

Abstract
Kinase inhibitors have played an increasingly prominent role in the treatment of cancer and other diseases. Currently, more than 25 oncology drugs that target kinases have been approved, and numerous additional therapeutics are in various stages of clinical evaluation. In this Review, we provide an in-depth analysis of activation mechanisms for kinases in cancer, highlight recent successes in drug discovery, and demonstrate the clinical impact of selective kinase inhibitors. We also describe the substantial progress that has been made in designing next-generation inhibitors to circumvent on-target resistance mechanisms, as well as ongoing strategies for combining kinase inhibitors in the clinic. Last, there are numerous prospects for the discovery of novel kinase targets, and we explore cancer immunotherapy as a new and promising research area for studying kinase biology.
Introduction
Protein phosphorylation is the most common form of reversible post-translational modification (1), with an estimated 50% of all proteins undergoing phosphorylation (2). The phosphorylation state of any given protein is controlled by the coordinated action of specific kinases and phosphatases that add and remove phosphate, respectively. There are at least 518 kinases (3) and 156 phosphatases (4) in the human genome, and despite intensive research in this area, the underlying biology of many of these enzymes remains to be fully elucidated. Notwithstanding, signaling networks that employ phosphorylation to modulate target activities have been shown to be critically involved in all aspects of cellular function, and in cancer, the abnormal activation of protein phosphorylation is frequently either a driver or direct consequence of the disease (5). For instance, kinase signaling pathways have been shown to drive many of the hallmark phenotypes of tumor biology (6), including proliferation, survival, motility, metabolism, angiogenesis, and evasion of antitumor immune responses (Figure 1).
Figure 1. Regulation of tumorigenic programming by activation of kinases.

Survival: Kinase pathways regulate programmed cell death by either directly modulating apoptosis regulators or altering their expression. Necroptosis is controlled by signaling pathways that intersect apoptosis regulators, including RIPK3. Motility: Cytoskeletal dynamics are mediated by RTKs and integrin clustering to promote the activation of FAK and GTPases. Effector kinases include ROCK1, MLCK (encoded by MYLK3), PAK1, and LIMK1. Evading antitumor immunity: Numerous cell types, including CTLs, Tregs, DCs, MDSCs, and macrophages, are actively involved in modulating the tumor immune response. MAP4K1, PI3K∂, MST1R, MERTK, and PKCη, represent potential targets for reversing the myriad mechanisms involved in tumor-mediated immunosuppression of the tumor microenvironment. Proliferation: MAPK pathway components increase levels of CCND1. CCND1 regulates the activity of CDK4/6 and, in turn, induces cell cycle progression via RB1 and E2F transcriptional regulators. DNA damage checkpoints: Following induction of double-strand DNA breaks by ionizing radiation or chemotherapeutic agents, ATM and ATR phosphorylate CHK1. Activated CHK1 subsequently prevents cell cycle progression and induces DNA repair mechanisms. Angiogenesis: Tumor cells modulate the microenvironment by secreting angiogenic factors (ligands for VEGFR, FGFRs, and the angiopoietin receptor TIE2 [encoded by TEK]) to stimulate endothelial cells and enhance vascularization. Pericytes are in turn stimulated by PDGF and TGF growth factors to contribute to endothelial cell proliferation and migration. Metabolism: PI3K/AKT pathway activity is directly correlated with increased glucose metabolism in cancer cells. Signaling is mediated through hexokinases, MTOR, and HIF1α (encoded by HIF1A) to increase oxidative phosphorylation, tricarboxylic acid cycle, and mitochondrial respiration.
Given this opportunity for therapeutic intervention, there has been considerable effort in developing pharmacological tools to inhibit protein phosphorylation and interrogate signaling (7). Currently, there are multiple examples of small molecule kinase inhibitors with both selectivity and suitable pharmaceutical properties that have produced meaningful clinical benefit. For instance, imatinib is utilized to inhibit BCR-ABL1 in chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia with the Philadelphia chromosome (8); crizotinib and other ALK kinase inhibitors for cancers driven by ALK fusions (9); lapatinib for ERBB2/HER2-amplified tumors (10); gefitinib and erlotinib for EGFR mutated tumors (10); and vemurafenib for BRAF mutant tumors (11). In each of these cases, superior clinical benefit was observed for the targeted agent in molecularly selected patients compared with prior standard-of-care regimens. Our evolving ability to genomically characterize tumors heralds a new era in which selective kinase inhibitors can be utilized to inactivate molecular drivers of the malignant state.
Despite these successes (12), the field is still just beginning to develop an understanding of how to fully exploit kinase inhibitors for therapeutic benefit. In this Review, we examine new trends in targeting protein and lipid kinases employing next-generation small molecule inhibitors and focus on four areas: (a) identification of novel, putative driver kinases as a result of advances in deep sequencing technologies and genomic analyses; (b) the use of increasingly potent and selective kinase inhibitors to further our understanding of resistance mechanisms; (c) potential combination strategies to improve antitumor efficacy and disrupt resistance mechanisms; and (d) novel kinase targets in the regulation of the tumor microenvironment and immune responses.
Kinase activation by genomic events
The abnormal oncogenic activation of protein and lipid kinases derives from multiple types of genetic and epigenetic changes (12, 13). These alterations result in increased specific activity of the kinase itself, its overexpression, or the loss of negative regulation. Most frequently, tumor cells harbor somatic point mutations at structurally conserved residues, or mutation hotspots, which constitutively upregulate kinase activity (14). Examples of these hotspots include M918T in RET (15) and M1268T in MET (16), which occur at a structurally equivalent position within the kinase domain. Another prevalent mutation hotspot conserved across several oncogenic kinases is BRAF V600 (17), which corresponds to KIT D816 (18) and FLT3 D835 (19). Recent efforts from large-scale consortia such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) have uncovered many new mutations in kinases and enabled a robust delineation of the spectrum of activating kinase mutations in cancer through careful statistical analysis (refs. 20, 21, Figure 2, and Table 1). In addition, genomic instability, a hallmark of cancer cells (6, 22), can also result in elevated kinase activity that enhances signaling through a number of distinct mechanisms. Defects in the surveillance pathways that maintain genomic integrity can produce amplifications of large chromosomal regions or complex chromosomal rearrangements, which in turn result in the mis-expression of a kinase or the expression of a constitutively activated chimeric form (kinase fusions) (Figure 2 and Table 1).
