

Abstract
The effects of mechanical cues on cell behaviors in 3D remain difficult to characterize as the ability to tune hydrogel mechanics often requires changes in the polymer density, potentially altering the material’s biochemical and physical characteristics. Additionally, with most PEG diacrylate (PEGDA) hydrogels, forming materials with compressive moduli less than ∼10 kPa has been virtually impossible. Here, we present a new method of controlling the mechanical properties of PEGDA hydrogels independent of polymer chain density through the incorporation of additional vinyl group moieties that interfere with the cross-linking of the network. This modification can tune hydrogel mechanics in a concentration dependent manner from <1 to 17 kPa, a more physiologically relevant range than previously possible with PEG-based hydrogels, without altering the hydrogel’s degradation and permeability. Across this range of mechanical properties, endothelial cells (ECs) encapsulated within MMP-2/MMP-9 degradable hydrogels with RGDS adhesive peptides revealed increased cell spreading as hydrogel stiffness decreased in contrast to behavior typically observed for cells on 2D surfaces. EC-pericyte cocultures exhibited vessel-like networks within 3 days in highly compliant hydrogels as compared to a week in stiffer hydrogels. These vessel networks persisted for at least 4 weeks and deposited laminin and collagen IV perivascularly. These results indicate that EC morphogenesis can be regulated using mechanical cues in 3D. Furthermore, controlling hydrogel compliance independent of density allows for the attainment of highly compliant mechanical regimes in materials that can act as customizable cell microenvironments.
Keywords: vasculogenesis, tissue engineering, biomimetic material, PEG hydrogel
Introduction
Mechanical cues are critical in influencing a variety of cellular behaviors, such as stem cell differentiation,1,2 satellite cell proliferation,3 cardiomyocyte maturation,4 and tumor cell migration.5 However, many of these mechanically regulated effects have only been studied in two-dimensional (2D) culture, often using materials that preclude three-dimensional (3D) cell studies such as polyacrylamide or poly(dimethylsiloxane) (PDMS). Because few materials both support 3D cell culture and have tunable mechanical properties in physiologically relevant regimes, relatively little is known about the effects of mechanical cues in 3D culture conditions. To address this, a variety of natural and synthetic materials have been developed to study cellular behaviors in 3D, yet the ability to independently assess mechanical effects has proven difficult. For example, naturally derived materials often encode innate biochemical cues, making it difficult to decouple the mechanical and biochemical effects. In addition, many hydrogel-forming materials require changes to the molecular weight of the polymer or polymer concentration/density to control mechanical properties. These changes, however, can affect the density of biochemical cues for natural materials as well as other physical properties of the hydrogel such as the cross-link density, degradation kinetics, permeability, and accumulation of biomolecules within the hydrogel.6−9
Poly(ethylene glycol) diacrylate (PEGDA) has been used extensively as a 3D hydrogel scaffold to study cellular behaviors as it is biologically inert, rapidly photopolymerizable, and permits cell encapsulation. By photopolymerizing PEGDA hydrogels in the presence of acrylate functionalized proteins and peptides, the hydrogel provides custom tailored biochemistry to probe the effects of various biochemical cues on stem cell differentiation,10 modulating inflammatory responses,11 and epithelial morphogenesis.12 Similarly, the effects of mechanical cues on extracellular matrix (ECM) production13 as well as cell adhesion and proliferation14 have been assessed using PEGDA hydrogel systems. However, like many synthetic scaffolds, PEGDA hydrogels generally require changes to their polymer density or polymer chain length to tune their mechanical properties,13,15,16 so it still suffers from many of the aforementioned drawbacks. To circumvent these issues, methods have been developed to control hydrogel mechanics through polymer valency (i.e., 2-arm or 4-arm macromers)6 or photoinitiation conditions,14,17,18 but these approaches often address only changes to degradative or diffusive properties and lead to other problems such as noncontinuous tuning of hydrogel mechanics or significant experiment-to-experiment variation.
Nonetheless, PEGDA has been an important material platform for the study of cell, and particularly endothelial cell (EC), behavior in both 2D and 3D settings due to its customizability and physiologically relevant mechanical properties. For 3D EC studies, PEGDA hydrogels can be rendered enzymatically degradable through the incorporation of peptide sequences within the polymer backbone that are degraded by cellular proteases such as the matrix metalloproteinases (MMPs). MMPs are actively secreted by ECs to degrade the basement membrane during angiogenesis, and thus these PEGDA-based materials are capable of creating biomimetic microenvironments that permit EC encapsulation and migration.15,19,20 While much of this existing work has examined the role of the ECM and the biochemical environment during vasculogenesis and angiogenesis, it has also revealed the importance of the local mechanical properties in regulating multiple aspects of EC behavior, including network assembly kinetics, tubule diameter, tubule length, and adhesion site morphologies.15,21−23 Because vascularized tissues with different local mechanical properties can exhibit vastly different vascular architectures, it is unsurprising that material compliance can have such dramatic effects on vessel phenotype. However, to better understand these effects of mechanical properties on EC morphology and behavior, new materials are required which are capable of spanning these physiologically relevant mechanical regimes and doing so independently of the diffusive and degradative properties of the hydrogel environment.
This study aims to develop new control over hydrogel mechanics by altering the formation of the cross-linked network independent of polymer density. To enable such control, noncanonical amino acids bearing vinyl moieties via allyloxycarbonyl (alloc) groups were incorporated adjacent to MMP-sensitive peptide sequences to act as weak competitive cross-linking sites with very low free radical propagation during photopolymerization. The mechanical effects of this cross-linking site were then assessed using both compressive and rheological testing. Because many changes to the hydrogel mesh structure and mechanical properties can result in modifications to other physical parameters of the hydrogel, the polymer degradation and diffusivity were examined. To test the ability of these materials to control EC morphological and vasculogenic behaviors, EC:pericyte cocultures were encapsulated in these hydrogels and allowed to assemble into vessel-like networks, permitting the direct investigation of the effects of polymer mechanics on EC network formation kinetics and overall network architecture.
