostridial species, 3–3 bonds are the primary form of cross-linking (Leyh-Bouille et al., 1970; Wietzerbin et al., 1974; Lavollay et al., 2008, 2011; Peltier et al., 2011; Brown et al., 2012; Kumar et al., 2012). The significance of cross-link positioning is not clear. Cameron et al. (2014) detected strong evidence for D,D-transpeptidase residence and activity at the A. tumefaciens septum but much weaker signatures at polar elongation sites. Based on the abundance of putative L,D-transpeptidases encoded in the A. tumefaciens genome and the high proportion of the 3–3 cross-links in A. tumefaciens peptidoglycan (Brown et al., 2012), they hypothesized that L,D-transpeptidases might play a key role in remodeling during polar growth. In support of this hypothesis, one of the L,D-transpeptidase homologs localized strongly to the growth pole (Cameron et al., 2014). To test whether L,D-transpeptidase activity was similarly concentrated at the elongation site, they performed D-alanine metabolic labeling. D-amino acid analogs are likely incorporated into A. tumefaciens peptidoglycan by L,D transpeptidation (Kuru et al., 2012). The labeling patterns produced by these probes (Kuru et al., 2012; Cameron et al., 2014) are consistent with known sites of nascent cell wall synthesis during division and polar elongation (Brown et al., 2012), suggesting that L,D-transpeptidases play a major role in remodeling new or nearly new peptidoglycan at these locations.
Since signal from metabolic labeling is a balance between what gets added to the cell surface and what gets removed, this technique can also shed insight on degradative enzymatic activity such as peptidoglycan carboxypeptidases and amidases. Olrichs et al. (2011) showed that a fluorophore-coupled L-ala D-glu L-lys tripeptide can access the peptidoglycan recycling pathway in E. coli and label both the cytoplasm and cell surface. Surprisingly, they found that the mid-cell regions of dividing cells appeared consistently less fluorescent than the sidewalls of bacteria. Other groups have observed septal labeling in wild-type E. coli using single D-amino acid analogs (Kuru et al., 2012; Fenton and Gerdes 2013), so a trivial explanation for this result could have been that the washes required to remove the excess label prior to imaging the live bacteria produced an inadvertent pulse-chase effect. Alternatively, it may be that the products of the recycling pathway do not contribute to new peptidoglycan at the division site, or that the label is rapidly removed from the septum by peptidoglycan hydrolases. To distinguish between these possibilities, the authors surveyed the labeling patterns of mutants defective at different stages of division. Bacteria deficient in FtsZ, the first protein to assemble at the E. coli division site, had a less pronounced difference in labeling intensity between mid-cell and sidewalls compared to wild-type, whereas those deficient in the later-recruited FtsQ or PBP3 had a similar or augmented difference compared to wild-type. These data suggested that the non-fluorescent bands at the division site are dependent on FtsZ but not FtsQ or PBP3. Pulse-chase experiments showed that cell surface fluorescence disappeared more quickly than expected from growth dilution alone, hinting that the label might be removed enzymatically. In support, a triple-amidase mutant, which grows as chains of cell connected by thick peptidoglycan bands, had strong fluorescence at nearly every incipient septa. Based on these data, the authors hypothesize that the presence of FtsZ is required to initiate concentrated amidase activity.
Secretion systems
The bacterial cell surface protects against various environmental insults. However, it also poses a physical barrier to the acquisition of nutrients and export of toxins. To overcome this challenge, bacteria employ both general and specialized systems to enable proteins to traverse the cell envelope. In recent years, researchers have used metabolic labeling by unnatural amino acids to probe the structure and function of export machines and their substrates (Table 2).
The well-conserved Sec pathway transfers proteins into and across the endoplasmic reticulum in eukaryotic cells and the inner membrane of bacteria (Beckwith 2013). The SecA protein plays an integral role in bacterial Sec export. It maintains substrates in a secretion-competent form, binds components of the SecYEG translocase and provides energy for translocation (Mandon, Trueman and Gilmore 2009). The genomes of some Gram-positive species encode two SecA homologs (Feltcher and Braunstein 2012). Whereas the SecA1 system performs housekeeping functions and is required for bacterial viability, the SecA2 accessory systems are more specialized and often not essential (Feltcher and Braunstein 2012). In E. coli, site-specific, in vivo metabolic labeling experiments utilizing photoreactive amino acid analogs (Pham, Parker and Kohler 2013; Preston and Wilson 2013) have mapped SecA-SecY (Mori and Ito 2006; Das and Oliver 2011) and SecA-SecG (Das and Oliver 2011) interactions as well as the SecA dimer interface (Yu et al., 2013) and binding between SecY and the YidC protein (Sachelaru et al., 2013). The same approach has been used to detect interactions between SecA2 and the GspB substrate protein of the accessory S. gordonii Sec secretion system reconstituted in E. coli (Bensing et al., 2012).
One of the strengths of the photo-cross-linking amino acid approach is that it may be used to capture transient contacts in a near-native environment. In addition to the Sec system, the technique has been used to map protein–protein interactions for the twin-arginine translocation (Tat) secretion system (Frobel, Rose and Muller 2011; Zoufaly et al., 2012), EspA autotransporter (Ieva et al., 2011; Pavlova, Ieva and Bernstein 2013), Lol lipoprotein chaperones (Okuda and Tokuda 2009) in E. coli, the HdeA acid chaperone (Lin et al., 2011; Zhang et al., 2011) in E. coli and Shigella flexneri and the Isd heme transporters in S. aureus (Abe et al., 2012). Researchers have also used an analogous strategy to trap complexes between proteins and polysaccharides in transit, for example, between Lpt proteins and LPS intermediates (Okuda, Freinkman and Kahne 2012) and between the Wza channel and capsular polysaccharide intermediates (Nickerson et al., 2014).