Figure 2. Activating genomic alterations of protein and lipid kinases.
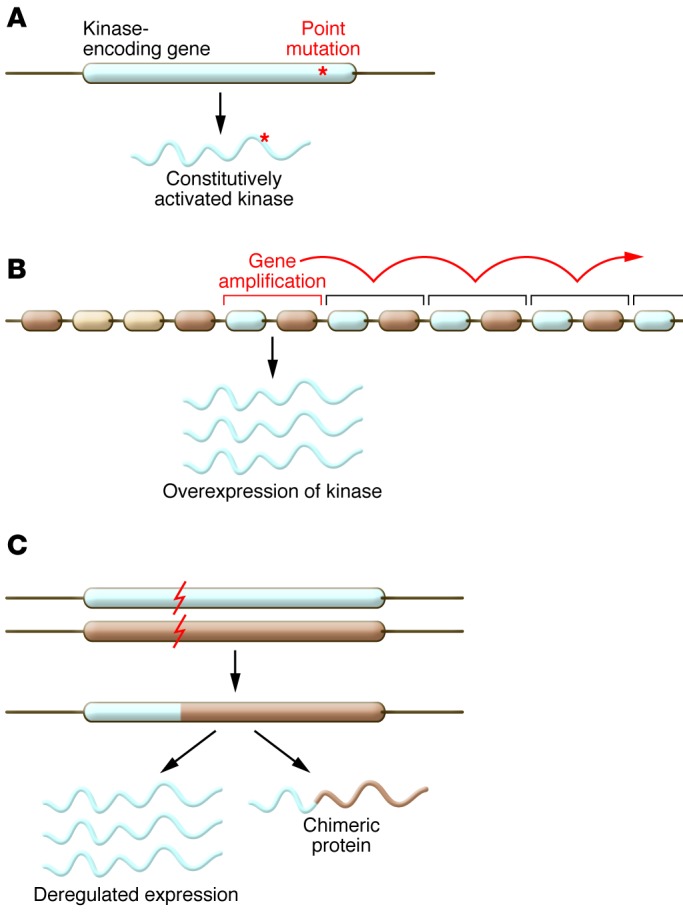
(A) Activating point mutations in genes coding for kinases lead to the expression of a constitutively activated kinase. Such mutations either lead to an amino acid substitution in the catalytic site, rendering it active; or change the general properties of the protein, for instance by disrupting the interaction with negative regulators, by releasing a mechanism of autoinhibition within the kinase itself, or by inducing constitutive dimerization. Last, they can cause changes in the splicing of the mRNA. Point mutations are the most common mechanism of kinase activation. (B) Chromosomal amplification of a region containing a kinase leads to its increased transcription and the production of an increased amount of protein in the cell. Consequently, the downstream pathway becomes overactivated. (C) Chromosomal alterations such as translocations or deletions can localize a kinase gene in proximity to another gene and lead to the expression of a constitutively activated chimeric or truncated kinase, or deregulate the expression of the kinase by putting it under the control of another promoter.
Table 1. Examples of known mechanisms of kinase activation in cancer.

Although the target gene(s) within an amplification that confers a growth advantage can be difficult to identify — as these genomic regions often include multiple genes — studies of large cohorts of tumors have facilitated the identification of several driver kinases from genomically amplified loci (refs. 23, 24, Figure 2, and Table 1). In particular, the receptor tyrosine kinases (RTKs) EGFR, ERBB2/HER2, and MET and the serine/threonine kinases CDK4 and CDK6 are most frequently overexpressed as a consequence of gene amplification.
Kinase fusions represent ideal targets for the development of cancer drugs because they often confer oncogenic dependency in hematopoietic and solid malignancies, as demonstrated by the success of several kinase inhibitors (refs. 25, 26, and Table 1). For example, imatinib induces remission in leukemia patients who are positive for BCR-ABL1 fusions (8), and crizotinib and ceritinib have produced significant clinical benefit in patients with lung adenocarcinomas and mesenchymal tumors harboring ALK fusions (9, 27). In certain solid tumors, such as thyroid carcinomas or glioblastoma, kinase fusions represent an important driver mechanism of the disease (occurring in 12% and 6% of patients, respectively; ref. 26). In other solid tumor types genomically profiled in TCGA, fusions in ALK, BRAF, EGFR, FGFR1–3, NTRK1–3, PDGFRA, RAF1, RET, and ROS1 collectively occur in 1%–3% of patients and constitute strong driver events of the disease. Moreover, oncogenes such as MET and PIK3CA, known to be activated by mutations or amplifications, have been recently shown to harbor fusions as well (26). In some instances, novel kinase fusions have been implicated as the sole drivers of tumorigenesis, as has been recently shown with PRKACA in fibrolamellar hepatocellular carcinoma (28). The mechanisms of kinase activation can be diverse depending on the fusions; however, for most RTKs, the acquisition of a dimerization domain contributed by the fusion partner promotes trans-autophosphorylation. Loss of a kinase autoinhibitory domain and overexpression of the kinase (such as observed in the context of gene amplification) have also been described, especially for non-RTKs (26).
Other mechanisms of kinase activation include overexpression as a consequence of epigenetic changes (29), activation of a kinase transcription factor (30), inactivating mutations of negative regulators (31, 32), modulation of regulatory proteases (33), alternative splicing (34), splice site mutations causing exon skipping (35), and the overexpression of a ligand or an upstream positive regulator by genetic or epigenetic regulation (ref. 36 and Figure 1). Taken together, the multitude of direct and indirect events that result in the activation of kinase pathways in tumors represent important and tractable opportunities for therapeutic intervention.