Results
PEG Macromer Design and Synthesis
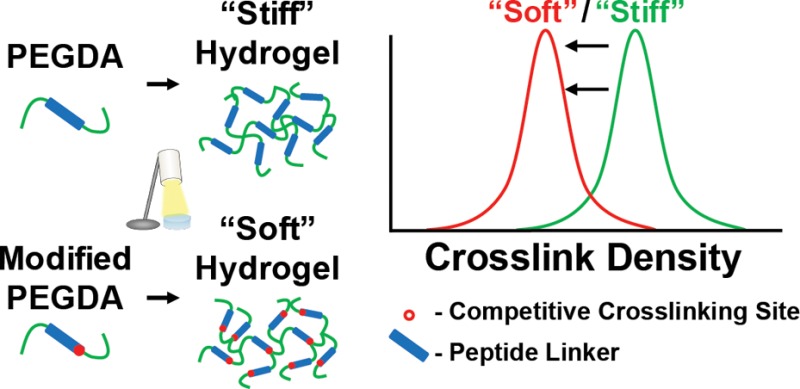
The GGGGGPQGIWGQGGGGK (PQ) peptide sequence was derived from a proteolytically degradable peptide sequence that is specifically recognized and cleaved by many members of the MMP family of proteinases.24,25 A second peptide sequence was designed incorporating an alloc protecting group through a Lys(alloc) amino acid adjacent to the degradable sequence, designated PQ(alloc). The alloc group (Figure 1a, red oval) bears a vinyl group that can weakly participate in hydrogel cross-linking, but with a low tendency for free radical propagation.26,27 Because the alloc group is not acid or base labile, it can be easily incorporated into the peptide sequences using normal fluorenylmethyloxycarbonyl (Fmoc) synthesis methods. These peptide sequences were synthesized and PEGylated at the N-terminus and a C-terminal lysine residue to create the acrylate-PEG-peptide-PEG-acrylate macromers (Figure 1a). The Lys(alloc) modification should permit the formation of both acrylate-alloc (low propagation) as well as acrylate-acrylate (high propagation) terminal junctions to alter the mesh structure of the overall hydrogel (Figure 1b).
Figure 1.

Lys(alloc) amino acids alter hydrogel cross-linking. (a) Both peptide sequences contained the MMPsensitive PQ sequence (green box and font). PQ(alloc) contained a Lys(alloc) (red circle and font) spaced from the PQ sequence and C-terminus by glycine residues. Terminal amine groups could be reacted with an acrylate-PEG-SVA to generate the PEG-peptide-PEG diacrylate macromer. (b) PEG−PQ macromers can undergo photopolymerization to form acrylate-based cross-links which impart mechanical stiffness to the hydrogel. However, PEG–PQ(alloc) macromers offer an extra cross-linking site within the peptide sequence, allowing the macromers to terminate at acrylate groups or alloc groups to control the hydrogel mechanics.
Mechanical Testing
To investigate the effects of the Lys(alloc) modification on hydrogel mechanical properties, PEG–PQ and PEG–PQ(alloc) hydrogels were analyzed via compression testing. Although 5% PEG–PQ yielded a compressive modulus of 15.63 ± 3.72 kPa, consistent with previous work,12 the PEG–PQ(alloc) hydrogels exhibit a >20 fold lower compressive modulus of 0.66 ± 0.52 kPa. Similar trends were observed at 7.5% and 10% macromer densities where PEG–PQ hydrogels displayed significantly higher compressive moduli of 28.99 ± 8.53 and 41.00 ± 3.69 kPa compared to those of PEG–PQ(alloc), 2.38 ± 0.84 and 7.05 ± 4.72 kPa, respectively (Figure 2a). As hydrogels can exhibit viscoelastic behaviors, the storage and loss moduli were measured to assess their elastic and viscous components. For PEG–PQ, the storage (G′) and loss (G″) moduli were measured as 5.31 ± 0.65 kPa and 6.1 ± 5.0 Pa, respectively. PEG–PQ(alloc), however, exhibited a significant drop in storage modulus at 1.64 ± 0.22 kPa compared to PEG–PQ, but retained a nearly identical loss modulus of 5.8 ± 0.6 Pa (Figure 2b,c).
Figure 2.

Incorporation of Lys(alloc) amino acids influences the resulting hydrogel mechanical properties. (a) Different polymer densities of PEG–PQ and PEG–PQ(alloc) hydrogels were mechanically tested to determine their compressive moduli. PEG–PQ(alloc) hydrogels exhibited significantly lower (∼10 fold) compressive moduli as compared to PEG–PQ. (b, c) In rheological measurements, PEG–PQ(alloc) had a significantly lower storage modulus (G′) compared to PEG–PQ yet displayed a nearly identical loss modulus (G″) to PEG–PQ. (d) Holding the overall prepolymer density at 5% PEG, the two macromers could be mixed at ratios of 100/0, 75/25, 50/50, 25/75, or 100/0 PEG–PQ/PEG–PQ(alloc) to create a continuum of mechanics between the minimal and maximal values, exhibiting an exponential relationship between the measured compressive modulus and PEG–PQ(alloc) content. (* indicates statistical significance, in all cases p < 0.001).
To assess the ability to continuously control hydrogel mechanics between the PEG–PQ and PEG–PQ(alloc) cases, the two macromers were copolymerized at 5% total polymer density and ratios of 100/0, 75/25, 50/50, 25/75, and 0/100 PEG–PQ/PEG–PQ(alloc). In these experiments, increasing amounts of PEG–PQ(alloc) resulted in decreases in compressive modulus (Figure 2d). The overall hydrogel mechanics scaled exponentially with the concentration of alloc groups in the prepolymer solution.
Assessing Collagenase-Dependent Degradation of PEG Hydrogels
Because amino acid substitutions to the PQ sequence can potentially lead to significantly different MMP cleavage kinetics,25,28 the Lys(alloc) amino acid was spaced from the MMP-sensitive sequence by multiple glycine residues. However, it was still necessary to verify the proteolytic degradation kinetics of the PEG–PQ(alloc) hydrogels as compared to PEG–PQ hydrogels. PEG–PQ(alloc) hydrogels were incubated with collagenase and demonstrated collagenase-dependent degradation similar to that of PEG–PQ. Both hydrogel formulations were fully degraded in 24 h at an enzyme concentration of 10 μg/mL, with roughly 80% of the degradation occurring in the first 4 h (Figure 3). Importantly, when collagenase was omitted, effectively no degradation was observed, indicating that degradation via hydrolysis or dissolution is not contributing to this response.
Figure 3.
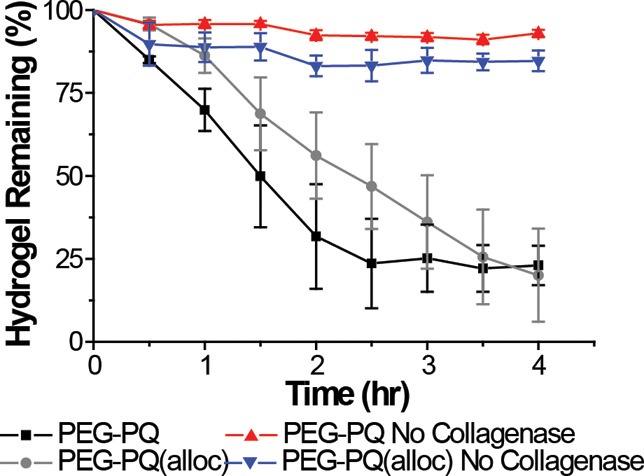
Collagenase-dependent degradation of PEG–PQ and PEG–PQ(alloc) hydrogels. The degradation of both PEG–PQ and PEG–PQ(alloc) hydrogels was monitored over 4 h via UV absorbance. The addition of the Lys(alloc) amino acid did not hinder the degradation of PEG–PQ(alloc) hydrogels as both hydrogels were ∼80% degraded in 4 h and exhibited similar rates of degradation. No degradation was observed when collagenase was omitted.
