

Abstract
Epithelial cells that line the conducting airways provide the initial barrier and innate immune responses to the abundant particles, microbes, and allergens that are inhaled throughout life. The transcription factors SPDEF and FOXA3 are both selectively expressed in epithelial cells lining the conducting airways, where they regulate goblet cell differentiation and mucus production. Moreover, these transcription factors are upregulated in chronic lung disorders, including asthma. Here, we show that expression of SPDEF or FOXA3 in airway epithelial cells in neonatal mice caused goblet cell differentiation, spontaneous eosinophilic inflammation, and airway hyperresponsiveness to methacholine. SPDEF expression promoted DC recruitment and activation in association with induction of Il33, Csf2, thymic stromal lymphopoietin (Tslp), and Ccl20 transcripts. Increased Il4, Il13, Ccl17, and Il25 expression was accompanied by recruitment of Th2 lymphocytes, group 2 innate lymphoid cells, and eosinophils to the lung. SPDEF was required for goblet cell differentiation and pulmonary Th2 inflammation in response to house dust mite (HDM) extract, as both were decreased in neonatal and adult Spdef–/– mice compared with control animals. Together, our results indicate that SPDEF causes goblet cell differentiation and Th2 inflammation during postnatal development and is required for goblet cell metaplasia and normal Th2 inflammatory responses to HDM aeroallergen.
Keywords: Development, Genetics, Immunology, Inflammation, Therapeutics
Introduction
The tracheobronchial tree is lined by a diversity of epithelial cells including basal, ciliated, serous, and goblet cells that make unique contributions to mucociliary clearance, innate immunity, and recruitment and activation of professional immune cells that mediate inflammatory responses to environmental exposures (1–5). The nature and extent of innate and acquired immune responses to respiratory pathogens and allergens are influenced genetically and developmentally and by exposure to specific pathogens and toxicants. There is increasing evidence that pulmonary immune responses are initiated early in development, in utero, and during infancy (6–10). Mechanisms by which early exposures of the lung to pathogens instruct the acquisition of innate and acquired immunity are of considerable interest. For example, wheezing following infection of infants to rhinovirus (RV) strongly predicts subsequent risks for development of asthma later in life (11, 12). The observations that the incidence of childhood asthma is lower in infants exposed to an abundance of bacterial and fungal microbes supports the concept that early environmental exposures influence the development of asthma (13). Mechanisms influencing the acquisition of Th1- and Th2-mediated immunity in infancy have important implications for host defense and allergic responses that mediate subsequent responses to environmental stimuli throughout life (14). Recent studies in gnotobiotic mice demonstrate an age-dependent programming of invariant NK cells that influences subsequent allergic responses in the lung (14). Respiratory epithelial cells are increasingly recognized as important modulators of innate immune responses that govern inflammatory and immune responses to pathogens and toxicants via their synthesis of chemokines and cytokines, growth factors that regulate the migration and activation of the diverse immune cells to sites of injury (refs. 15–17, for review). Airway epithelial cells mount rapid and robust responses to a diversity of pathogen-associated molecular pattern molecules (PAMPs), damage-associated molecular pattern molecules (DAMPs) that influence both innate immune and repair processes that maintain pulmonary homeostasis. The airway epithelial barrier is maintained by a diversity of specific epithelial cell types that each contribute uniquely in response to airway injury. Basal and nonciliated progenitor cells serve as precursors from which ciliated and goblet cells are derived (2, 18). Airway goblet cells synthesize and secrete a variety of mucins and other proteins that play important roles in mucociliary clearance and host defense (19). While the activation of goblet cells and mucus secretion are normal responses to acute injury and infection, chronic goblet cell hyperplasia and metaplasia are features of common chronic pulmonary diseases, including asthma, cystic fibrosis (CF), and chronic obstructive pulmonary disease (COPD), wherein mucus hyperproduction contributes to the mortality and morbidity associated with these disorders (19–21).