Overcoming therapeutic resistance
The genomically informed application of selective kinase inhibitors in clinical oncology has shown tremendous promise in improving patient outcomes (37). For example, prior to the development of imatinib, CML was treated with a combination of hydroxyurea and interferon, a regimen that allowed disease control but did not prevent its progression to more advanced stages (38). In contrast, imatinib therapy has produced remarkably durable responses, with an 8-year survival rate of 85% (39). Despite the significant clinical benefit of some agents, patients who initially respond to targeted therapeutics commonly relapse. Efforts to elucidate the molecular mechanisms underlying this process have been greatly aided by the genomic characterization of paired samples of pre-treatment and post-relapse tumors. These studies have revealed that resistance to kinase inhibitors can occur through a variety of processes: (a) “on-target” resistance via mutations, amplifications, or altered splicing of the target kinase (40, 41); (b) “on-pathway” resistance via activation of downstream components or disruption of negative feedback loops; (c) bypass mechanisms that upregulate parallel signaling pathways (42, 43); (d) epigenetic alterations and histological transformations that modify a cell’s phenotypic state (44, 45); and (e) modulation of cellular processes that influence drug transport, distribution, or stability (40, 46, 47).
On-target mutations that reactivate a kinase are a predominant mechanism of resistance to small molecule inhibitors. For example, resistance mutations in the molecular targets of imatinib, BCR-ABL1 and KIT, have been described in both indications for which this agent is approved as therapy, namely CML and gastrointestinal stromal tumors (GISTs) (48–51). Similarly, on-target resistance mutations have been documented in the kinases FLT3 (52) ALK (53, 54), ROS1 (55), EGFR (56, 57), KIT (51), and PDGFRA (58), as well as in kinases that are not directly genetically deregulated, such as BTK (59, 60) and MTOR (61). In terms of molecular mechanisms, BCR-ABL1 mutations conferring resistance to imatinib provide a useful paradigm to understand the structural basis for diminished inhibitor potency by kinase mutations. By reducing the inherent affinity of the compound for BCR-ABL1 either by elimination of critical interactions, such as mutation of the active site gatekeeper residue, or by induction of a conformational change in the kinase, these mutations reduce imatinib binding to BCR-ABL1 and therefore its clinical efficacy (62). For other kinases, such as EGFR, resistance mutations can increase the kinase’s affinity for ATP and thereby lower the relative cellular potency of ATP-competitive inhibitors (63). Mutational reactivation of kinases in relapsed tumors firmly establishes their roles as disease drivers and highlights the prominent role of this mechanism in mediating on-target resistance.
The specific mechanisms of resistance that arise in a tumor are influenced by several factors, including the molecular features of the compound, the kinase target itself, and the underlying genomic characteristics of the tumor. For instance, imatinib-resistant CML patients who have progressed on second-line dasatinib treatment harbor mutations that are distinct from those patients having relapsed following second-line treatment with nilotinib (64). This phenomenon has also been documented in KIT-driven GISTs, where primary and secondary KIT mutations have been observed upon relapse to either first- or second-line treatments, respectively (65, 66). In this regard, in vitro mutagenesis or in vivo resistance screens combined with structural analysis and genomic characterization of resistant cells can delineate the mechanisms that tumors utilize to evade pharmacological interventions. These approaches have tremendous value in designing second-generation therapies that can resensitize tumors to targeted agents. Moreover, the occurrence of secondary or tertiary mutations in a kinase following multiple rounds of targeted therapy suggests that some tumors exhibit a near-obligate dependence on a driver kinase. In support of this notion, in CML clones from patients who have undergone multiple rounds of kinase inhibitor therapies, the number of silent mutations in BCR-ABL1 increased disproportionately compared with the total number of coding substitutions (64). This finding suggests there may be a fundamental limit to the number of drug-resistance mutations that BCR-ABL1 can tolerate while retaining its catalytic activity (64) and supports the development of a repertoire of selective kinase inhibitors — including both ATP-competitive and allosteric inhibitors (67) — to target all substitutions that disrupt drug binding to a given oncogenic kinase.
Combination therapies
Drug combinations within a single kinase pathway.
Preclinical and clinical data demonstrate that therapeutic combinations enhance primary antitumor responses and delay the onset of resistance (68), and combinations of kinase inhibitors are actively being explored for the treatment of cancer. Prioritization of kinase inhibitor combinations has frequently been based on either robust synthetic lethality in model systems (69) or identification of putative resistance mechanisms in tumor tissues following relapse in patients on monotherapy (10). The multipronged approaches to targeting the MAPK pathway in melanoma represent an emerging example of the effective use of combination therapies within a single pathway to circumvent compensatory signaling as well as genetic resistance mechanisms. Activating, oncogenic mutations of BRAF occur in 50% of malignant melanomas, and catalytic inhibition using vemurafenib or dabrafenib has elicited positive clinical responses (70). However, patient benefit is generally short lived (11), and there is a need to understand the molecular events driving patient relapse. To date, the majority of putative resistance mechanisms involve reactivation or hyperactivation of the MAPK pathway itself (on-pathway resistance). The molecular mechanisms include amplification, mutation, or gene fusions of upstream (NRAS), parallel (CRAF; encoded by RAF1), or downstream (MEK1 and MEK2; MAP2K1 and MAP2K2, respectively) pathway components (71–73). As such, combined blockade of BRAF together with its immediate effectors, MEK1 and MEK2, has been examined in advanced clinical trials (74, 75). For example, patients receiving dabrafenib and trametinib combination treatment had improved progression-free survival (PFS) compared with those receiving dabrafenib monotherapy (76). These results are consistent with preclinical studies showing that sustained >80% pathway inhibition and relief of ERK1- and ERK2-dependent (encoded by MAPK3 and MAPK1, respectively) feedback results in enhanced antitumor efficacy (77–79). Furthermore, preclinical and clinical evidence suggests that additional MAPK pathway alterations, including activating mutations in MEK1 and MEK2, confer resistance to combined BRAF and MEK1/2 inhibition (80–82). The emergence of these additional mechanisms of resistance supports the development of novel inhibitors of multiple upstream and downstream kinase components for the next generation of combination therapy treatments.
Drug combinations targeting parallel kinase pathways.