Determining Diffusive Behaviors of PEG Hydrogels
Because the prepolymer densities were identical yet yielded significantly different compressive properties, the effects of the Lys(alloc) amino acid on the permeability of the hydrogels were examined. Hydrogels were loaded with proteins of varying sizes (insulin, trypsin inhibitor, carbonic anhydrase, or ovalbumin) to create a range of molecular weights that spanned the vast majority of soluble factors in media (Figure 4a). All proteins in both hydrogels displayed similar release profiles and fits, indicating very similar diffusive behaviors for PEG–PQ and PEG–PQ(alloc) hydrogels (Figure S1). Statistical comparisons of these diffusivity values indicated no significant differences between hydrogels composed of PEG–PQ or PEG–PQ(alloc) (Figure 4b).
Figure 4.

Protein release via diffusion from PEG–PQ and PEG–PQ(alloc) hydrogels. (a) For each protein, the hydrodynamic radius and diffusivity in water (DW) were calculated from their molecular weight. (b) The release profiles for all proteins were fitted to estimate their diffusivity for PEG–PQ and PEG–PQ(alloc) hydrogels (DH). No significant difference was observed for any of the proteins measured (p > 0.05).
Mechanically Regulated EC Spreading
Although the different hydrogel formulations were capable of exhibiting diverse mechanical properties, it was necessary to evaluate the ability of cells to sense and respond to these changes. Human umbilical vein endothelial cells (HUVECs) were encapsulated at low cell density to minimize cell–cell interactions and any associated signaling and allowed to spread over 24 h. Individual cells were scored according to their circularity, where high circularity corresponds to minimal or no cell spreading and low circularity indicates highly spread cells. Both 25/75 (2.10 ± 1.02 kPa) and 0/100 (0.90 ± 0.19 kPa) PEG–PQ/PEG–PQ(alloc) exhibited significantly more spread HUVEC morphologies than the 100/0 (17.50 ± 6.28 kPa), 75/25 (9.43 ± 1.17 kPa), and 50/50 (4.77 ± 1.48 kPa) PEG–PQ/PEG–PQ(alloc) ratios with the 100% PEG–PQ(alloc) exhibiting the lowest average circularity values (Figure 5a). Histograms of cells in each of the treatment groups depict a depletion of the highly rounded cell population (circularity ∼0.8) as the hydrogels become increasingly compliant (Figure 5b–f). The circularity trends seen in Figure 5 can be visualized qualitatively by the nuclear localization and the actin cytoskeletal arrangement of HUVECs in each gel formulation (Figure 5b–f, insets; and Figure S2). To ensure the observed cell behaviors were not due to differential incorporation of RGDS, we prepared hydrogels without cells and incorporated fluorescently tagged RGDS. After complete degradation of the gels, the fluorescence was measured indicating similar incorporation of PEG-RGDS in both PEG–PQ and PEG–PQ(alloc) hydrogels (Figure S3).
Figure 5.
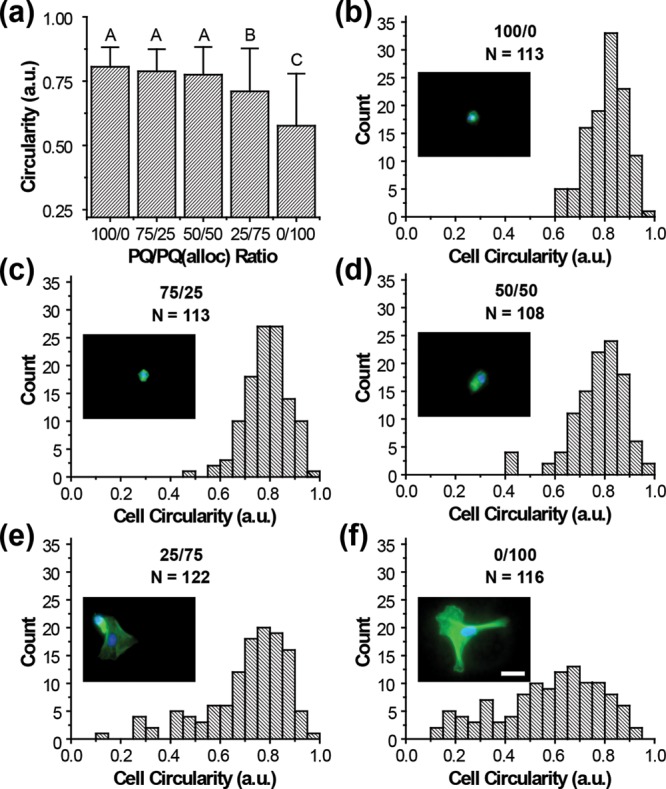
Effects of hydrogel compliance on EC spreading. (a) Average circularity values for cells encapsulated in 5% PEG hydrogels with ratios of 100/0, 75/25, 50/50, 25/75, or 0/100 PEG–PQ/PEGPQ(alloc). Cell spreading, as assessed by lower circularity, increased as the hydrogels became more compliant. 25/75 and 0/100 PEG–PQ/PEG–PQ(alloc) exhibited significantly lower circularities than the other formulations and ECs in the 100% PEG–PQ(alloc) hydrogel were the most spread (p < 0.005). (b-f) Histograms that summarize the measured circularity for all the cells assessed in each hydrogel formulation. While a population of highly rounded cells (circularity ∼0.8) can be found in all hydrogels, populations of spread cells begin to emerge as the compliance decreases. Nuclear (DAPI, blue) and actin (green) staining of characteristic cells show more developed actin networks with identifiable stress fibers in highly spread cells as compared to diffuse bands surrounding the nucleus in rounded cells (inset). Scale bar represents 30 μm.
Mechanical Effects on EC Network Formation
Previous work has demonstrated the ability of EC:pericyte cocultures to undergo tubulogenesis in 3D using RGDS-functionalized PEG–PQ hydrogels in vitro.15,29 This coculture model was used to analyze the effects of hydrogel compliance on EC network formation. Using platelet endothelial cell adhesion molecule (PECAM) and α-smooth muscle actin (αSMA) staining to indicate HUVECs and human brain pericyte cells (HBVPs), respectively, the cells and their resulting vessel-like networks were visualized via fluorescence microscopy after 1, 3, or 6 days of culture. HUVECs in PEG–PQ hydrogels remained rounded with minimal spreading or cell–cell connections after 1 day, whereas HUVECs in PEG–PQ(alloc) hydrogels were already spread and contained extensive cell–cell connections (Figure 6a, top). After 3 days of coculture, HUVECs in PEG–PQ began to form cell clusters, but exhibited very few tubule-like structures. HUVECs in PEG–PQ(alloc) hydrogels, however, exhibited mature tubule-like networks and began to show pericyte incorporation (Figure 6a, middle). Only after 6 days of culture did HUVECs in PEG–PQ hydrogels form tubule-like networks that incorporated pericytes. At the same time, HUVECs in PEG–PQ(alloc) hydrogels retained their tubule-like structures through all 6 days of culture (Figure 6a, bottom).
Figure 6.