In addition to the general Sec and Tat secretion systems, bacteria employ a variety of specialized export mechanisms. These systems tend to be inducible and required under particular conditions. Effector proteins exported by specialized secretion systems must traverse many layers of the cell envelope to reach the outside environment. When the target of the effector is intracellular, the protein must additionally cross eukaryotic membranes (Galan 2009) or the cell envelope of another bacterium (Ho, Dong and Mekalanos 2014; Russell, Peterson and Mougous 2014). The spatial constraints on protein delivery restrict the choice and position of genetically encoded tags and have hampered efforts to visualize effectors in vivo. The small chemical reporters used in metabolic labeling offer less perturbing alternatives to bulky protein tags. For example, Lin et al. (2011) were able to detect secretion of the S. flexneri type III secretion system (TTSS) substrate OspF after site-specific unnatural amino acid incorporation but not after fusion to GFP. Furthermore, the function of unnatural amino acid-labeled OspF was preserved in an in vitro assay of mitogen-activated protein kinase dephosphorylation. OspF was observed in S. flexneri supernatants by immunoblot and in-gel fluorescence. Recently, Mahdavi et al. (2014) took a more general approach and labeled the proteomes of both extracellular and intracellular Yersinia enterocolitica with azide-functionalized amino acids. They compared the secretomes of wild-type bacteria to a TTSS mutant to identify effector proteins, which they visualized by both in-gel fluorescence and microscopy. In addition to distinguishing between effectors secreted by intracellular and extracellular Y. enterocolitica, the researchers were able to define the order of TTSS substrates injected into cultured host cells by pulse labeling.
The interplay between secreted bacterial proteins and host targets is complex. Some bacterial pathogens can modify host proteins directly while others co-opt host machinery to modify the function of their own proteins. Dynamic, substoichiometric protein modifications can be difficult to detect by conventional means but in many cases are readily monitored by metabolic labeling (Grammel and Hang 2013). For example, in vitro assays have been used to track AMPylation and cleavage of N-myristoyl groups of mammalian cell proteins by bacterial effectors (Grammel et al., 2011; Burnaevskiy et al., 2013). Host lipidation and delipidation of bacterial proteins have also been characterized by metabolic labeling in vivo. The Salmonella TTSS effector proteins SspH2 and SseI home to the plasma membrane of host cells, a process that depends on the S-palmitoyl post-translational modification. Lipidation and subsequent subcellular localization depends on host enzymes (Hicks et al., 2011). By surveying a panel of mutants for plasma membrane residence and the ability to incorporate an alkynyl palmitic acid reporter, Hicks and coworkers were able to identify the cysteine attachment site. Ivanov et al. (2010) used alkynyl farnesol reporters to demonstrate that lipidation of some Legionella pneumophila type IV/Dot secretion effectors likewise is dependent on host enzymes. Perturbation of these host systems by different small molecule inhibitors caused delocalization of the effectors and interfered with L. pneumophila vacuole remodeling.
FUTURE OUTLOOK
Metabolic labeling of the bacterial cell surface has yielded unique insights into growth, division and secretion. The technique has been particularly powerful when applied to organisms with less tractable life cycles or genetics, or to biomolecules that are not amenable to tagging by other methods. The embedded metabolite enables precise tracking in time and space, and the versatility of chemoselective detection offers the ability to pursue biochemical and imaging experiments in tandem. Given the right building block, uptake pathway and enzymatic activity, chemists can target nearly any macromolecule in the cell envelope, from proteins to non-genetically-encoded glycans and lipids. Likewise, biologists can use labeled metabolites to ask questions that were previously inaccessible by conventional techniques.
Tremendous opportunities remain for discovery and innovation via metabolic cell surface tagging. For example, one area that is unexplored by non-radioactive labeling is innate immune detection of bacterial cell wall pathogen-associated molecular patterns (PAMPs). In the case of peptidoglycan, the current methods may not be ideal because they do not tag the portion of the polymer that is recognized by host receptors (Philpott et al., 2014). Investigating new locations for appending chemical reporters to cell wall fragments released by pathogenic and commensal bacteria in vivo might enable researchers to track the PAMPS by microscopy and to identify binding partners by cross-linking. Another opportunity for metabolic labeling is the development of more sophisticated diagnostics. While current efforts are generally directed towards identifying the disease-causing pathogen, an additional metabolic labeling step might enable detection of enzymatic activity that is susceptible to therapeutic intervention. Because the tagging strategies rely on metabolic activity of the bacterium for probe uptake and incorporation, they may also facilitate discrimination of actively growing and quiescent subpopulations from patient samples. Multidirectional collaborations between chemists, biologists and medical researchers will be essential for realizing the basic and translational potential of the bacterial cell surface.
FUNDING
This work was supported by a grant to C.R.B. from N.I.H. (1RO1 AI051622). M.S.S. received a postdoctoral fellowship from the American Cancer Society (119087-PF-10-258-01-MPC). B.M.S. is supported by Central Michigan University and a Cottrell College Science Award from the Research Corporation for Science Advancement (#22525).
Conflict of Interest Statement. None declared.