Kinase inhibitor sensitivity can also be influenced by the broader network of signaling events, including crosstalk and feedback to parallel pathways. For instance, several factors that have been implicated in conferring resistance to MAPK pathway therapeutics fall within the PI3K pathway (Figure 3). These events include the mutation of PIK3CA, loss of the PTEN and INPP4B tumor suppressors, increased basal activity of the PI3K effector kinase AKT, induction of AKT activation in response to MAPK inhibition, and signaling via TORC1 (83–86). Given that the MAPK and PI3K signaling pathways also act on key regulators of proliferation and survival in normal cells, intermittent scheduling regimens and the targeting of different nodes within the signaling network are being evaluated to improve tolerability and efficacy in the clinic (87, 88). In addition to direct activation of the PI3K pathway, increased expression of RTKs, such as EGFR, PDGFRB, and IGF1R, has also been proposed as a mediator of resistance to MAPK pathway inhibitors (89–92). In the case of EGFR-mediated resistance to MAPK pathway inhibition, elevated EGFR expression has been proposed to occur via an epigenetic adaptive response (89). The non-genetic nature of this mechanism suggests that resistance may be readily reversible, and there is early evidence that melanoma patients who have developed resistance to BRAF inhibitors can regain sensitivity following a prolonged drug holiday (93). Taken together, these results suggest that combinations of kinase inhibitors that target multiple nodes within a complex signaling network will require extensive preclinical and clinical characterization to identify the critical pathway interdependencies and reveal the best targets and optimal dosing regimens to maximize efficacy and tolerability for patients.
Figure 3. Potential therapeutic combinations to overcome resistance to BRAF inhibitors.

Dual blockade within the MAPK pathway or between the MAPK and parallel PI3K pathways improves clinical outcome by reducing compensatory signaling and delaying the onset of resistance. Mut, mutation; OE, overexpression; LOH, loss of heterozygosity; ↓Exp, decreased expression.
Drug combinations converging on a hallmark characteristic of tumors.
Kinase inhibitors can also be used in combination to regulate a hallmark phenotype of tumorigenesis (Figure 1). For instance, even though many kinase inhibitors function as cytostatic agents, kinase inhibition has been utilized to modulate the activity of cytotoxic agents; however, these combinations have often been opportunistic, with kinase inhibitors simply added to standard-of-care chemotherapeutic regimens approved for a particular tumor indication (94). An emerging rational strategy for improving the clinical benefit of DNA-damaging agents is the combined inhibition of kinases that regulate cell cycle checkpoints and phase transitions (95). For instance, the serine/threonine kinase CHK1 is activated via ATR-mediated phosphorylation during DNA repair and is required for the S- and G2/M-phase DNA damage checkpoints (96). Accordingly, CHK1 inhibition potentiates antitumor efficacy of chemotherapies via inactivation of these checkpoints, resulting in premature entry into mitosis with damaged DNA and subsequent mitotic catastrophe and cell death (97, 98). Kinase inhibitors can also be used to mitigate the effects of cytotoxic agents on rapidly proliferating normal tissue. For example, preclinical studies have utilized palbociclib to inhibit the G1/S-phase checkpoint kinases CDK4 and CDK6 and ameliorate the dose-limiting toxicities of chemotherapies (99). Inhibition of CDK4/6 induced G1 arrest and pharmacological quiescence of hematopoietic cells to mitigate apoptosis and myelosuppression (100). Moreover, treatment of genetically engineered mouse models with palbociclib reduced irradiation-induced toxicity without limiting therapeutic responses (101). Taken together, these approaches demonstrate that cell cycle kinases can be selectively targeted to both enhance antitumor efficacy and reduce certain normal tissue toxicities induced by standard-of-care chemotherapeutics.
Combination of kinase inhibitors with other therapies is also being explored to more effectively abrogate tumor immune evasion, which has recently emerged as a cancer hallmark in its own right, as well as an important therapeutic modality (Figure 1 and ref. 6). The cancer cells’ propensity to incur and accumulate genetic alterations results in the synthesis of mutant polypeptides, ultimately leading to the generation of neo-epitopes that enable the host’s immune system to recognize these cells as foreign and eliminate them (102). However, tumor cells can exert a substantial influence on their local environment to suppress these antitumor immune responses and escape this critical defense mechanism. Accordingly, therapeutic agents that interfere with these suppressive effects have elicited profound and durable clinical responses, with this line of research culminating in the approval of ipilimumab, an antibody that targets a negative regulator of T cell activation (CTLA4) and, more recently pembrolizumab, an antibody that inhibits an alternative T cell–negative regulator programmed death–1 (PD-1; encoded by PDCD1), for the treatment of advanced melanoma (103). Despite these successes, there remains a significant opportunity to improve upon the often-restricted response rates of these therapies, and a number of kinase inhibitors already approved for specific oncology indications are being clinically evaluated in combination with antitumor immune activators. In fact, there are currently more than 20 active or planned clinical trials exploring the combination and scheduling of various kinase inhibitors with ipilimumab, pembrolizumab, or agents targeting alternative negative immune regulators (103, 104). The overarching rationale behind the application of kinase inhibitors in this context is to directly target the tumor and reduce its immune-suppressive influences in order to shift the local microenvironment toward a proinflammatory state and thereby enhance the activity of the immune activators (105). For example, there are a number of combination trials assessing the relative benefit of BRAF and MEK1/2 inhibition in patients with BRAF-mutant melanoma in combination with ipilimumab or anti–PD-1 antibodies (106). While inhibition of these kinases alone has been shown to yield significant clinical responses and enhance progression-free and overall survival (74, 75), combination with agents that enhance antitumor immune activation reduction in tumor burden is expected to produce therapeutic benefit in addition to a reduction in tumor burden. However, as in the case of MEK, these kinases often play important roles in immune cell function that can abrogate or limit any therapeutic benefit of the combination. As a consequence, adjustments to clinical studies such as dosing holidays or staggered dosing regimens may be necessary (106). Accordingly, while the current deployment of kinase inhibitors in combination with immune activators is an important opportunistic approach to rapidly expand the utility of immune modulation in oncology, much more needs to be elucidated about the numerous pathways and processes involved before additional kinase inhibitors can be developed and appropriately leveraged to maximize their impact in this setting.
Future directions
Kinase inhibitor selectivity.