Visualization and analysis of HUVEC:HBVP coculture in PEG–PQ or PEG–PQ(alloc) hydrogels. (a) Gels were fixed and stained for PECAM (green) and αSMA (red) then counterstained with DAPI (blue) after 1, 3, or 6 days of culture. Within 24 h of coculture, PEG–PQ(alloc) hydrogels had begun to form HUVEC networks as can be seen by the PECAM staining. The PEG–PQ hydrogels, however, exhibited no networks and minimal cell–cell contacts. After 3 days in coculture, the PEG–PQ hydrogels showed increasing cell–cell contacts and short networks with some HBVP incorporation (as seen by αSMA staining), whereas PEGPQ(alloc) hydrogels had highly developed HUVEC networks with HBVP support. At the 6 day time point, both PEG–PQ and PEG–PQ(alloc) hydrogels had well developed HUVEC networks that had directly interacting HBVPs. Scale bar represents 100 μm. (b) Total tubule length was measured for z-projection for each time point in both hydrogels. At all time points, PEG–PQ(alloc) exhibited significantly more tubule-like networks. (c) The number of branch points was counted for each field-of-view (image). Significantly more branch points were identified after 1 and 6 days of culture. (d) Comparisons of the total cell volume to cell network volume indicated significantly more cellular integration in PEG–PQ(alloc) hydrogels at all time points. (*indicates statistical significance: * p < 0.05, ** p < 0.01, *** p < 0.001).
These images were then used to quantify tubule lengths, number of branch points, and cell/vessel densities. The total length of tubules in each image was measured, indicating a significantly higher total tubule length per imaging volume (6.075 × 10–3 mm3) in the PEG–PQ(alloc) hydrogels with lengths of 5.86 ± 1.22 mm (day 1), 6.02 ± 1.01 mm (day 3), and 7.84 ± 1.67 mm (day 6) as compared to 1.78 ± 0.91 mm, 3.87 ± 1.59 mm, and 5.48 ± 1.16 mm for PEG–PQ hydrogels at 1, 3, and 6 days of culture, respectively (Figure 6b). Using these tubule networks, the number of tubule branch points for each z-projection was counted with PEG–PQ hydrogels exhibiting significantly less branching compared to PEG–PQ(alloc) hydrogels at days 1 and 6 (Figure 6c). To evaluate the extent of cell integration into network structures, the total cell volume for each z-stack was calculated and compared to the total network volumes identified in the z-projections. PEG–PQ hydrogels exhibited significantly lower cell integration ratios at all time points than PEG–PQ(alloc) hydrogels, suggesting that the cells continue to spread throughout the culture period to form new connections and nonintegrated cells are rapidly pruned (Figure 6d).
Because the PEG–PQ(alloc) hydrogels and their cell networks were able to persist through 6 days of culture, the length of time that the hydrogels and EC networks could persist in in vitro culture conditions was investigated. Similarly, as ECs are known to deposit ECM proteins during tubulogenesis to construct basement membrane, the ability of HUVECs to deposit these ECM proteins in the highly compliant PEG–PQ(alloc) environments was evaluated. To assess this, we maintained cultures for 4 weeks and stained for laminin and collagen IV, two primary constituents of the basal lamina, which should surround vasculature.30 Both collagen IV and laminin deposition were observed in close proximity to and largely colocalized with the PECAM signal. While some thinning of the vessel networks was observed at later time points, we observed tubule structures throughout the entire 4 weeks of culture (Figure 7).
Figure 7.

Extended culture and ECM deposition of HUVEC:HBVP cocultures in PEG–PQ(alloc) hydrogels. From left to right, PECAM fluorescence merged with (a) collagen IV and (b) laminin can be seen after 1, 2, and 4 weeks. These images indicate the localization of laminin and collagen IV deposition at the periphery of the vessel-like networks as well as the ability for these networks to persist out to 4 weeks of in vitro culture. Scale bar represents 100 μm.
Discussion
Although a multitude of peptide modifications have been incorporated into PEG-based hydrogels to control enzymatic cleavage kinetics,24,28 exploit photouncaging chemistries,31 or act as functional sites for the introduction of new biochemical cues,32 our findings are the first that employ peptide modifications to alter the cross-linking behavior for mechanical control. Here the use of Lys(alloc) amino acids in the peptide sequence reduces the overall mechanics of the resulting photopolymerized PEG-peptide hydrogels in a concentration dependent manner. The use of the alloc protecting group within the polymer backbone effectively creates a trivalent macromer capable of photopolymerization, and increases in macromer valency generally translate to increases in hydrogel stiffness.6 However, Matsumoto et al. and Iio et al. demonstrated that alloc groups generally form short polymers or oligomers during photopolymerization due to the “degradative monomer chain transfer” effect. In these cases, the free radical from the allyl/alloc group has a decreased tendency to propagate during polymerization as compared to other free radical polymerizing groups (i.e., vinyl acetates, methacrylate, or acrylates) and often results in termination.26,27 Consequently, when the PEG–PQ(alloc) macromer is incorporated, predominantly single junctions are formed (i.e., one acrylate to one alloc) as opposed to the large number of acrylate groups that can be present at a single junction during a PEG-diacrylate or dimethacrylate polymerization.33 These cross-links would then contribute significantly less to the overall mechanics of the hydrogel network by decreasing the number of available acrylate groups and size of the poly(acrylate) cross-linking centers during the photopolymerization reaction. These effects lead to the synthesis of photopolymerizable PEG-based hydrogels that exhibit highly compliant mechanical properties at high density capable of forming self-supporting structures. This basic capability overcomes the lower mechanical limits generally established by the need for a minimal amount of polymer to form a hydrogel. Furthermore, combining the PEG–PQ and PEG–PQ(alloc) macromers at different ratios and constant density permitted control over the compressive modulus between the 100% PEG–PQ (∼17 kPa) and 100% PEG–PQ(alloc) (<1 kPa) cases, suggesting tailorable material stiffnesses can be achieved within the ranges explored. This is a critical range of material compliance as it encompasses the local mechanical properties of many brain, liver, cardiac, and other muscle tissues,34,35 permitting functional investigations of ECs encapsulated within these mechanically matched hydrogel environments.
Similar proteolytic degradation trends across PEG–PQ and PEG–PQ(alloc) hydrogels were observed, indicating the degradation kinetics are coupled to the polymer density as opposed to the hydrogel’s mechanical properties. Furthermore, the alloc side group and its photopolymerization products did not create any significant steric barriers or noticeable changes to the MMP-dependent cleavage over the time periods investigated. In our protein diffusivity experiments, it was found that hydrogels consisting of either 100% PEG–PQ or 100% PEG–PQ(alloc) exhibit similar diffusivities for proteins ranging from 5 to 50 kDa. Here, it is likely that the incorporation of Lys(alloc) groups alters the mesh structure of the hydrogel through the incorporation of acrylate-alloc cross-links, decreasing the average number of chains terminating at a cross-linking center. Yet, although this shift in cross-link density is capable of greatly decreasing the mechanical properties of the hydrogel, it is not enough to significantly alter protein diffusion in the resulting hydrogel environment because the overall polymer density is unchanged.