REFERENCES
- Abe R, Caaveiro JM, Kozuka-Hata H, et al. Mapping ultra-weak protein–protein interactions between heme transporters of Staphylococcus aureus. J Biol Chem. 2012;287:16477–87. doi: 10.1074/jbc.M112.346700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–7. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Aldridge BB, Fernandez-Suarez M, Heller D, et al. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science. 2012;335:100–4. doi: 10.1126/science.1216166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MM, Kang DK, Tsang K, et al. Cell-surface sensors: lighting the cellular environment. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4:547–61. doi: 10.1002/wnan.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez L, Espaillat A, Hermoso JA, et al. Peptidoglycan remodeling by the coordinated action of multispecific enzymes. Microb Drug Resist. 2014;20:190–8. doi: 10.1089/mdr.2014.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonczak AK, Simova Z, Tippmann EM. A critical examination of Escherichia coli esterase activity. J Biol Chem. 2009;284:28795–800. doi: 10.1074/jbc.M109.027409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KM, Boshoff HI, Barry CS, et al. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat Chem Biol. 2011;7:228–35. doi: 10.1038/nchembio.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal-Mutalik R, Nikaido H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. P Natl Acad Sci USA. 2014;111:4958–63. doi: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardou F, Quemard A, Dupont MA, et al. Effects of isoniazid on ultrastructure of Mycobacterium aurum and Mycobacterium tuberculosis and on production of secreted proteins. Antimicrob Agents Ch. 1996;40:2459–67. doi: 10.1128/aac.40.11.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreteau H, Kovac A, Boniface A, et al. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- Barry CE, 3rd, Boshoff HI, Dartois V, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–55. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin JM, Prescher JA, Laughlin ST, et al. Copper-free click chemistry for dynamic in vivo imaging. P Natl Acad Sci USA. 2007;104:16793–7. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith J. The Sec-dependent pathway. Res Microbiol. 2013;164:497–504. doi: 10.1016/j.resmic.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belisle JT, Vissa VD, Sievert T, et al. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–2. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- Bensing BA, Yen YT, Seepersaud R, et al. A specific interaction between SecA2 and a region of the preprotein adjacent to the signal peptide occurs during transport via the accessory Sec system. J Biol Chem. 2012;287:24438–47. doi: 10.1074/jbc.M112.378059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besanceney-Webler C, Jiang H, Wang W, et al. Metabolic labeling of fucosylated glycoproteins in Bacteroidales species. Bioorg Med Chem Lett. 2011;21:4989–92. doi: 10.1016/j.bmcl.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsell DC, Cota-Robles EH. Production and ultrastructure of lysozyme and ethylenediaminetetraacetate-lysozyme spheroplasts of Escherichia coli. J Bacteriol. 1967;93:427–37. doi: 10.1128/jb.93.1.427-437.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman ML, Royzen M, Fox JM. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J Am Chem Soc. 2008;130:13518–9. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneca IG. The role of peptidoglycan in pathogenesis. Curr Opin Microbiol. 2005;8:46–53. doi: 10.1016/j.mib.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Bouley DM, Ghori N, Mercer KL, et al. Dynamic nature of host-pathogen interactions in Mycobacterium marinum granulomas. Infect Immun. 2001;69:7820–31. doi: 10.1128/IAI.69.12.7820-7831.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PJ, de Pedro MA, Kysela DT, et al. Polar growth in the Alphaproteobacterial order Rhizobiales. P Natl Acad Sci USA. 2012;109:1697–701. doi: 10.1073/pnas.1114476109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunschoten A, Welling MM, Termaat MF, et al. Development and prospects of dedicated tracers for the molecular imaging of bacterial infections. Bioconjugate Chem. 2013;24:1971–89. doi: 10.1021/bc4003037. [DOI] [PubMed] [Google Scholar]
- Burnaevskiy N, Fox TG, Plymire DA, et al. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature. 2013;496:106–9. doi: 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G, Peregrin-Alvarez JM, Li J, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–7. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- Cabeen MT, Jacobs-Wagner C. Bacterial cell shape. Nat Rev Microbiol. 2005;3:601–10. doi: 10.1038/nrmicro1205. [DOI] [PubMed] [Google Scholar]
- Cameron TA, Anderson-Furgeson J, Zupan JR, et al. Peptidoglycan synthesis machinery in Agrobacterium tumefaciens during unipolar growth and cell division. mBio. 2014;5:e01219–14. doi: 10.1128/mBio.01219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparros M, Pisabarro AG, de Pedro MA. Effect of D-amino acids on structure and synthesis of peptidoglycan in Escherichia coli. J Bacteriol. 1992;174:5549–59. doi: 10.1128/jb.174.17.5549-5559.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carel C, Nukdee K, Cantaloube S, et al. Mycobacterium tuberculosis proteins involved in mycolic acid synthesis and transport localize dynamically to the old growing pole and septum. PLoS One. 2014;9:e97148. doi: 10.1371/journal.pone.0097148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cava F, de Pedro MA, Lam H, et al. Distinct pathways for modification of the bacterial cell wall by non-canonical D-amino acids. EMBO J. 2011;30:3442–53. doi: 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cava F, Kuru E, Brun YV, et al. Modes of cell wall growth differentiation in rod-shaped bacteria. Curr Opin Microbiol. 2013;16:731–7. doi: 10.1016/j.mib.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cava F, Lam H, de Pedro MA, et al. Emerging knowledge of regulatory roles of D-amino acids in bacteria. Cell Mol Life Sci. 2011;68:817–31. doi: 10.1007/s00018-010-0571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champasa K, Longwell SA, Eldridge AM, et al. Targeted identification of glycosylated proteins in the gastric pathogen Helicobacter pylori (Hp) Mol Cell Proteomics. 2013;12:2568–86. doi: 10.1074/mcp.M113.029561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Dodani SC, Chang CJ. Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nat Chem. 2012;4:973–84. doi: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Lofton H, Maloney E, et al. Interference of Mycobacterium tuberculosis cell division by Rv2719c, a cell wall hydrolase. Mol Microbiol. 2006;62:132–47. doi: 10.1111/j.1365-2958.2006.05333.x. [DOI] [PubMed] [Google Scholar]