The discovery of small molecule kinase inhibitors was first reported more than 30 years ago (107). These early compounds bound in the ATP-binding pocket residing in the cleft between the N- and C-lobes of kinases. Even today, the majority of kinase inhibitors bind via a similar mechanism and mimic the hydrogen bond interactions normally formed by the adenosine ring of ATP (7). Of the current generation of small molecules, kinase inhibitors can vary with respect to certain properties, including the activation loop conformation that is preferentially recognized (type 1/DFG-in or type 2/DFG-out) (108), ATP displacement (competitive or non-competitive/allosteric), and the mechanism of inhibitor binding (reversible or irreversible/covalent). Significant clinical benefit can then be achieved by treating tumors harboring specific genetic alterations with highly selective targeted agents (8, 10, 109). One of the prominent themes of the past decade has therefore been to improve the overall kinome selectivity of small molecule inhibitors. There are a number of advantages to this approach, including (a) improvements in tolerability and therapeutic index, as well as enabling (b) the development of drug combination regimens, (c) validation of new targets and molecular dependencies of tumors, and (d) assessment of mechanisms of action. For instance, next-generation EGFR inhibitors that selectively target the inhibitor-sensitizing and resistance (T790M) mutations exhibit a dramatically improved safety profile (with reduced rash and diarrhea) by sparing wild-type EGFR activity in normal cells (110). In addition, concomitant inhibition of structurally related kinases using less-selective inhibitors, such as sunitinib or dasatinib, can have opposing and confounding effects in modulating the immune system (111), which could limit antitumor efficacy and complicate the interpretation of molecular mechanisms. Despite the considerable technical challenges in developing highly selective kinase inhibitors, recent efforts have been aided by using specialized chemical libraries and structure-informed design (7, 112). Importantly, these efforts are meeting with overall success, such as for ATP-competitive small molecules that specifically target PAK4 (113), as well as cysteine-targeted covalent inhibitors of FGFR4 and CDK7 that selectively perturb proliferation and transcriptional regulation (114, 115). Additional highly selective kinase inhibitors are expected to be available in the near future to test new clinical hypotheses in the new era of precision medicine.
Kinases in cancer immunotherapy.
Much of past and current efforts to discover kinase inhibitors for oncology indications have focused on targets with well-established roles in the regulation of cell proliferation and survival or tumor angiogenesis, three of the fundamental hallmarks of cancer. However, potential kinase targets involved in the regulation of the remaining tumor hallmarks (Figure 1) have yet to be thoroughly elucidated, either preclinically or clinically. One such opportunity may exist in the realm of tumor immunity, which, as mentioned above, represents a key mechanism in the development of cancer and an important therapeutic strategy that has recently demonstrated significant promise across a range of diverse tumor types (103, 116). As outlined in greater detail elsewhere (117), tumor immunity is initiated by antigen-presenting cells (APCs; DCs and macrophages) displaying neo-epitopes on their surface derived from their engulfment and/or proteolytic processing of cancer cells. The APCs engage naive T cells that recognize these neo-epitopes via their T cell receptor (TCR), driving their expansion and maturation to cytotoxic T lymphocytes (CTLs), which can recognize and eradicate the cancerous cells. Given their critical role in tumor immunity and potential for triggering autoimmunity (118), CTL functions are tightly regulated to enable a robust, targeted, and limited immune response. Tumors often subvert the immune system’s attack by engaging the negative regulatory mechanisms that limit CTL function (118). These mechanisms are not entirely CTL autonomous, but involve other cell types, including Tregs (119, 120), regulatory or immature DCs (121, 122), M2 macrophages (123), and myeloid-derived suppressor cells (MDSCs) (124, 125), which are all present within the tumor microenvironment. As detailed below, kinases play a role in essentially all processes that constitute tumor immunity and tumor immune evasion. Accordingly, significant opportunities exist for the development of kinase inhibitors to reactivate the immune system to eradicate the tumor via mechanisms that are far beyond the current trials evaluating their use in combination with the antibody-based therapies.
Kinases are known to play pivotal roles in the resolution of an immune response and could therefore serve as relevant therapeutic intervention points. For example, multiple kinases are involved in limiting the extent and duration of TCR-mediated signaling in CTLs. One pathway involves the kinase PKA (encoded by PRKACA), which, when activated by cAMP (126), phosphorylates and activates c-SRC kinase (CSK), a negative regulator of SRC family kinases (127). CSK then phosphorylates and inhibits lymphocyte-specific protein tyrosine kinase (LCK), a kinase critical for transducing proximal TCR signals. LCK also limits the duration of its own signaling through activation of another non-RTK, MAP4K1, which phosphorylates the key adapter protein SLP76 (encoded by LCP2), thereby blocking LCK’s ability to recruit downstream signaling molecules (128). As an additional example, the α and ξ diacylglycerol kinases (DGKs; encoded by DGKA and DGKE, respectively) have been shown to be involved in limiting the activation of PKCθ (PRKCQ), another positive effector of TCR signaling, thereby blunting CTL proliferation and effector function (129). With the exception of PKA and CSK, the expression of these kinases is predominantly restricted to immune cells, suggesting selective roles in immune modulation. Accordingly, genetically engineered mouse models enabling the abrogation of PKA, CSK, MAP4K1, or DGKξ functions exhibit hallmarks of excessive immune activation and/or TCR hypersensitivity (128, 130–132). At least in the case of Dgka–/–, Dgke–/–, and Map4k1–/– mice, the role of these kinases in tumor immune surveillance has already been established, confirming the viability of these approaches for restoring antitumor immunity (131, 133). In contrast, ubiquitous expression and pleiotropic functions of CSK and PKA may limit the viability of these targets in this context.
As mentioned above, a number of immune cell types act on CTLs to limit their capacity to carry out their effector functions, especially Tregs. This specialized T cell subtype acts via several mechanisms, including secretion of suppressive signaling molecules, titration of CTL activating factors (such as IL-2), and direct cell-cell interactions with CTLs and APCs, to blunt antitumor immune responses (119, 134). Notably, several kinases have been implicated in either the regulation of Treg development and homeostasis or their suppressor functions. In addition to suppressing CTL responses, the PKA/CSK pathway has been shown to be important for both the development and suppressive activity of Tregs (135). Likewise, the lipid kinase PI3Kδ (encoded by PIK3CD) has been shown to play a crucial role in the maturation and homeostasis of Tregs (136). In particular, a recent report demonstrated that loss of PI3Kδ activity, either by genetic knockout or small molecule inhibition, markedly decreased growth and reduced metastasis in immune-competent syngeneic mouse tumor models (137). This positive activity was associated with a substantial increase in CTL proliferation and activation in the tumor microenvironment. Another report implicates PKCη (encoded by PRKCH) and PAK2 as important modulators of Treg suppressor functions via modulation of Treg-APC interactions (138). The authors demonstrated that efficient Treg-mediated suppression of APC-driven CTL activation was dependent on these kinases. Upon Treg-APC engagement, CTLA4 and PKCη were shown to facilitate the disassembly of the structural complexes in Tregs that mediate these cell-cell interactions, thereby enabling the Tregs to release the APC to re-engage other cognate APCs in a serial process of immune suppression. One important unanswered question concerns the absolute requirement of the catalytic activity of either PKCη or PAK2 versus a scaffolding function for these essential Treg functions.