In 2D studies of mechanical effects on cellular morphological behaviors, it has generally been observed that stiff materials lead to highly spread morphologies with extensive actin stress fibers as compared to ECs cultured on more compliant matrices.36 However, matrix dimensionality is believed to greatly affect how cells sense their mechanical environment because of changes in cell–matrix interactions.37 In our 3D studies, more compliant materials yielded more highly spread cells. This result is consistent with other short-term 3D cell remodeling experiments in proteolytically degradable PEG-fibrin hydrogels where, after 30 h of culture, smooth muscle cells took on spindled morphologies in compliant hydrogels as compared to rounded morphologies in stiffer hydrogels.21 Similar behaviors have also been observed in MMP-sensitive PEG hydrogels, where murine preosteoblastic cells (MC3T3-E1) were able to spread and migrate faster in more compliant hydrogels.38 However, both of these studies required the precise tailoring of mechanical properties by control of polymer chain length21 or density,38 which can affect the degradative and diffusive properties of the hydrogel. In several recent 3D studies, matrix degradability was used to control cell spreading and morphology in 3D, suggesting that matrix degradation could be tuned to regulate local ECM remodeling events.39−41 Here, the hydrogel mechanics, independent of degradative properties, allowed similar levels of control using a minimal synthetic hydrogel environment.
During angiogenesis and vasculogenesis, ECs sense and respond to each other as well as their environment to organize and form vascular networks. These networks can then be stabilized by pericytes while nonfunctional networks regress.42 In this work, the HUVECs alone were able to respond to the changes in hydrogel compliance by spreading, but no network formation was observed. In the coculture model, this heightened ability to spread in softer hydrogels permitted faster assembly into tubule-like networks. The enhanced ability of the coculture to form networks in the PEG–PQ(alloc) hydrogels is very apparent at early time points, but appears to converge with the PEG–PQ hydrogels at later time points. This suggests that the matrix compliance is able to accelerate network formation without altering the long-term behaviors of the networks. Several groups have assessed the rate of network formation based upon the material compliance in 2D, showing more compliant matrices to be more amenable to network formation.43−45 Matrix dimensionality greatly affects how cells sense their local environment, yet far less work has been done to assess the mechanical regulation of cellular behaviors in 3D.
After longer in vitro culture times, we assessed changes in the vessel network, deposition of ECM proteins, and the overall persistence of the PEG–PQ(alloc) hydrogels. Although the networks seen after 1 and 2 weeks loosely resemble the end points (6 day) of the short-term studies, the networks seen after 4 weeks have taken on a more contractile phenotype characterized by the thinner networks as compared to earlier time points. Throughout this period, the cell networks exhibited positive staining for both laminin and collagen IV, suggesting the ECs ability to produce and secrete ECM proteins is unaffected over time. Importantly, though, the compliant PEG–PQ(alloc) hydrogels remained intact and exhibited sustained support for cellular networks through all 4 weeks of culture. This is a critical result, as the ability to use highly compliant materials for in vitro and in vivo studies is generally limited by the breakdown of the scaffolding material, often necessitating the use of denser, stiffer materials. In our experiments, both the hydrogels and cell networks remained intact, indicating that these compliant hydrogels are a viable option for long-term studies of EC behavior in vitro.
Conclusions
This new method to alter and precisely tune the mechanical properties of photopolymerized PEG hydrogels using engineered peptide sequences is capable of decoupling changes in mechanical properties from the hydrogel’s degradative and diffusive properties. This independent control of hydrogel mechanical properties was then capable of regulating the spreading and network formation kinetics of ECs in vitro. We have demonstrated that EC behaviors in 3D can be modulated similarly to 2D studies through mechanical control. Although biochemical cues and degradability are still critical determinants of EC responses to their local environments, these data suggest mechanical properties of the cell environment play equally important roles in influencing many 3D cell fate decisions. Thus, the ability to span large ranges of mechanical properties and encompass these highly compliant mechanical regimes using constant polymer density, synthetic systems would permit better understandings of the independent effects of biochemical and mechanical cues on EC behaviors during morphogenic processes such as angiogenesis and vasculogenesis.
In addition, the unique mechanical capabilities of PEG–PQ(alloc) hydrogels should lend their use to a multitude of other applications where sufficiently compliant mechanical properties have been difficult to achieve in many photopolymerizable synthetic systems. Tissue niches such as the brain, lung, and a variety of tumors have been difficult to recapitulate in 3D because of their highly compliant and complex local environments. Therefore, customizable materials that can achieve such mechanical regimes and persist for long periods of time in culture should be of interest for functional cell studies which mimic these difficult tissues. This demonstrated ability to achieve long-term culture and material persistence through highly compliant and dense materials may allow the effective translation of many of these in vitro investigations to in vivo settings.
Lastly, for many tissue engineering applications, the role of pro-angiogenic materials is to permit the formation of new vasculature, ideally restoring the affected tissue’s function or inducing perfusion of an implanted engineered tissue. Often, the affected tissues are avascular (i.e., artificial tissues or organs) or necrotic (i.e., ischemic regions). Therefore, the chosen material must permit rapid integration of cells and vessel networks. The findings that the soft PEG–PQ(alloc) materials permit fast EC assembly into vessel-like networks with similar end point phenotypes to their stiffer counterparts suggest that more compliant materials, such as those reported here, may enhance tissue and vessel integration to improve therapeutic outcomes.
Materials and Methods
Cell Maintenance
HUVECs (pooled donors; Lonza, Walkersville, MD) were used at passage three and cultured in endothelial growth medium (EGM-2, Lonza) supplemented with 2 mM l-glutamine, 1000 U/mL penicillin, and 0.1 mg/mL streptomycin (Sigma; St. Louis, MO). HBVPs (ScienCell, San Diego, CA) were cultured in Pericyte Media (ScienCell) in poly-l-lysine (2 μg/cm2) coated tissue culture plastic flasks and used at passage three. All cell lines were maintained at 37 °C with 5% CO2.