Airway goblet cell differentiation is influenced by the transcription factors SAM-pointed domain–containing ETS-like factor (SPDEF) and forkhead ortholog A3 (FOXA3), which are selectively expressed in airway goblet cells and induced by toxicants, aeroallergens, viral infections, and cytokines. SPDEF is sufficient and required for goblet cell differentiation and mucus production in the mouse airway (22, 23). SPDEF and FOXA3 are induced following exposure to RV, aeroallergens, and Th2 cytokines, including IL-13, that enhance goblet cell differentiation and regulate a network of genes involved in the synthesis and packaging of airway mucins (22–25). SPDEF induces FOXA3 in airway epithelial cells, the latter also being sufficient to cause goblet cell metaplasia in the adult mouse lung (24). The present study was designed to test whether goblet cell differentiation induced by SPDEF or FOXA3 in the perinatal period influences the development of pulmonary innate immune responses. We demonstrated that expression of either SPDEF or FOXA3 in nonciliated airway epithelial cells of the developing mouse lung caused extensive goblet cell differentiation and Th2-mediated pulmonary inflammation. SPDEF was required for Th2 inflammatory responses to house dust mite (HDM) extract in neonatal and adult mice and was necessary for FOXA3-induced goblet cell differentiation and Th2 inflammation.
Results
SPDEF causes Th2-mediated inflammation and airway hyperresponsiveness.
To assess the potential role of SPDEF in the regulation of goblet cell metaplasia and inflammation in the developing lung, Spdef mRNA was conditionally expressed in nonciliated respiratory epithelial cells under the control of the Scgb1a1 promoter induced by administration of doxycycline to the dam from E16 to P15. On P15, SPDEF caused extensive goblet cell metaplasia in conducting airways, increasing Foxa3, Muc5ac, and Muc5b mRNAs and immunostaining (Figure 1A and Table 1). In contrast to the lack of inflammation following transient expression of SPDEF in adult mice (22, 23), SPDEF caused widespread inflammatory infiltrates consisting primarily of eosinophils in the neonatal mice (Figure 1, A and C). Airway hyperresponsiveness (AHR), determined by methacholine challenge tests, was augmented in association with increased αSMA (Acta2) mRNA (Figure 1B and Table 1). Consistent with the observed eosinophilic inflammation, mRNAs encoding cytokines and chemokines that mediate Th2 inflammation were significantly increased, including those mediating DC activation (Il33, Il25, Csf2); likewise, Th2 cytokines (Il13, Ccl17) and eosinophilic chemoattractants (Ccl24 and Ccl20) were increased. SPDEF induced Ear11 mRNA, which was associated with the eosinophilic inflammation noted histologically (Table 1).
Figure 1. SPDEF causes goblet cell differentiation, pulmonary inflammation, and AHR.

Dams of Scgb1ab-rtTA TRE-Spdef (black bars) and control littermate single-transgenic mice (white bars) were placed on doxycycline from E16.5 to P15. (A) At P15, lung histology of the pups was assessed by H&E (n = 8) and BALF (n = 4) staining with Diff-Quik. Immunostaining for SPDEF, FOXA3, and MUC5B demonstrated extensive goblet cell differentiation (n = 8). (A and C) Cells isolated from BALF were stained with Diff-Quik and showed eosinophilic infiltrates. Data represent the mean ± SEM. *P < 0.05 compared with controls using an unpaired, 2-tailed Student’s t test. (B) AHR is represented as Penh in response to methacholine. Data represent the mean ± SEM of 6 mice per group. *P < 0.05 by 2-way ANOVA. (E) Flow cytometric analysis of lung cells obtained at P15 demonstrated increased numbers of SiglecF+CCR3+ in Scgb1a1-rtTA Spdef mice (black bar) compared with cell numbers detected in littermate control mice (white bar). (D and E) ST2+, IL-17RB+, and ICOS+ ILCs and total CD3+ and CD3+ IL-4–producing T cells were increased, and IFN-γ–producing CD3+ T cells were unaltered. Data represent the mean ± SEM. *P < 0.05 compared with controls using an unpaired, 2-tailed Student’s t test. n = 4/group. Examples of FACS analyses are provided in Supplemental Figure 1.
Table 1. SPDEF induced mRNAs related to goblet cell differentiation and Th2 inflammation.