In addition to Tregs, macrophages, MDSCs and DCs play key roles in the suppression of antitumor immunity, and numerous kinases are critically involved in the modulation of these processes. The RTKs CSF1R and KIT regulate the proliferation, survival, and differentiation of myeloid precursor cells, including monocytes and macrophages (123, 139). Small molecule inhibition of CSF1R alone or in combination with adoptive cell therapy, gemcitabine, or antibodies against the T cell regulatory receptors PD-1 or CTLA4 has been shown to markedly inhibit tumor growth and metastasis in syngeneic mouse tumor models, with a corresponding improvement in overall survival (140–143). Inhibition of KIT activity has also been shown to both reduce MDSC numbers and activate DCs in the tumor microenvironment, resulting in enhanced tumor immunity (139, 144). As another example, MST1 (encoded by RON), an RTK predominantly expressed on macrophages, is involved in the resolution of macrophage proinflammatory responses, and a number of tumor types express this receptor’s ligand (MSP; MSMB), thereby contributing to an immunosuppressive tumor microenvironment (145). Small molecule inhibition of MST1 was shown to profoundly limit outgrowth in a syngeneic mouse tumor model of metastasis (146). Finally, as a member of the TAM RTK family, MERTK is important for both macrophage and DC suppressive activity (147). These cells are known to express the type I interferon receptor to which the TAMs heterodimerize and transduce signals to dampen inflammatory signaling. Accordingly, a recent report employing the Mertk–/– mouse demonstrated that loss of this receptor significantly enhanced the latency and reduced the growth and metastasis of various syngeneic mouse tumor models (148).
Beyond the illustrative examples provided here, numerous other cell types and signaling pathways outside the scope of this Review can influence the tumor immune response. While modulation of tumor immunity via immune checkpoint modulation has been clinically validated, this field is still in its infancy, and much remains to be elucidated around the various regulatory mechanisms controlling the processes involved in activating and resolving immune responses to the tumor. Accordingly, it is anticipated that additional kinase targets will be identified, providing further therapeutic entry points and unique mechanisms to modulate this important cancer hallmark.
Conclusions
Kinases represent a large proportion of actionable, cancer gene alterations, and in the past decade there has been significant progress in generating novel therapeutic inhibitors. Given the recent advances described here in genomics and tumor biology, there is a vast landscape of new kinase targets and emerging inhibitors that warrant further investigation to benefit patients (20, 26). These exciting developments are generating increased optimism within the oncology community that selective targeted therapies will continue to improve response rates and significantly extend the lives of molecularly selected patient populations.
Acknowledgments
We thank Philina Lee, Steve Wenglowsky, Nancy Kohl, and Lan Xu for insightful discussions and feedback on the manuscript.
Footnotes
Conflict of interest: The authors are employees and shareholders of Blueprint Medicines.
Reference information:J Clin Invest. 2015;125(5):1780–1789. doi:10.1172/JCI76094.
References
- 1.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep. 2011;1(90):srep00090. doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139(3):468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Cohen P. Protein kinases — the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kantarjian H, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 9.Shaw AT, et al. Ceritinib in ALK-rearranged non–small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303. doi: 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torkamani A, Verkhivker G, Schork NJ. Cancer driver mutations in protein kinase genes. Cancer Lett. 2009;281(2):117–127. doi: 10.1016/j.canlet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knowles PP, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem. 2006;281(44):33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 16.Miller M, Ginalski K, Lesyng B, Nakaigawa N, Schmidt L, Zbar B. Structural basis of oncogenic activation caused by point mutations in the kinase domain of the MET proto-oncogene: modeling studies. Proteins. 2001;44(1):32–43. doi: 10.1002/prot.1069. [DOI] [PubMed] [Google Scholar]
- 17.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 18.Ferrao PT, Gonda TJ, Ashman LK. Constitutively active mutant D816VKit induces megakayocyte and mast cell differentiation of early haemopoietic cells from murine foetal liver. Leuk Res. 2003;27(6):547–555. doi: 10.1016/S0145-2126(02)00272-2. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. doi: 10.1182/blood.V97.8.2434. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396(6712):643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 23.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zack TI, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medves S, Demoulin J-B. Tyrosine kinase gene fusions in cancer: translating mechanisms into targeted therapies. J Cell Mol Med. 2012;16(2):237–248. doi: 10.1111/j.1582-4934.2011.01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw AT, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 28.Honeyman JN, et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science. 2014;343(6174):1010–1014. doi: 10.1126/science.1249484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shames DS, Minna JD, Gazdar AF. DNA methylation in health, disease, and cancer. Curr Mol Med. 2007;7(1):85–102. doi: 10.2174/156652407779940413. [DOI] [PubMed] [Google Scholar]
- 30.Shern JF, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirschner LS, et al. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26(1):89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 32.Kirschner LS, et al. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005;65(11):4506–4514. doi: 10.1158/0008-5472.CAN-05-0580. [DOI] [PubMed] [Google Scholar]
- 33.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirschi B, Kolligs FT. Alternative splicing of BRAF transcripts and characterization of C-terminally truncated B-Raf isoforms in colorectal cancer. Int J Cancer. 2013;133(3):590–596. doi: 10.1002/ijc.28061. [DOI] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawey ET, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell. 2011;19(3):347–358. doi: 10.1016/j.ccr.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vucic EA, et al. Translating cancer “omics” to improved outcomes. Genome Res. 2012;22(2):188–195. doi: 10.1101/gr.124354.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Comert M, Baran Y, Saydam G. Changes in molecular biology of chronic myeloid leukemia in tyrosine kinase inhibitor era. Am J Blood Res. 2013;3(3):191–200. [PMC free article] [PubMed] [Google Scholar]
- 39. Deininger M, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Presented at: 51st ASH Annual Meeting and Exposition; December 5, 2009; New Orleans, Louisiana, USA. Abstract 1126. [Google Scholar]