PEG Macromer and Hydrogel Synthesis
The peptides GGGGGPQGIWGQGGGGK and GGGGGPQGIWGQGG-Lys(alloc)-GK were synthesized using standard Fmoc chemistry using an Apex 396 parallel synthesizer (Aapptec, Louisville, KY). Peptide products were verified using a DE-Pro MALDI-MS (Applied Biosystems). The resulting peptides were PEGylated by reacting with 2.1 mol equiv of acrylate-PEG-succinimidyl valerate (acryl-PEG-SVA; Laysan Bio, Arab, AL) in dimethyl sulfoxide (DMSO; Sigma) with 2:1 molar excess N,N-diisopropylethylamine (DIPEA; Sigma) to acryl-PEG-SVA. The reactions were performed overnight (∼18 h) on a rocker plate at room temperature under inert atmosphere. PEGylated peptides were then dialyzed and lyophilized. Purity was assessed via gel permeation chromatography (GPC) using an evaporative light scattering detector (Polymer Laboratories; Amherst, MA), yield >95%. RGDS peptides (American Peptide, Sunnyvale, CA) were conjugated similarly with acryl-PEG-SVA (1.5:1 RGDS:acryl-PEG-SVA) in DMSO with DIPEA. PEGylated RGDS was dialyzed and lyophilized. Purity was assessed via GPC, yield >85%. PEG-RGDS could then be fluorescently tagged by reacting overnight at 1:1 a ratio with Alexafluor488-NHS and 2:1 molar excess DIPEA in DMSO, which was then dialyzed and lyophilized to form PEG-RGDS-A488. All PEGylated peptides were protected from light and stored at −80 °C under inert atmosphere until use. To form hydrogels, PEG–PQ or PEG–PQ(alloc) macromers were dissolved at identical PEG densities (w/v) in hepes-buffered saline with 1.5% (v/v) triethanolamine (HBS-TEOA) containing 10 μM eosin Y photoinitiator and 0.35% (v/v) n-vinylpyrrolidone (NVP; Sigma). To account for differences in peptide molecular weight, the PEG-peptide macromers concentrations were normalized for PEG-acrylate content. Here, 5%, 7.5%, and 10% PEG w/v corresponded to 61.1, 91.65, and 122.2 mg/mL PEG–PQ macromer and 62.3, 93.45, and 124.6 mg/mL PEG–PQ(alloc) macromer, respectively. The prepolymer solution was then dropped between PDMS spacers on to clean glass slides treated with Sigmacote (Sigma) per the manufacturer’s protocol. The droplet was capped with methacrylate-functionalized cover glass (no. 1.5). Methacrylation was performed by submerging clean cover glass in ethanol with 2% (v/v) 3-(trimethoxysilyl)propyl methacrylate for 48 h. Hydrogels were then polymerized under a white light lamp (Dolan-Jenner, Boxborough, MA) set to 195 mW/cm2 at 514 nm. For PEG-RGDS incorporation studies, the hydrogels were polymerized either in the presence of 7.5 μM PEG-RGDS-A488 alone or 3.5 mM PEG-RGDS with 7.5 μM PEG-RGDS-A488. Hydrogels were soaked overnight in PBS at 37 °C and washed several times to remove any unreacted materials. Hydrogels were then completely degraded with 100 μg/mL colagenase in 0.36 mM CaCl2 containing 0.2 mg/mL NaN3. The fluorescence from each solution was then assessed using an Infinite M200 Pro microplate reader (Tecan, Männedorf, Switzerland).
Mechanical Testing
One millimeter thick hydrogels were prepared at 5%, 7.5%, or 10% (w/v) then soaked overnight in PBS at 37 °C and briefly rinsed with PBS to remove any unpolymerized material before mechanical testing. Compression testing was performed on a RSA III microstrain analyzer (TA Instruments, New Castle, DE). Samples were compressed at a constant rate of 0.003 mm/s. The slope of the initial linear range was taken to be the compressive modulus (N = 4). For rheology, 1 mm thick hydrogels were cast between two Sigmacote-treated glass slides. Gels were then cut out into 10 mm diameter disks with a biopsy punch and soaked in PBS at 37 °C overnight prior to testing. Rheological measurements were performed using coarse stainless steel platens to eliminate slippage on an AR-G2 (TA Instruments) with 10% axial strain. Samples were swept to establish the linear regions where G′ and G″ were reported.
Collagenase-Dependent Degradation
One millimeter thick hydrogels (5% PEG w/v) were photopolymerized and soaked overnight at 37 °C in phosphate buffered saline (PBS). Gels were rinsed briefly with PBS then transferred to an ultralow attachment 24-well plate (Corning, Corning, NY) in 0.36 mM CaCl2 containing 0.2 mg/mL NaN3 and 10 μg/mL collagenase. Control gels were incubated with buffer lacking collagenase. Buffer was exchanged every 30 min for 4 h and degradation was monitored via absorption at 280 nm to assess the production of tryptophan-containing degradation fragments using an Infinite M200 Pro microplate reader. A final measurement was taken after 24 h and was used to normalize the previous absorption values.
Diffusivity Measurements
Diffusivity was determined as previously described.7 Briefly, 380 μm thick hydrogels (5% PEG w/v, N = 3) were cast between two Sigmacote-treated glass slides and punched into 10 mm disks. The gels were then transferred to 24 well plates and soaked overnight at 4 °C in the presence of 1 mg/mL protein (insulin, trypsin inhibitor, carbonic anhydrase, or ovalbumin) in PBS. Gels were transferred into PBS and incubated at 37 °C. Buffer was exchanged at time points of 8, 15, 30, 60, and 120 min. The recovered solution’s protein concentration was evaluated using a Micro BCA protein assay (Thermo Scientific, Rockford, IL). Standards were prepared for each individual protein and treated identically to samples. Given the high aspect ratio of top and bottom areas to the side area of the hydrogel disks (∼26:1), the hydrogel could be modeled as a sheet using and calculated by fitting the resulting data points as a flat sheet with eq 1.
| 1 |
Here, Mt is the mass of protein released at time, t, M∞ is the total mass of protein released, DH is the diffusivity of the protein in the hydrogel, and l is the thickness of the hydrogel (380 μm).7 The hydrodynamic radius and protein diffusivity in water were estimated by eqs 2 and 3, respectively.
| 2 |
| 3 |
Here, a is the hydrodynamic radius, MW is the molecular weight, ρ is the density (∼1 g/cm2), NA is Avogadro’s number, DW is the protein diffusivity in water at 37 °C, R is the ideal gas constant, T is the temperature, and μ is the viscosity of water (0.0076 g/(cm s) at 37 °C).46
3D Cell Spreading Assay
PEG–PQ and PEG–PQ(alloc) macromers were dissolved at different ratios (100/0, 75/25, 50/50. 25/75, and 0/100 PEG–PQ/PEG–PQ(alloc), N = 4) at a constant 5% w/v PEG, then polymerized with 1 × 106 HUVECs/mL and 3.5 mM PEG-RGDS to encapsulate HUVECs in hydrogels exhibiting a range of mechanical properties. In past work, 3.5 mM PEG-RGDS has been shown to enhance HUVEC spreading and network assembly in 3D more so than higher or lower concentrations.15,47 After polymerization, gels were cultured in ultralow attachment 24 well plates with EGM-2 media. Phase images were taken at 24 h on a Zeiss Axiovert 135 (Carl Zeiss Microscopy GMBH, Jena, Germany) with a Zeiss 40× plan-neofluoar phase objective (NA = 0.6). Individual cells (>100/gel formulation, ∼25/gel) were traced and scored for circularity using ImageJ (NIH; Bethesda, MD). Afterward, the gels were stained with DAPI and phalloidin conjugated to Alexa Fluor 488, then imaged again to visualize cells’ actin cytoskeleton and nuclear arrangement using a Zeiss 63× plan-neofluoar phase objective (NA = 0.75).