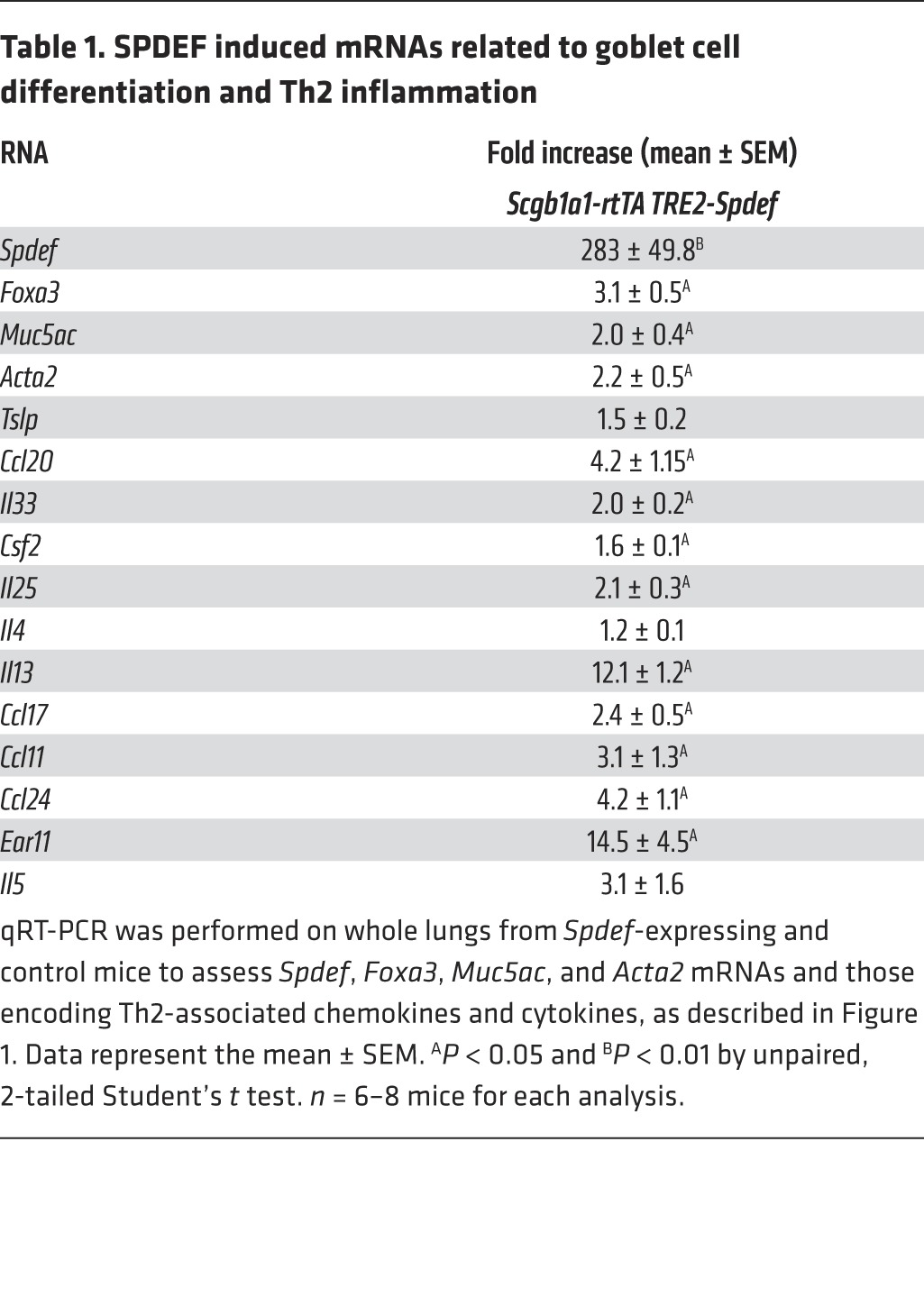
Consistent with increased expression of Th2 cytokines and chemokines, the numbers of ST2+ and IL-17RB+ innate lymphoid cells (ILCs), CD3+ T lymphocytes, and IL-4+ T cells were increased by SPDEF, while IFN-γ CD3+ cell numbers were unaltered (Figure 1, D and E, and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI79422DS1). Taken together, expression of SPDEF in nonciliated respiratory epithelial cells in the newborn period caused extensive goblet cell metaplasia, spontaneous Th2 pulmonary inflammation, and increased AHR, findings likely mediated by airway epithelial cell elaboration of the cytokines and chemokines that recruit and activate DCs, ILCs, and lymphocytes to mediate a Th2 inflammatory cascade in the neonatal-postnatal lung.