- Check Hayden E. ‘Killer application’ for protein synthesis is retracted. Nature. 2009;462:707. doi: 10.1038/462707a. [DOI] [PubMed] [Google Scholar]
- Chopra I, Storey C, Falla TJ, et al. Antibiotics, peptidoglycan synthesis and genomics: the chlamydial anomaly revisited. Microbiology. 1998;144(Pt 10):2673–8. doi: 10.1099/00221287-144-10-2673. [DOI] [PubMed] [Google Scholar]
- Crawford JM, Portmann C, Zhang X, et al. Small molecule perimeter defense in entomopathogenic bacteria. P Natl Acad Sci USA. 2012;109:10821–6. doi: 10.1073/pnas.1201160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins GT, Isberg RR. Analyzing microbial disease at high resolution: following the fate of the bacterium during infection. Curr Opin Microbiol. 2012;15:23–7. doi: 10.1016/j.mib.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JL. Electron microscopy analysis of Mycobacterium tuberculosis cell division. FEMS Microbiol Lett. 2004;240:15–20. doi: 10.1016/j.femsle.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Daniel RA, Errington J. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell. 2003;113:767–76. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- Das S, Oliver DB. Mapping of the SecA.SecY and SecA.SecG interfaces by site-directed in vivo photocross-linking. J Biol Chem. 2011;286:12371–80. doi: 10.1074/jbc.M110.182931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pedro MA, Grunfelder CG, Schwarz H. Restricted mobility of cell surface proteins in the polar regions of Escherichia coli. J Bacteriol. 2004;186:2594–602. doi: 10.1128/JB.186.9.2594-2602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pedro MA, Quintela JC, Holtje JV, et al. Murein segregation in Escherichia coli. J Bacteriol. 1997;179:2823–34. doi: 10.1128/jb.179.9.2823-2834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmarais SM, De Pedro MA, Cava F, et al. Peptidoglycan at its peaks: how chromatographic analyses can reveal bacterial cell wall structure and assembly. Mol Microbiol. 2013;89:1–13. doi: 10.1111/mmi.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommerholt J, Schmidt S, Temming R, et al. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew Chem Int Edit. 2010;49:9422–5. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube DH, Champasa K, Wang B. Chemical tools to discover and target bacterial glycoproteins. Chem Commun (Camb) 2011;47:87–101. doi: 10.1039/c0cc01557a. [DOI] [PubMed] [Google Scholar]
- Dumont A, Malleron A, Awwad M, et al. Click-mediated labeling of bacterial membranes through metabolic modification of the lipopolysaccharide inner core. Angew Chem Int Edit. 2012;51:3143–6. doi: 10.1002/anie.201108127. [DOI] [PubMed] [Google Scholar]
- Egan AJ, Vollmer W. The physiology of bacterial cell division. Ann NY Acad Sci. 2013;1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- Eun YJ, Zhou M, Kiekebusch D, et al. Divin: a small molecule inhibitor of bacterial divisome assembly. J Am Chem Soc. 2013;135:9768–76. doi: 10.1021/ja404640f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltcher ME, Braunstein M. Emerging themes in SecA2-mediated protein export. Nat Rev Microbiol. 2012;10:779–89. doi: 10.1038/nrmicro2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AK, Gerdes K. Direct interaction of FtsZ and MreB is required for septum synthesis and cell division in Escherichia coli. EMBO J. 2013;32:1953–65. doi: 10.1038/emboj.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleurie A, Manuse S, Zhao C, et al. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet. 2014;10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss MH, Eun YJ, Weibel DB. Chemical-biological studies of subcellular organization in bacteria. Biochemistry. 2011;50:7719–34. doi: 10.1021/bi200940d. [DOI] [PubMed] [Google Scholar]
- Frirdich E, Gaynor EC. Peptidoglycan hydrolases, bacterial shape, and pathogenesis. Curr Opin Microbiol. 2013;16:767–78. doi: 10.1016/j.mib.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Frobel J, Rose P, Muller M. Early contacts between substrate proteins and TatA translocase component in twin-arginine translocation. J Biol Chem. 2011;286:43679–89. doi: 10.1074/jbc.M111.292565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fura JM, Sabulski MJ, Pires MM. D-amino acid mediated recruitment of endogenous antibodies to bacterial surfaces. ACS Chem Biol. 2014;9:1480–9. doi: 10.1021/cb5002685. [DOI] [PubMed] [Google Scholar]
- Galan JE. Common themes in the design and function of bacterial effectors. Cell Host Microbe. 2009;5:571–9. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam S, Gniadek TJ, Kim T, et al. Exterior design: strategies for redecorating the bacterial surface with small molecules. Trends Biotechnol. 2013;31:258–67. doi: 10.1016/j.tibtech.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Young KD. Helical disposition of proteins and lipopolysaccharide in the outer membrane of Escherichia coli. J Bacteriol. 2005;187:1913–22. doi: 10.1128/JB.187.6.1913-1922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghuysen JM, Goffin C. Lack of cell wall peptidoglycan versus penicillin sensitivity: new insights into the chlamydial anomaly. Antimicrob Agents Ch. 1999;43:2339–44. doi: 10.1128/aac.43.10.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs KA, Isaac DD, Xu J, et al. Complex spatial distribution and dynamics of an abundant Escherichia coli outer membrane protein, LamB. Mol Microbiol. 2004;53:1771–83. doi: 10.1111/j.1365-2958.2004.04242.x. [DOI] [PubMed] [Google Scholar]