3D Tubulogenesis Assay
PEG–PQ and PEG–PQ(alloc) prepolymers were prepared at 5% (PEG w/v) and polymerized in the presence of 2.4 × 107 HUVECs/mL, 6 × 106 HBVPs/mL, and 3.5 mM PEG-RGDS. After polymerization, gels were cultured in ultralow attachment 24-well plates with EGM-2 media. Media was changed after 4 h, then every other day. Gels were immediately fixed after 1, 3, or 6 days of culture with 4% paraformaldehyde in PBS. Gels were then permeabilized with 2.5% Triton-X, blocked with 5% donkey serum in PBS, and stained for PECAM and αSMA using goat anti-PECAM (Santa Cruz Biotechnology; Santa Cruz, CA) and mouse anti-α-smooth muscle actin (Abcam, Cambridge, MA). For long-term culturing (out to 4 weeks), gels were fixed after 1, 2, or 4 weeks then stained for PECAM as well as laminin and collagen IV deposition using mouse anticollagen IV (Abcam) and rabbit antilaminin (Abcam). Prior to imaging, cells were counterstained with DAPI. Imaging was performed on a Zeiss LSM 510 inverted confocal microscope using a 20× plan-apochromat objective (NA = 0.80) taking 30 × 2 μm thick slices with 1 μm overlap, yielding 30 μm thick z-stacks (three gels/polymer/time point with four z-stacks/gel). Tubule lengths, tubule segments, and branch points were estimated using the Angiogenesis application in Metamorph (Molecular Devices, Sunnyvale, CA). Cell and network volumes were created using the Imaris (Bitplane, Zurich, Switzerland) software package.
Statistical Analyses
Statistical analyses were performed using the software package JMP Pro 11 (SAS Institute, Cary, NC). Data sets were analyzed using either a two-tailed Student’s t test or one-way analysis of variance (ANOVA), followed by Tukey’s Honest Significant Difference test for multiple comparisons. In all cases, p-values less than 0.05 were considered significant and all values are reported as mean ± standard deviation.
Acknowledgments
The authors thank Dr. Lori Setton for use of the AR-G2 instrument and Dr. Aubrey Francisco for help with rheological measurements.
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.5b00064.
Release data for protein diffusion studies (Figure S1), additional fluorescence images for the 3D cell spreading study (Figure S2), and analysis of PEG-RGDS incorporation into PEG–PQ and PEG–PQ(alloc) hydrogels (Figure S3) (PDF)
This work was supported by grants from the National Institutes of Health F32HL120650 (to RMS) and R01HL097520 (to JLW).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Engler A.; Bacakova L.; Newman C.; Hategan A.; Griffin M.; Discher D. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 2004, 86, 617–628 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K.; Keung A. J.; Irwin E. F.; Li Y.; Little L.; Schaffer D. V.; Healy K. E. Substrate modulus directs neural stem cell behavior. Biophys. J. 2008, 95, 4426–4438 10.1529/biophysj.108.132217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonen K. J.; Rosaria-Chak K. Y.; Baaijens F. P.; van der Schaft D. W.; Post M. J. Essential environmental cues from the satellite cell niche: optimizing proliferation and differentiation. Am. J. Phys. Cell Phys. 2009, 296, C1338–1345 10.1152/ajpcell.00015.2009. [DOI] [PubMed] [Google Scholar]
- Jacot J. G.; McCulloch A. D.; Omens J. H. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophys. J. 2008, 95, 3479–3487 10.1529/biophysj.107.124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A.; Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 10334–10339 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning M. B.; Wilems T.; Hahn M.; Cosgriff-Hernandez E. Compositional control of poly(ethylene glycol) hydrogel modulus independent of mesh size. J. Biomed. Mater. Res., Part A 2011, 98A, 268–273 10.1002/jbm.a.33109. [DOI] [PubMed] [Google Scholar]
- Weber L. M.; Lopez C. G.; Anseth K. S. Effects of PEG hydrogel crosslinking density on protein diffusion and encapsulated islet survival and function. J. Biomed. Mater. Res., Part A 2009, 90A, 720–729 10.1002/jbm.a.32134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X.; Yang F. Engineering interpenetrating network hydrogels as biomimetic cell niche with independently tunable biochemical and mechanical properties. Biomaterials 2014, 35, 1807–1815 10.1016/j.biomaterials.2013.11.064. [DOI] [PubMed] [Google Scholar]
- Lee S.; Tong X.; Yang F. The effects of varying poly(ethylene glycol) hydrogel crosslinking density and the crosslinking mechanism on protein accumulation in three-dimensional hydrogels. Acta Biomater. 2014, 10, 4167–4174 10.1016/j.actbio.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Hwang N. S.; Varghese S.; Zhang Z.; Elisseeff J. Chondrogenic differentiation of human embryonic stem cell-derived cells in arginine-glycine-aspartate modified hydrogels. Tissue Eng. 2006, 12, 2695–2706 10.1089/ten.2006.12.2695. [DOI] [PubMed] [Google Scholar]
- Lin C. C.; Metters A. T.; Anseth K. S. Functional PEG-peptide hydrogels to modulate local inflammation induced by the pro-inflammatory cytokine TNF alpha. Biomaterials 2009, 30, 4907–4914 10.1016/j.biomaterials.2009.05.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill B. J.; Gibbons D. L.; Roudsari L. C.; Saik J. E.; Rizvi Z. H.; Roybal J. D.; Kurie J. M.; West J. L. A Synthetic Matrix with Independently Tunable Biochemistry and Mechanical Properties to Study Epithelial Morphogenesis and EMT in a Lung Adenocarcinoma Model. Cancer Res. 2012, 72, 6013–6023 10.1158/0008-5472.CAN-12-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H. M.; Munoz-Pinto D.; Qu X.; Hu Y. P.; Grunlan M. A.; Hahn M. S. Influence of hydrogel mechanical properties and mesh size on vocal fold fibroblast extracellular matrix production and phenotype. Acta Biomater. 2008, 4, 1161–1171 10.1016/j.actbio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turturro M. V.; Sokic S.; Larson J. C.; Papavasiliou G. Effective tuning of ligand incorporation and mechanical properties in visible light photopolymerized poly(ethylene glycol) diacrylate hydrogels dictates cell adhesion and proliferation. Biomed. Mater. 2013, 8, 025001. 10.1088/1748-6041/8/2/025001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J. J.; Saik J. E.; Poche R. A.; Leslie-Barbick J. E.; Lee S. H.; Smith A. A.; Dickinson M. E.; West J. L. Biomimetic hydrogels with pro-angiogenic properties. Biomaterials 2010, 31, 3840–3847 10.1016/j.biomaterials.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durst C. A.; Cuchiara M. P.; Mansfield E. G.; West J. L.; Grande-Allen K. J. Flexural characterization of cell encapsulated PEGDA hydrogels with applications for tissue engineered heart valves. Acta Biomater. 2011, 7, 2467–2476 10.1016/j.actbio.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant S. J.; Bender R. J.; Durand K. L.; Anseth K. S. Encapsulating Chondrocytes in degrading PEG hydrogels with high modulus: Engineering gel structural changes to facilitate cartilaginous tissue production. Biotechnol. Bioeng. 2004, 86, 747–755 10.1002/bit.20160. [DOI] [PubMed] [Google Scholar]