FOXA3 causes goblet cell metaplasia and Th2 inflammation in neonatal mice.
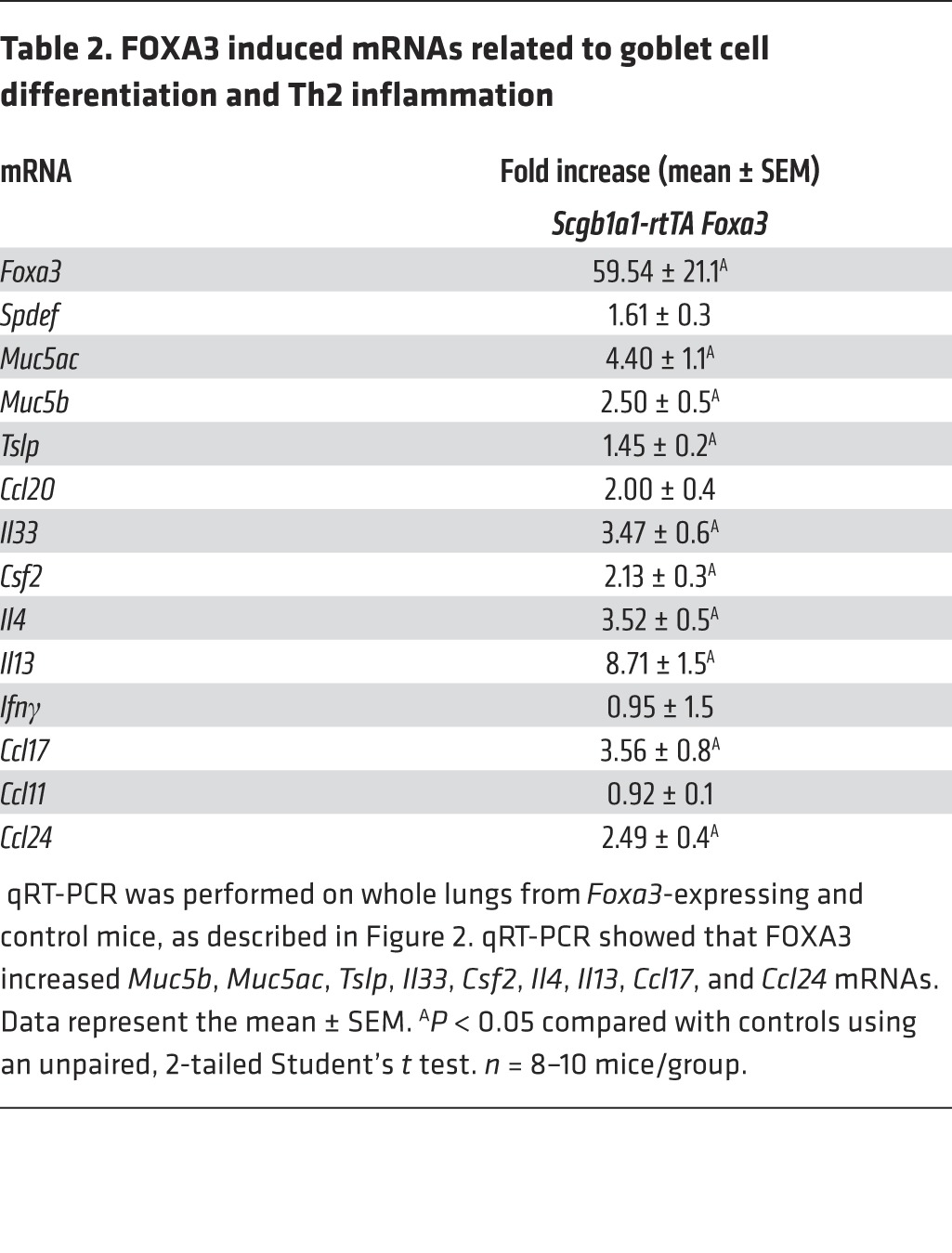
In adult mice and in human airway epithelial cells in vitro, FOXA3 induced SPDEF and caused goblet cell metaplasia (24). To test whether FOXA3 was sufficient to cause goblet cell metaplasia in neonatal mice, FOXA3 was conditionally expressed in nonciliated airway epithelial cells under the control of the same Scgb1a1 promoter by placing the dams on doxycycline from E16.5 to P15. When FOXA3 was selectively expressed in Club cells, SPDEF was induced and SCBG1A1 (CCSP) staining decreased but was still present in the goblet cells induced by FOXA3, where it was coexpressed with MUC5B (Figure 2A and Supplemental Figure 2). In contrast, ciliated cells did not express MUC5B or SCGB1A1, consistent with the specificity of the Scgb1a1 promoter in Club cells. FOXA3 caused extensive eosinophilic inflammation and increased AHR in response to methacholine, findings similar to those observed after expression of SPDEF (Figure 2, A–C). FOXA3 caused goblet cell differentiation associated with increased expression of SPDEF, MUC5AC, and MUC5B (Figure 2A and Supplemental Figure 2). As observed in SPDEF-expressing mice, pulmonary infiltrates consisted of increased numbers of eosinophils and ILCs, including ST2+, IL-17RB+, and ICOS+ lineage–negative cells; CD103+ DCs and IL-4+ CD3 lymphocytes were also increased (Figure 2, D–G). mRNAs, including Tslp, Il33, and Csf2, that encode factors mediating DC chemotaxis and activation were significantly increased by