- 40.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 41.Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26(7):641–650. doi: 10.1101/gad.186965.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6(294):re6. doi: 10.1126/scisignal.2004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandarlapaty S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012;2(4):311–319. doi: 10.1158/2159-8290.CD-12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–481. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 46.Bardin C, et al. Therapeutic drug monitoring in cancer — are we missing a trick? Eur J Cancer. 2014;50(12):2005–2009. doi: 10.1016/j.ejca.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 47.Widmer N, et al. Review of therapeutic drug monitoring of anticancer drugs part two — targeted therapies. Eur J Cancer. 2014;50(12):2020–2036. doi: 10.1016/j.ejca.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 48.Shah NP, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–125. doi: 10.1016/S1535-6108(02)00096-X. [DOI] [PubMed] [Google Scholar]
- 49.Gorre ME, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 50.Antonescu CR, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 51.Heinrich MC, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heidel F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 53.Doebele RC, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasaki T, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051–6060. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Awad MM, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–2401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 57.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Debiec-Rychter M, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128(2):270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 59.Woyach JA, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furman RR, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352–2354. doi: 10.1056/NEJMc1402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagle N, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371(15):1426–1433. doi: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sawyers CL. The 2011 Gordon Wilson Lecture: overcoming resistance to targeted cancer drugs. Trans Am Clin Climatol Assoc. 2012;123:114–123. [PMC free article] [PubMed] [Google Scholar]
- 63.Yun CH, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khorashad JS, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor–resistant CML: frequency and clonal relationships. Blood. 2013;121(3):489–498. doi: 10.1182/blood-2012-05-431379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serrano C, George S. Recent advances in the treatment of gastrointestinal stromal tumors. Ther Adv Med Oncol. 2014;6(3):115–127. doi: 10.1177/1758834014522491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo T, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15(22):6862–6870. doi: 10.1158/1078-0432.CCR-09-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adrián FJ, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2(2):95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- 68.Dancey JE, Chen HX. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov. 2006;5(8):649–659. doi: 10.1038/nrd2089. [DOI] [PubMed] [Google Scholar]
- 69.Carver BS, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–467. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Antony R, Emery CM, Sawyer AM, Garraway LA. C-RAF mutations confer resistance to RAF inhibitors. Cancer Res. 2013;73(15):4840–4851. doi: 10.1158/0008-5472.CAN-12-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Allen EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Larkin J, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 75.Robert C, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 76.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 78.Hoeflich KP, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69(7):3042–3051. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 79.Wong H, et al. Pharmacodynamics of 2-[4-[(1E)-1-(hydroxyimino)-2,3-dihydro-1H-inden-5-yl]-3-(pyridine-4-yl)-1H-pyrazol-1-yl]ethan-1-ol (GDC-0879), a potent and selective B-Raf kinase inhibitor: understanding relationships between systemic concentrations, phosphorylated mitogen-activated protein kinase kinase 1 inhibition, and efficacy. J Pharmacol Exp Ther. 2009;329(1):360–367. doi: 10.1124/jpet.108.148189. [DOI] [PubMed] [Google Scholar]
- 80.Hatzivassiliou G, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11(5):1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- 81.Villanueva J, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4(6):1090–1099. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wagle N, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoeflich KP, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 84.Gewinner C, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16(2):115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gopal YN, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Corcoran RB, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. 2013;5(196):196ra98. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.She QB, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18(1):39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoeflich KP, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012;72(1):210–219. doi: 10.1158/0008-5472.CAN-11-1515. [DOI] [PubMed] [Google Scholar]
- 89.Sun C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–122. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 90.Corcoran RB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71(15):5067–5074. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seghers AC, Wilgenhof S, Lebbé C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012;22(6):466–472. doi: 10.1097/CMR.0b013e3283541541. [DOI] [PubMed] [Google Scholar]
- 94.Savage P, Stebbing J, Bower M, Crook T. Why does cytotoxic chemotherapy cure only some cancers? Nat Clin Pract Oncol. 2009;6(1):43–52. doi: 10.1038/ncponc1260. [DOI] [PubMed] [Google Scholar]
- 95.Bruyère C, Meijer L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr Opin Cell Biol. 2013;25(6):772–779. doi: 10.1016/j.ceb.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 96.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Del Nagro CJ, et al. Chk1 inhibition in p53-deficient cell lines drives rapid chromosome fragmentation followed by caspase-independent cell death. Cell Cycle Georget. 2014;13(2):303–314. doi: 10.4161/cc.27055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xiao Y, et al. Identification of preferred chemotherapeutics for combining with a CHK1 inhibitor. Mol Cancer Ther. 2013;12(11):2285–2295. doi: 10.1158/1535-7163.MCT-13-0404. [DOI] [PubMed] [Google Scholar]
- 99.Fry DW, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438. [PubMed] [Google Scholar]
- 100.Roberts PJ, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476–487. doi: 10.1093/jnci/djs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Johnson SM, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120(7):2528–2536. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Savage PA. Tumor antigenicity revealed. Trends Immunol. 2014;35(2):47–48. doi: 10.1016/j.it.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Antonia SJ, Larkin J, Ascierto PA. Immuno-oncology combinations: a review of clinical experience and future prospects. Clin Cancer Res. 2014;20(24):6258–6268. doi: 10.1158/1078-0432.CCR-14-1457. [DOI] [PubMed] [Google Scholar]