- Gill WP, Harik NS, Whiddon MR, et al. A replication clock for Mycobacterium tuberculosis. Nat Med. 2009;15:211–4. doi: 10.1038/nm.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–89. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzy A, Larrouy-Maumus G, Wu TD, et al. Mycobacterium tuberculosis nitrogen assimilation and host colonization require aspartate. Nat Chem Biol. 2013;9:674–6. doi: 10.1038/nchembio.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel M, Hang HC. Chemical reporters for biological discovery. Nat Chem Biol. 2013;9:475–84. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel M, Luong P, Orth K, et al. A chemical reporter for protein AMPylation. J Am Chem Soc. 2011;133:17103–5. doi: 10.1021/ja205137d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzenpichler R, Scheller S, Tavormina PL, et al. In situ visualization of newly synthesized proteins in environmental microbes using amino acid tagging and click chemistry. Environ Microbiol. 2014;16:2568–90. doi: 10.1111/1462-2920.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helaine S, Holden DW. Heterogeneity of intracellular replication of bacterial pathogens. Curr Opin Microbiol. 2013;16:184–91. doi: 10.1016/j.mib.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Hett EC, Rubin EJ. Bacterial growth and cell division: a mycobacterial perspective. Microbiol Mol Biol Rev. 2008;72:126–56. doi: 10.1128/MMBR.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks SW, Charron G, Hang HC, et al. Subcellular targeting of Salmonella virulence proteins by host-mediated S-palmitoylation. Cell Host Microbe. 2011;10:9–20. doi: 10.1016/j.chom.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BT, Dong TG, Mekalanos JJ. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe. 2014;15:9–21. doi: 10.1016/j.chom.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking J, Priyadarshini R, Takacs CN, et al. Osmolality-dependent relocation of penicillin-binding protein PBP2 to the division site in Caulobacter crescentus. J Bacteriol. 2012;194:3116–27. doi: 10.1128/JB.00260-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf PS, Ford RS, Zebian N, et al. rotein glycosylation in Helicobacter pylori: beyond the flagellins? PLoS One. 2011;6:e25722. doi: 10.1371/journal.pone.0025722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Janga SC, Babu M, et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009;7:e96. doi: 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieva R, Tian P, Peterson JH, et al. Sequential and spatially restricted interactions of assembly factors with an autotransporter beta domain. P Natl Acad Sci USA. 2011;108:E383–391. doi: 10.1073/pnas.1103827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrigo J, Hunter RL, Jr, Actor JK. Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. 2003;149:2049–59. doi: 10.1099/mic.0.26226-0. [DOI] [PubMed] [Google Scholar]
- Ivanov SS, Charron G, Hang HC, et al. Lipidation by the host prenyltransferase machinery facilitates membrane localization of Legionella pneumophila effector proteins. J Biol Chem. 2010;285:34686–98. doi: 10.1074/jbc.M110.170746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier N, Frandi A, Pillonel T, et al. Cell wall precursors are required to organize the chlamydial division septum. Nat Commun. 2014;5:3578. doi: 10.1038/ncomms4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett JC, Sletten EM, Bertozzi CR. Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J Am Chem Soc. 2010;132:3688–90. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Brown PJ, Ducret A, et al. Sequential evolution of bacterial morphology by co-option of a developmental regulator. Nature. 2014;506:489–93. doi: 10.1038/nature12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce G, Williams KJ, Robb M, et al. Cell division site placement and asymmetric growth in mycobacteria. PLoS One. 2012;7:e44582. doi: 10.1371/journal.pone.0044582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice SS, Hunstad DA, Cegelski L, et al. Morphological plasticity as a bacterial survival strategy. Nat Rev Microbiol. 2008;6:162–8. doi: 10.1038/nrmicro1820. [DOI] [PubMed] [Google Scholar]
- Kaewsapsak P, Esonu O, Dube DH. Recruiting the host's immune system to target Helicobacter pylori's surface glycans. Chembiochem. 2013;14:721–6. doi: 10.1002/cbic.201300006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser KJ, Rubin EJ. How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol. 2014;12:550–62. doi: 10.1038/nrmicro3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigs MB, Richardson EA, Dube DH. Metabolic profiling of Helicobacter pylori glycosylation. Mol Biosyst. 2009;5:909–12. doi: 10.1039/b902178g. [DOI] [PubMed] [Google Scholar]
- Kumar P, Arora K, Lloyd JR, et al. Meropenem inhibits D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol Microbiol. 2012;86:367–81. doi: 10.1111/j.1365-2958.2012.08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, Hughes HV, Brown PJ, et al. In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Edit. 2012;51:12519–23. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kysela DT, Brown PJ, Huang KC, et al. Biological consequences and advantages of asymmetric bacterial growth. Annu Rev Microbiol. 2013;67:417–35. doi: 10.1146/annurev-micro-092412-155622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H, Oh DC, Cava F, et al. D-amino acids govern stationary phase cell wall remodeling in bacteria. Science. 2009;325:1552–5. doi: 10.1126/science.1178123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavollay M, Arthur M, Fourgeaud M, et al. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J Bacteriol. 2008;190:4360–6. doi: 10.1128/JB.00239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavollay M, Fourgeaud M, Herrmann JL, et al. The peptidoglycan of Mycobacterium abscessus is predominantly cross-linked by L,D-transpeptidases. J Bacteriol. 2011;193:778–82. doi: 10.1128/JB.00606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebar MD, May JM, Meeske AJ, et al. Reconstitution of peptidoglycan cross-linking leads to improved fluorescent probes of cell wall synthesis. J Am Chem Soc. 2014;136:10874–7. doi: 10.1021/ja505668f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J, St Clair J. Protoplasts and L-type growth of Escherichia coli. J Bacteriol. 1958;75:143–60. doi: 10.1128/jb.75.2.143-160.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyh-Bouille M, Bonaly R, Ghuysen JM, et al. LL-diaminopimelic acid containing peptidoglycans in walls of Streptomyces sp. and of Clostridium perfringens (type A) Biochemistry. 1970;9:2944–52. doi: 10.1021/bi00817a002. [DOI] [PubMed] [Google Scholar]