FOXA3. Il13, Il4, Ccl17, and Ccl24 mRNAs were increased, consistent with the increased Th2-mediated inflammation induced by FOXA3 (Table 2). Taken together, FOXA3 expression in respiratory epithelial cells of postnatal mice induced SPDEF and caused goblet cell metaplasia and Th2-dominated pulmonary inflammation, both of which are essentially phenocopying effects of SPDEF.
Figure 2. Conditional expression of FOXA3 in airway epithelial cells causes pulmonary inflammation and goblet cell metaplasia.

(A–C) Control littermates (white bars) and Scgb1ab-rtTA OTet7-Foxa3 dams (black bars) were placed on doxycycline from E16.5 to P15. Lung tissue was obtained from the pups at P15. Scgb1ab-rtTA OTet7-Foxa3 pups (black bars) developed spontaneous eosinophilic inflammation, goblet cell metaplasia, and AHR. Diff-Quik staining of BALF (n = 4) demonstrated eosinophilic infiltration. (E) Flow cytometric analysis of lung cells obtained at P15 demonstrated increased numbers of SiglecF+CCR3+, (D–F) ST2+, IL-17RB+, and ICOS+ ILCs and CD3+ IL-4–producing T cells. IFN-γ–producing CD3+ T cells were unaltered. (G) pDCs (CD11b–CD11c+B220+), CD103+ mDCs (CD11b+CD11c+B220–) were increased and mDCs were unchanged in lung from Scgb1a1-rtTA OTet7-Foxa3 pups. Activation markers (I-A/E and CD86) on pDCs and mDCs are shown in G. Data represent the mean ± SEM. *P < 0.05 compared with controls using an unpaired, 2-tailed Student’s t test. n = 4/group.
Table 2. FOXA3 induced mRNAs related to goblet cell differentiation and Th2 inflammation.
SPDEF is required for HDM-induced pulmonary inflammation and AHR.