- Huang G.; Wang L.; Wang S.; Han Y.; Wu J.; Zhang Q.; Xu F.; Lu T. J. Engineering three-dimensional cell mechanical microenvironment with hydrogels. Biofabrication 2012, 4, 042001. 10.1088/1758-5082/4/4/042001. [DOI] [PubMed] [Google Scholar]
- Miller J. S.; Shen C. J.; Legant W. R.; Baranski J. D.; Blakely B. L.; Chen C. S. Bioactive hydrogels made from step-growth derived PEG-peptide macromers. Biomaterials 2010, 31, 3736–3743 10.1016/j.biomaterials.2010.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps E. A.; Landazuri N.; Thule P. M.; Taylor W. R.; Garcia A. J. Bioartificial matrices for therapeutic vascularization. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 3323–3328 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikovsky D.; Bianco-Peled H.; Seliktar D. Defining the role of matrix compliance and proteolysis in three-dimensional cell spreading and remodeling. Biophys. J. 2008, 94, 2914–2925 10.1529/biophysj.107.105841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. K.; Seliktar D.; Putnam A. J. Capillary morphogenesis in PEG-collagen hydrogels. Biomaterials 2013, 34, 9331–9340 10.1016/j.biomaterials.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura N.; Sudo R.; Ikeda M.; Tanishita K. Effects of the mechanical properties of collagen gel on the in vitro formation of microvessel networks by endothelial cells. Tissue Eng. 2007, 13, 1443–1453 10.1089/ten.2006.0333. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Lauer-Fields J. L.; Schmoekel H. G.; Metters A. T.; Weber F. E.; Fields G. B.; Hubbell J. A. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5413–5418 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H.; Fields G. B. Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Biopolymers 1996, 40, 399–416. [DOI] [PubMed] [Google Scholar]
- Iio K.; Kobayashi K.; Matsunaga M. Radical polymerization of allyl alcohol and allyl acetate. Polym. Adv. Technol. 2007, 18, 953–958 10.1002/pat.870. [DOI] [Google Scholar]
- Matsumoto A.; Kumagai T.; Aota H.; Kawasaki H.; Arakawa R. Reassessment of Free-Radical Polymerization Mechanism of Allyl Acetate Based on End-Group Determination of Resulting Oligomers by MALDI-TOF-MS Spectrometry. Polym. J. 2009, 41, 26–33 10.1295/polymj.PJ2008174. [DOI] [Google Scholar]
- Patterson J.; Hubbell J. A. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials 2010, 31, 7836–7845 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- Cuchiara M. P.; Gould D. J.; McHale M. K.; Dickinson M. E.; West J. L. Integration of Self-Assembled Microvascular Networks with Microfabricated PEG-Based Hydrogels. Adv. Funct. Mater. 2012, 22, 4511–4518 10.1002/adfm.201200976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie G. W.; Leblond C. P.; Martin G. R. Localization of Type-Iv Collagen, Laminin, Heparan-Sulfate Proteoglycan, and Fibronectin to the Basal Lamina of Basement-Membranes. J. Cell Biol. 1982, 95, 340–344 10.1083/jcb.95.1.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeForest C. A.; Anseth K. S. Photoreversible Patterning of Biomolecules within Click-Based Hydrogels. Angew. Chem., Int. Ed. 2012, 51, 1816–1819 10.1002/anie.201106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeForest C. A.; Anseth K. S. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 2011, 3, 925–931 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Gibson S.; Jones R. L.; Washburn N. R.; Horkay F. Structure-property relationships of photopolymerizable poly(ethylene glycol) dimethacrylate hydrogels. Macromolecules 2005, 38, 2897–2902 10.1021/ma0487002. [DOI] [Google Scholar]
- Discher D. E.; Mooney D. J.; Zandstra P. W. Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324, 1673–1677 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemir S.; West J. L. Synthetic Materials in the Study of Cell Response to Substrate Rigidity. Ann. Biomed. Eng. 2010, 38, 2–20 10.1007/s10439-009-9811-1. [DOI] [PubMed] [Google Scholar]
- Byfield F. J.; Reen R. K.; Shentu T. P.; Levitan I.; Gooch K. J. Endothelial actin and cell stiffness is modulated by substrate stiffness in 2D and 3D. J. Biomech 2009, 42, 1114–1119 10.1016/j.jbiomech.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E.; Pankov R.; Stevens D. R.; Yamada K. M. Taking cell-matrix adhesions to the third dimension. Science 2001, 294, 1708–1712 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Ehrbar M.; Sala A.; Lienemann P.; Ranga A.; Mosiewicz K.; Bittermann A.; Rizzi S. C.; Weber F. E.; Lutolf M. P. Elucidating the Role of Matrix Stiffness in 3D Cell Migration and Remodeling. Biophys. J. 2011, 100, 284–293 10.1016/j.bpj.2010.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanjaya-Putra D.; Wong K. T.; Hirotsu K.; Khetan S.; Burdick J. A.; Gerecht S. Spatial control of cell-mediated degradation to regulate vasculogenesis and angiogenesis in hyaluronan hydrogels. Biomaterials 2012, 33, 6123–6131 10.1016/j.biomaterials.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khetan S.; Burdick J. A. Patterning network structure to spatially control cellular remodeling and stem cell fate within 3-dimensional hydrogels. Biomaterials 2010, 31, 8228–8234 10.1016/j.biomaterials.2010.07.035. [DOI] [PubMed] [Google Scholar]
- Khetan S.; Guvendiren M.; Legant W. R.; Cohen D. M.; Chen C. S.; Burdick J. A. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat. Mater. 2013, 12, 458–465 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R. K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Saunders R. L.; Hammer D. A. Assembly of Human Umbilical Vein Endothelial Cells on Compliant Hydrogels. Cell. Mol. Bioeng. 2010, 3, 60–67 10.1007/s12195-010-0112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano J. P.; Reinhart-King C. A. A Balance of Substrate Mechanics and Matrix Chemistry Regulates Endothelial Cell Network Assembly. Cell. Mol. Bioeng. 2008, 1, 122–132 10.1007/s12195-008-0022-x. [DOI] [Google Scholar]
- Hanjaya-Putra D.; Yee J.; Ceci D.; Truitt R.; Yee D.; Gerecht S. Vascular endothelial growth factor and substrate mechanics regulate in vitro tubulogenesis of endothelial progenitor cells. J. Cell Mol. Med. 2010, 14, 2436–2447 10.1111/j.1582-4934.2009.00981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier R. L.Basic Transport Phenomena in Biomedical Engineering, 1st ed.; Taylor & Francis: Philadelphia, PA, 1999; p 312. [Google Scholar]
- Gobin A. S.; West J. L. Cell migraton through defined, synthetic ECM analogs. FASEB J. 2002, 16, 751–753 10.1096/fj.01-0759fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.