- Li N, Overkleeft HS, Florea BI. Activity-based protein profiling: an enabling technology in chemical biology research. Curr Opin Chem Biol. 2012;16:227–33. doi: 10.1016/j.cbpa.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Liechti GW, Kuru E, Hall E, et al. A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature. 2013;506:507–10. doi: 10.1038/nature12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Zhang Z, Xu H, et al. Site-specific incorporation of photo-cross-linker and bioorthogonal amino acids into enteric bacterial pathogens. J Am Chem Soc. 2011;133:20581–7. doi: 10.1021/ja209008w. [DOI] [PubMed] [Google Scholar]
- Link AJ, Tirrell DA. Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3+2] cycloaddition. J Am Chem Soc. 2003;125:11164–5. doi: 10.1021/ja036765z. [DOI] [PubMed] [Google Scholar]
- Link AJ, Vink MK, Agard NJ, et al. Discovery of aminoacyl-tRNA synthetase activity through cell-surface display of noncanonical amino acids. P Natl Acad Sci USA. 2006;103:10180–5. doi: 10.1073/pnas.0601167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Vink MK, Tirrell DA. Presentation and detection of azide functionality in bacterial cell surface proteins. J Am Chem Soc. 2004;126:10598–602. doi: 10.1021/ja047629c. [DOI] [PubMed] [Google Scholar]
- Liu F, Aubry AJ, Schoenhofen IC, et al. The engineering of bacteria bearing azido-pseudaminic acid-modified flagella. Chembiochem. 2009;10:1317–20. doi: 10.1002/cbic.200900018. [DOI] [PubMed] [Google Scholar]
- Luchansky SJ, Hang HC, Saxon E, et al. Constructing azide-labeled cell surfaces using polysaccharide biosynthetic pathways. Method Enzymol. 2003;362:249–72. doi: 10.1016/S0076-6879(03)01018-8. [DOI] [PubMed] [Google Scholar]
- Lupoli TJ, Tsukamoto H, Doud EH, et al. Transpeptidase-mediated incorporation of D-amino acids into bacterial peptidoglycan. J Am Chem Soc. 2011;133:10748–51. doi: 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Maurelli AT. Building the invisible wall: updating the chlamydial peptidoglycan anomaly. Trends Microbiol. 2006;14:70–7. doi: 10.1016/j.tim.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Mahdavi A, Szychowski J, Ngo JT, et al. Identification of secreted bacterial proteins by noncanonical amino acid tagging. P Natl Acad Sci USA. 2014;111:433–8. doi: 10.1073/pnas.1301740111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V, Manina G, Mikusova K, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–4. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandon EC, Trueman SF, Gilmore R. Translocation of proteins through the Sec61 and SecYEG channels. Curr Opin Cell Biol. 2009;21:501–7. doi: 10.1016/j.ceb.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manina G, McKinney JD. A single-cell perspective on non-growing but metabolically active (NGMA) bacteria. Curr Top Microbiol Immunol. 2013;374:135–61. doi: 10.1007/82_2013_333. [DOI] [PubMed] [Google Scholar]
- Mas Pons J, Dumont A, Sautejeau G, et al. Identification of living Legionella pneumophila using species-specific metabolic lipopolysaccharide labeling. Angew Chem Int Edit. 2014;53:1275–8. doi: 10.1002/anie.201309072. [DOI] [PubMed] [Google Scholar]
- Memmel E, Homann A, Oelschlaeger TA, et al. Metabolic glycoengineering of Staphylococcus aureus reduces its adherence to human T24 bladder carcinoma cells. Chem Commun (Camb) 2013;49:7301–3. doi: 10.1039/c3cc43424a. [DOI] [PubMed] [Google Scholar]
- Meniche X, Otten R, Siegrist MS, et al. Subpolar addition of new cell wall is directed by DivIVA in mycobacteria. P Natl Acad Sci USA. 2014;111:E3243–51. doi: 10.1073/pnas.1402158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P, Gutierrez J, Pogliano K, et al. Cell wall synthesis is necessary for membrane dynamics during sporulation of Bacillus subtilis. Mol Microbiol. 2010;76:956–70. doi: 10.1111/j.1365-2958.2010.07155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi T, Breukink E. The chlamydial anomaly clarified? Chembiochem. 2014;15:1391–2. doi: 10.1002/cbic.201402143. [DOI] [PubMed] [Google Scholar]
- Mori H, Ito K. Different modes of SecY-SecA interactions revealed by site-directed in vivo photo-cross-linking. P Natl Acad Sci USA. 2006;103:16159–64. doi: 10.1073/pnas.0606390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder JW. Why is Chlamydia sensitive to penicillin in the absence of peptidoglycan? Infect Agent Dis. 1993;2:87–99. [PubMed] [Google Scholar]
- Muhlradt PF, Menzel J, Golecki JR, et al. Outer membrane of salmonella. Sites of export of newly synthesised lipopolysaccharide on the bacterial surface. Eur J Biochem. 1973;35:471–81. doi: 10.1111/j.1432-1033.1973.tb02861.x. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Chamessian AG, McEnaney PJ, et al. A biosynthetic strategy for re-engineering the Staphylococcus aureus cell wall with non-native small molecules. ACS Chem Biol. 2010;5:1147–55. doi: 10.1021/cb100195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JT, Tirrell DA. Noncanonical amino acids in the interrogation of cellular protein synthesis. Accounts Chem Res. 2011;44:677–85. doi: 10.1021/ar200144y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson NN, Mainprize IL, Hampton L, et al. Trapped translocation intermediates establish the route for export of capsular polysaccharides across Escherichia coli outer membranes. P Natl Acad Sci USA. 2014;111:8203–8. doi: 10.1073/pnas.1400341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning X, Guo J, Wolfert MA, et al. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew Chem Int Edit. 2008;47:2253–5. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Freinkman E, Kahne D. Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science. 2012;338:1214–7. doi: 10.1126/science.1228984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Tokuda H. Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. P Natl Acad Sci USA. 2009;106:5877–82. doi: 10.1073/pnas.0900896106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olrichs NK, Aarsman ME, Verheul J, et al. A novel in vivo cell-wall labeling approach sheds new light on peptidoglycan synthesis in Escherichia coli. Chembiochem. 2011;12:1124–33. doi: 10.1002/cbic.201000552. [DOI] [PubMed] [Google Scholar]