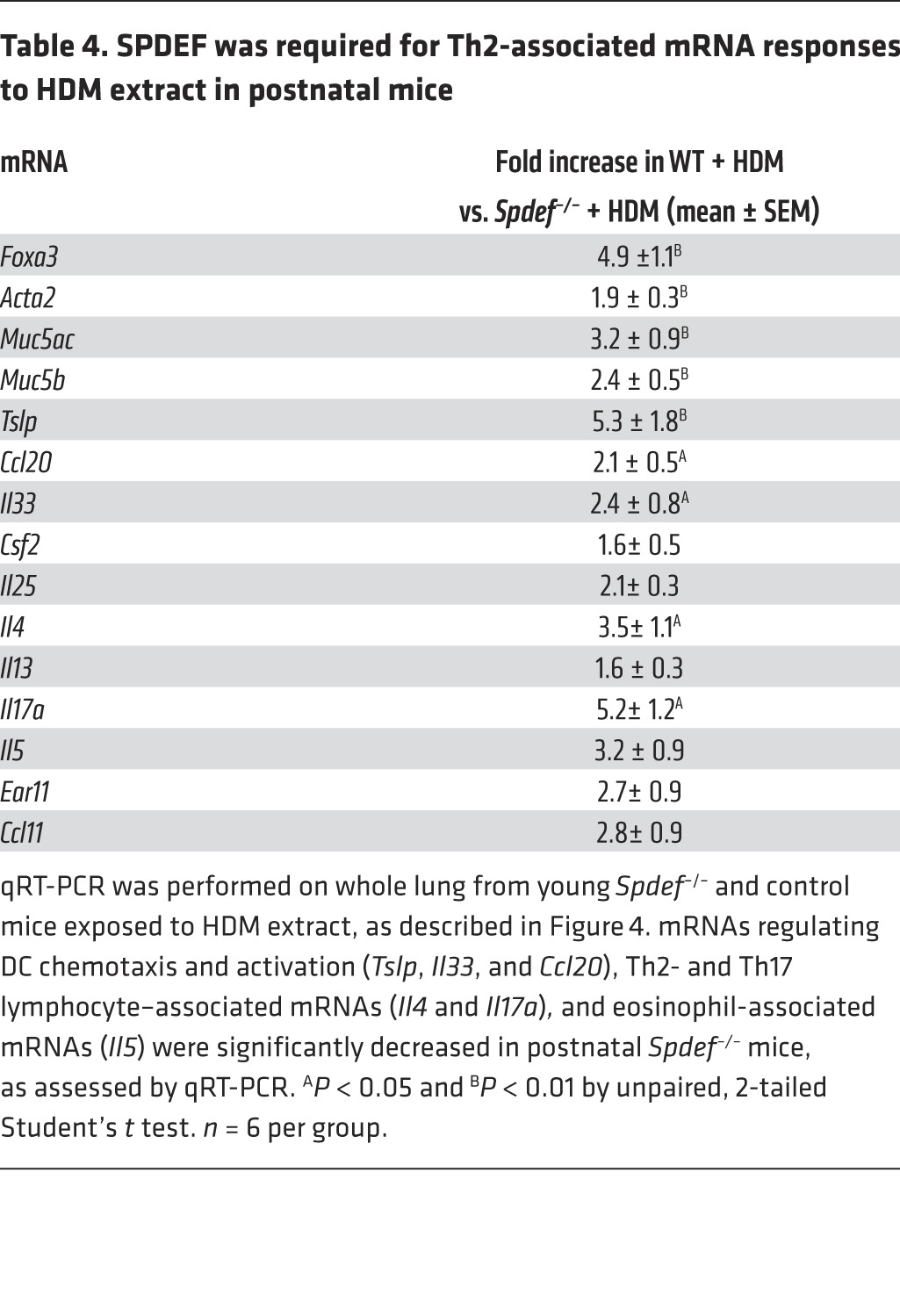
Since SPDEF and FOXA3 were sufficient to induce Th2-mediated pulmonary inflammation in the postnatal period, we tested whether the absence of SPDEF influenced goblet cell metaplasia and pulmonary inflammation following exposure to HDM extract in both neonatal and adult mice. Adult Spdef+/+ and Spdef–/– littermates were treated i.n. daily for 3 days with 100 μg HDM extract. AHR, goblet cell differentiation, eosinophilic infiltration, and Th2 inflammatory markers were markedly suppressed in the Spdef–/– mice (Figure 3, A–C, and Table 3). In control mice, HDM caused Th2-associated lung inflammation, goblet cell differentiation, and increased AHR. Induction of Foxa3, Muc5b, and Muc5ac mRNAs and those associated with goblet cell differentiation seen after HDM exposure were inhibited in the Spdef–/– mice (Table 3). Likewise, expression of mRNAs encoding the DC-related chemoattractants Il33 and Ccl20 and Th2 lymphocyte–associated Ccl17, Il4, Il13 were suppressed, as was eosinophilic infiltration (Table 3). Consistent with the latter findings, the induction of Ear11 and Ccl24 mRNAs after HDM treatment was inhibited in the Spdef–/– mice. Il17 mRNA and IL-17+CD3+ cells were decreased, and recruitment of ST2+ ILCs and T cells expressing IL-4, IL-13, or IL-17 were reduced in mice lacking SPDEF following HDM exposure (Figure 3, D and E, and Table 3). To assess whether effector T cells were deficient in Spdef–/– mice, anti-IgD serum was administered to directly induce T cell activation independently of pulmonary sensitization (26). Anti-IgD directly induced IL-4 expression and caused lymph node and spleen enlargement, demonstrating intact effector T cell responses in Spdef–/– mice. Consistent with intact T cell effector functions, serum IgE and IgG1 levels were similar in Spdef–/– and Spdef+/+ mice (Supplemental Figure 3). Taken together, SPDEF is required for HDM-induced goblet cell differentiation, Th2 inflammation, and pulmonary AHR responses in adult mice. To test whether SPDEF is required for inflammatory responses to HDM in the developing lung, neonatal Spdef+/+ and Spdef–/– mice were treated with 50 μg HDM extract. Consistent with our findings in adult mice, SPDEF was required for normal Th2 responses, mucous metaplasia, eosinophilic inflammation, and AHR in the neonatal mice exposed to HDM (Figure 4 and Table 4).