- 104.Luke JJ, Ott PA. Kinase inhibitors and immune check-point blockade for the treatment of metastatic melanoma and advanced cancer: synergistic or antagonistic? Expert Opin Pharmacother. 2013;14(18):2457–2462. doi: 10.1517/14656566.2013.849244. [DOI] [PubMed] [Google Scholar]
- 105.Casey SC, Li Y, Fan AC, Felsher DW. Oncogene withdrawal engages the immune system to induce sustained cancer regression. J Immunother. 2014;2:24. doi: 10.1186/2051-1426-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim DW, Trinh VA, Hwu W-J. Ipilimumab in the treatment of advanced melanoma — a clinical update. Expert Opin Biol Ther. 2014;14(11):1709–1718. doi: 10.1517/14712598.2014.963053. [DOI] [PubMed] [Google Scholar]
- 107.Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23(21):5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 108.Hari SB, Merritt EA, Maly DJ. Sequence determinants of a specific inactive protein kinase conformation. Chem Biol. 2013;20(6):806–815. doi: 10.1016/j.chembiol.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Finlay MRV, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57(20):8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 111.Nishioka Y, Aono Y, Sone S. Role of tyrosine kinase inhibitors in tumor immunology. Immunotherapy. 2011;3(1):107–116. doi: 10.2217/imt.10.79. [DOI] [PubMed] [Google Scholar]
- 112.Hubbard RE, Murray JB. Experiences in fragment-based lead discovery. Methods Enzymol. 2011;493:509–531. doi: 10.1016/B978-0-12-381274-2.00020-0. [DOI] [PubMed] [Google Scholar]
- 113.Staben ST, et al. Back pocket flexibility provides group II p21-activated kinase (PAK) selectivity for type I 1/2 kinase inhibitors. J Med Chem. 2014;57(3):1033–1045. doi: 10.1021/jm401768t. [DOI] [PubMed] [Google Scholar]
- 114.Hagel M, et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. doi: 10.1158/2159-8290.CD-14-1029. [published online ahead of print March 16, 2015]. doi: 10.1158/2159-8290.CD-14-1029. [DOI] [PubMed] [Google Scholar]
- 115.Kwiatkowski N, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ott PA, Fritsch EF, Wu CJ, Dranoff G. Vaccines and melanoma. Hematol Oncol Clin North Am. 2014;28(3):559–569. doi: 10.1016/j.hoc.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 117.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 118.Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, Teixeira MM. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther. 2013;139(2):189–212. doi: 10.1016/j.pharmthera.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 119.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127(4):759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 120.Huynh A, Zhang R, Turka LA. Signals and pathways controlling regulatory T cells. Immunol Rev. 2014;258(1):117–131. doi: 10.1111/imr.12148. [DOI] [PubMed] [Google Scholar]
- 121.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shurin GV, Ma Y, Shurin MR. Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron. 2013;6(2):159–167. doi: 10.1007/s12307-013-0133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol. 2014;5:489. doi: 10.3389/fimmu.2014.00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176(1):284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 125.Schmid MC, Varner JA. Myeloid cells in the tumor microenvironment: modulation of tumor angiogenesis and tumor inflammation. J Oncol. 2010;2010:201026. doi: 10.1155/2010/201026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sreeramkumar V, Fresno M, Cuesta N. Prostaglandin E2 and T cells: friends or foes? Immunol Cell Biol. 2012;90(6):579–586. doi: 10.1038/icb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mosenden R, Taskén K. Cyclic AMP-mediated immune regulation — overview of mechanisms of action in T cells. Cell Signal. 2011;23(6):1009–1016. doi: 10.1016/j.cellsig.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 128.Shui JW, et al. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat Immunol. 2007;8(1):84–91. doi: 10.1038/ni1416. [DOI] [PubMed] [Google Scholar]
- 129.Zhong XP, Guo R, Zhou H, Liu C, Wan CK. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol Rev. 2008;224:249–264. doi: 10.1111/j.1600-065X.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Funderud A, et al. Isoform-specific regulation of immune cell reactivity by the catalytic subunit of protein kinase A (PKA). Cell Signal. 2009;21(2):274–281. doi: 10.1016/j.cellsig.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 131.Joshi RP, Koretzky GA. Diacylglycerol kinases: regulated controllers of T cell activation, function, and development. Int J Mol Sci. 2013;14(4):6649–6673. doi: 10.3390/ijms14046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tan YX, et al. Inhibition of the kinase Csk in thymocytes reveals a requirement for actin remodeling in the initiation of full TCR signaling. Nat Immunol. 2014;15(2):186–194. doi: 10.1038/ni.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sawasdikosol S, Zha R, Yang B, Burakoff S. HPK1 as a novel target for cancer immunotherapy. Immunol Res. 2012;54(1–3):262–265. doi: 10.1007/s12026-012-8319-1. [DOI] [PubMed] [Google Scholar]
- 134.De la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34(9):2480–2488. doi: 10.1002/eji.200425274. [DOI] [PubMed] [Google Scholar]
- 135.Yaqub S, Taskén K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumor immune responses by regulatory T cells. Crit Rev Oncog. 2008;14(1):57–77. doi: 10.1615/CritRevOncog.v14.i1.40. [DOI] [PubMed] [Google Scholar]
- 136.Patton DT, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177(10):6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- 137.Ali K, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510(7505):407–411. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kong KF, et al. Protein kinase C-η controls CTLA-4-mediated regulatory T cell function. Nat Immunol. 2014;15(5):465–472. doi: 10.1038/ni.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pan PY, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111(1):219–228. doi: 10.1182/blood-2007-04-086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mok S, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mitchem JB, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhu Y, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pittoni P, Piconese S, Tripodo C, Colombo MP. Tumor-intrinsic and -extrinsic roles of c-Kit: mast cells as the primary off-target of tyrosine kinase inhibitors. Oncogene. 2011;30(7):757–769. doi: 10.1038/onc.2010.494. [DOI] [PubMed] [Google Scholar]
- 145.Wang X, Hankey PA. The ron receptor tyrosine kinase: a key regulator of inflammation and cancer progression. Crit Rev Immunol. 2013;33(6):549–574. doi: 10.1615/CritRevImmunol.2013007953. [DOI] [PubMed] [Google Scholar]
- 146.Eyob H, Ekiz HA, Derose YS, Waltz SE, Williams MA, Welm AL. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013;3(7):751–760. doi: 10.1158/2159-8290.CD-12-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11):a009076. doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.MerTK inhibition in tumor leukocytes decreases tumor growth metastasis J Clin Invest. 2013;123(8):3231–3242. doi: 10.1172/JCI67655. [DOI] [PMC free article] [PubMed] [Google Scholar]