- Park JT. Identification of a dedicated recycling pathway for anhydro-N-acetylmuramic acid and N-acetylglucosamine derived from Escherichia coli cell wall murein. J Bacteriol. 2001;183:3842–7. doi: 10.1128/JB.183.13.3842-3847.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson DM, Nazarova LA, Prescher JA. Finding the right (bioorthogonal) chemistry. ACS Chem Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Patterson DM, Nazarova LA, Xie B, et al. Functionalized cyclopropenes as bioorthogonal chemical reporters. J Am Chem Soc. 2012;134:18638–43. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- Pavlova O, Ieva R, Bernstein HD. Monitoring the assembly of a secreted bacterial virulence factor using site-specific crosslinking. J Vis Exp. 2013;17:e51217. doi: 10.3791/51217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier J, Courtin P, El Meouche I, et al. Clostridium difficile has an original peptidoglycan structure with a high level of N-acetylglucosamine deacetylation and mainly 3–3 cross-links. J Biol Chem. 2011;286:29053–62. doi: 10.1074/jbc.M111.259150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham ND, Parker RB, Kohler JJ. Photocrosslinking approaches to interactome mapping. Curr Opin Chem Biol. 2013;17:90–101. doi: 10.1016/j.cbpa.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott DJ, Sorbara MT, Robertson SJ, et al. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- Pilhofer M, Aistleitner K, Biboy J, et al. Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ. Nat Commun. 2013;4:2856. doi: 10.1038/ncomms3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho MG, Kjos M, Veening JW. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol. 2013;11:601–14. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- Preston GW, Wilson AJ. Photo-induced covalent cross-linking for the analysis of biomolecular interactions. Chem Soc Rev. 2013;42:3289–301. doi: 10.1039/c3cs35459h. [DOI] [PubMed] [Google Scholar]
- Rafelski SM, Theriot JA. Mechanism of polarization of Listeria monocytogenes surface protein ActA. Mol Microbiol. 2006;59:1262–79. doi: 10.1111/j.1365-2958.2006.05025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan KJ, Yang YY, Charron G, et al. Rapid visualization and large-scale profiling of bacterial lipoproteins with chemical reporters. J Am Chem Soc. 2010;132:10628–9. doi: 10.1021/ja101387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjit DK, Young KD. The Rcs stress response and accessory envelope proteins are required for de novo generation of cell shape in Escherichia coli. J Bacteriol. 2013;195:2452–62. doi: 10.1128/JB.00160-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, et al. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew Chem Int Edit. 2002;41:2596–9. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Russell AB, Peterson SB, Mougous JD. Type VI secretion system effectors: poisons with a purpose. Nat Rev Microbiol. 2014;12:137–48. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachelaru I, Petriman NA, Kudva R, et al. YidC occupies the lateral gate of the SecYEG translocon and is sequentially displaced by a nascent membrane protein. J Biol Chem. 2013;288:16295–307. doi: 10.1074/jbc.M112.446583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi I, Dhar N, Bousbaine D, et al. Single-cell dynamics of the chromosome replication and cell division cycles in mycobacteria. Nat Commun. 2013;4:2470. doi: 10.1038/ncomms3470. [DOI] [PubMed] [Google Scholar]
- Sathyamoorthy N, Takayama K. Purification and characterization of a novel mycolic acid exchange enzyme from Mycobacterium smegmatis. J Biol Chem. 1987;262:13417–23. [PubMed] [Google Scholar]
- Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–10. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Schindler M, Osborn MJ, Koppel DE. Lateral diffusion of lipopolysaccharide in the outer membrane of Salmonella typhimurium. Nature. 1980;285:261–3. doi: 10.1038/285261a0. [DOI] [PubMed] [Google Scholar]
- Schirm M, Soo EC, Aubry AJ, et al. Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori. Mol Microbiol. 2003;48:1579–92. doi: 10.1046/j.1365-2958.2003.03527.x. [DOI] [PubMed] [Google Scholar]
- Sham LT, Tsui HC, Land AD, et al. Recent advances in pneumococcal peptidoglycan biosynthesis suggest new vaccine and antimicrobial targets. Curr Opin Microbiol. 2012;15:194–203. doi: 10.1016/j.mib.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh P, Siegrist MS, Cullen AJ, et al. Imaging bacterial peptidoglycan with near-infrared fluorogenic azide probes. P Natl Acad Sci USA. 2014;111:5456–61. doi: 10.1073/pnas.1322727111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist MS, Whiteside S, Jewett JC, et al. (D)-amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol. 2013;8:500–5. doi: 10.1021/cb3004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Nitharwal RG, Ramesh M, et al. Asymmetric growth and division in Mycobacterium spp.: compensatory mechanisms for non-medial septa. Mol Microbiol. 2013;88:64–76. doi: 10.1111/mmi.12169. [DOI] [PubMed] [Google Scholar]
- Sjollema KA, Schnell U, Kuipers J, et al. Correlated light microscopy and electron microscopy. Method Cell Biol. 2012;111:157–73. doi: 10.1016/B978-0-12-416026-2.00009-1. [DOI] [PubMed] [Google Scholar]
- Solomon JM, Leung GS, Isberg RR. Intracellular replication of Mycobacterium marinum within Dictyostelium discoideum: efficient replication in the absence of host coronin. Infect Immun. 2003;71:3578–86. doi: 10.1128/IAI.71.6.3578-3586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spargo BJ, Crowe LM, Ioneda T, et al. Cord factor (alpha,alpha-trehalose 6,6′-dimycolate) inhibits fusion between phospholipid vesicles. P Natl Acad Sci USA. 1991;88:737–40. doi: 10.1073/pnas.88.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts BM, Holsclaw CM, Jewett JC, et al. Probing the mycobacterial trehalome with bioorthogonal chemistry. J Am Chem Soc. 2012;134:16123–6. doi: 10.1021/ja3062419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swulius MT, Jensen GJ. The helical MreB cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-Terminal yellow fluorescent protein tag. J Bacteriol. 2012;194:6382–6. doi: 10.1128/JB.00505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takade A, Takeya K, Taniguchi H, et al. Electron microscopic observations of cell division in Mycobacterium vaccae V1. J Gen Microbiol. 1983;129:2315–20. doi: 10.1099/00221287-129-7-2315. [DOI] [PubMed] [Google Scholar]