Figure 3. SPDEF is required for Th2 inflammatory responses, AHR, and recruitment of ST2+ ILCs and IL-4+, IL-13+, and IL-17+ T cells in response to HDM extract in adult mice.

HDM extract (100 μg) was administered i.n. daily for 3 days to adult control and Spdef–/– mice, which were sacrificed 2 days later. (A) Decreased mucous metaplasia and inflammatory responses to HDM were seen in Spdef–/– mice. (A and C) BALF cells (n = 4) were stained with Diff-Quik, which showed eosinophilic and neutrophilic infiltrates, and differential cell counts were quantified. Goblet cell differentiation (A) and the increase in Acta2 mRNA (Table 3) caused by HDM challenge were modestly decreased in Spdef–/– mice. (B) AHR was decreased in Spdef–/– mice. Data represent the mean ± SEM. Significance was analyzed using 2-way ANOVA with Bonferroni’s post tests. n = 6/group. (D and E) Flow analysis of lung cells from Spdef+/+ (white bars) and Spdef–/– mice (black bars) after HDM challenge. CD45+ and CD3+ cell numbers were not altered, and ST2+ ILC numbers were decreased. (E) Numbers of IL-4+, IL-13+, and IL-17+ T cells were decreased in Spdef–/– mice. Gating strategies are depicted in Supplemental Figure 1. Data represent the mean ± SEM. *P < 0.05 compared with controls using an unpaired, 2-tailed Student’s t test. n = 4/group.
Table 3. SPDEF was required for Th2 responses to HDM extract in adult mice.
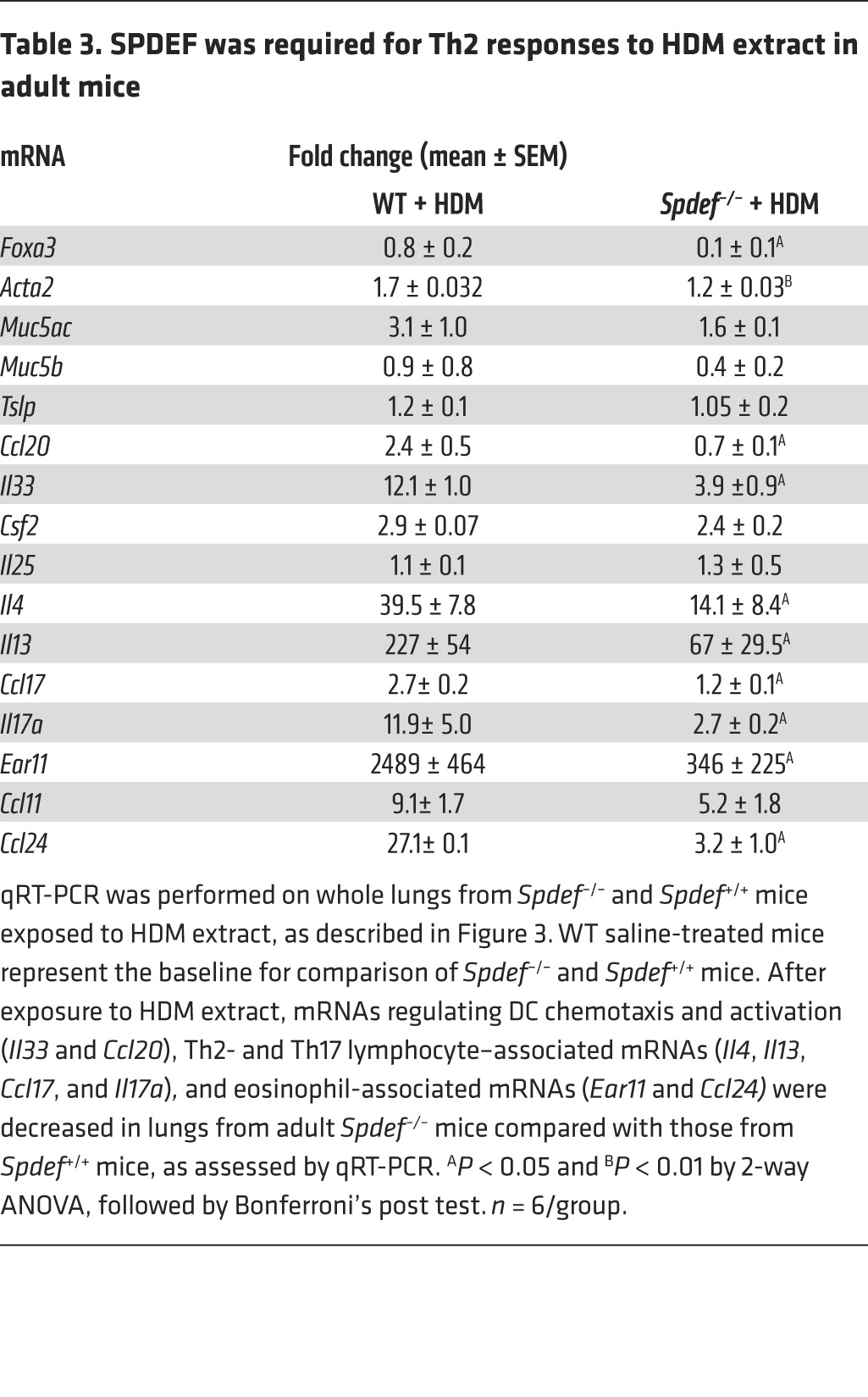
Figure 4. SPDEF is required for AHR, Th2, and TH17 inflammatory responses to HDM in postnatal mice.