- Tan Q, Awano N, Inouye M. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol Microbiol. 2011;79:109–18. doi: 10.1111/j.1365-2958.2010.07433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanrikulu IC, Schmitt E, Mechulam Y, et al. Discovery of Escherichia coli methionyl-tRNA synthetase mutants for efficient labeling of proteins with azidonorleucine in vivo. P Natl Acad Sci USA. 2009;106:15285–90. doi: 10.1073/pnas.0905735106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanky NR, Young DB, Robertson BD. Unusual features of the cell cycle in mycobacteria: polar-restricted growth and the snapping-model of cell division. Tuberculosis (Edinb) 2007;87:231–6. doi: 10.1016/j.tube.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Bouwer HG, Portnoy DA, et al. Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires D-alanine for growth. Infect Immun. 1998;66:3552–61. doi: 10.1128/iai.66.8.3552-3561.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiyanont K, Doan T, Lazarus MB, et al. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. P Natl Acad Sci USA. 2006;103:11033–8. doi: 10.1073/pnas.0600829103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocheva EI, Lopez-Garrido J, Hughes HV, et al. Peptidoglycan transformations during Bacillus subtilis sporulation. Mol Microbiol. 2013;88:673–86. doi: 10.1111/mmi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67:3057–64. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Tra VN, Dube DH. Glycans in pathogenic bacteria–potential for targeted covalent therapeutics and imaging agents. Chem Commun (Camb) 2014;50:4659–73. doi: 10.1039/c4cc00660g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruoka T, Tamura A, Miyata A, et al. Penicillin-insensitive incorporation of D-amino acids into cell wall peptidoglycan influences the amount of bound lipoprotein in Escherichia coli. J Bacteriol. 1984;160:889–94. doi: 10.1128/jb.160.3.889-894.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, et al. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2012;10:123–36. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek BL, Wing DC, Haislop KS, et al. Chemoenzymatic synthesis of trehalose analogues: rapid access to chemical probes for investigating mycobacteria. Chembiochem. 2014;15:2066–70. doi: 10.1002/cbic.201402288. [DOI] [PubMed] [Google Scholar]
- Ursell TS, Trepagnier EH, Huang KC, et al. Analysis of surface protein expression reveals the growth pattern of the gram-negative outer membrane. PLoS Comput Biol. 2012;8:e1002680. doi: 10.1371/journal.pcbi.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam V, Olrichs N, Breukink E. Specific labeling of peptidoglycan precursors as a tool for bacterial cell wall studies. Chembiochem. 2009;10:617–24. doi: 10.1002/cbic.200800678. [DOI] [PubMed] [Google Scholar]
- Wakamoto Y, Dhar N, Chait R, et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science. 2013;339:91–5. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- Watrous JD, Dorrestein PC. Imaging mass spectrometry in microbiology. Nat Rev Microbiol. 2011;9:683–94. doi: 10.1038/nrmicro2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous JD, Phelan VV, Hsu CC, et al. Microbial metabolic exchange in 3D. ISME J. 2013;7:770–80. doi: 10.1038/ismej.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Hu F, Shen Y, et al. Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering. Nat Methods. 2014;11:410–2. doi: 10.1038/nmeth.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietzerbin J, Das BC, Petit JF, et al. Occurrence of D-alanyl-(D)-meso-diaminopimelic acid and meso-diaminopimelyl-meso-diaminopimelic acid interpeptide linkages in the peptidoglycan of Mycobacteria. Biochemistry. 1974;13:3471–6. doi: 10.1021/bi00714a008. [DOI] [PubMed] [Google Scholar]
- Winther T, Xu L, Berg-Sorensen K, et al. Effect of energy metabolism on protein motility in the bacterial outer membrane. Biophys J. 2009;97:1305–12. doi: 10.1016/j.bpj.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Hanson SR, Zhang Z, et al. Site-specific incorporation of the mucin-type N-acetylgalactosamine-alpha-O-threonine into protein in Escherichia coli. J Am Chem Soc. 2004;126:15654–5. doi: 10.1021/ja044711z. [DOI] [PubMed] [Google Scholar]
- Yang J, Seckute J, Cole CM, et al. Live-cell imaging of cyclopropene tags with fluorogenic tetrazine cycloadditions. Angew Chem Int Edit. 2012;51:7476–9. doi: 10.1002/anie.201202122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Song Y, Zhang M, et al. Converting a solvatochromic fluorophore into a protein-based pH indicator for extreme acidity. Angew Chem Int Edit. 2012;51:7674–9. doi: 10.1002/anie.201204029. [DOI] [PubMed] [Google Scholar]
- Yi W, Liu X, Li Y, et al. Remodeling bacterial polysaccharides by metabolic pathway engineering. P Natl Acad Sci USA. 2009;106:4207–12. doi: 10.1073/pnas.0812432106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KD. The selective value of bacterial shape. Microbiol Mol Biol Rev. 2006;70:660–703. doi: 10.1128/MMBR.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Wowor AJ, Cole JL, et al. Defining the Escherichia coli SecA dimer interface residues through in vivo site-specific photo-cross-linking. J Bacteriol. 2013;195:2817–25. doi: 10.1128/JB.02269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Lin S, Song X, et al. A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat Chem Biol. 2011;7:671–7. doi: 10.1038/nchembio.644. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gildersleeve J, Yang YY, et al. A new strategy for the synthesis of glycoproteins. Science. 2004;303:371–3. doi: 10.1126/science.1089509. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Smith BA, Wang L, et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry. 2003;42:6735–46. doi: 10.1021/bi0300231. [DOI] [PubMed] [Google Scholar]
- Zinder ND, Arndt WF. Production of protoplasts of Escherichia Coli by lysozyme treatment. P Natl Acad Sci USA. 1956;42:586–90. doi: 10.1073/pnas.42.9.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoufaly S, Frobel J, Rose P, et al. Mapping precursor-binding site on TatC subunit of twin arginine-specific protein translocase by site-specific photo cross-linking. J Biol Chem. 2012;287:13430–41. doi: 10.1074/jbc.M112.343798. [DOI] [PMC free article] [PubMed] [Google Scholar]