HDM (50 μg) was administered i.n. daily for 3 days to PND15 control and Spdef–/– mice, and the mice were sacrificed 2 days later. (A) Decreased mucous metaplasia and inflammatory responses to HDM were observed in Spdef–/– mice. (A and C) BALF cells (n = 4) were stained with Diff-Quik, demonstrating eosinophilic and neutrophilic infiltrates. Goblet cell differentiation (A) and increased Acta2 mRNA caused by HDM challenge (Table 4) were modestly decreased in Spdef–/– mice. (B) Spdef–/– mice were less responsive to HDM (green) than were controls (red line). Data represent the mean ± SEM, using 2-way Anova with Bonferroni’s post tests. n = 6/group. (D and E) Flow analysis of lung cells from Spdef+/+ (white bars) and Spdef–/– mice (black bars) after HDM challenge. SiglecF+CCR3+ eosinophils were decreased, and ST2+, IL-17RB+, ICOS+ ILCs and CD3+ cells were unaltered. CD3+ IL-4+ and CD3+IL-17+ T cells were decreased in Spdef–/– mice, and CD3+IFN-γ+ T cells were unaltered. Gating strategies are depicted in Supplemental Figure 1. Data represent the mean ± SEM. *P < 0.05 compared with controls using an unpaired, 2-tailed Student’s t test. n = 4/group.
Table 4. SPDEF was required for Th2-associated mRNA responses to HDM extract in postnatal mice.
SPDEF is required for FOXA3-induced pulmonary inflammation.
Since expression of either SPDEF or FOXA3 was sufficient to induce Th2 inflammatory responses in neonatal mice, we tested whether SPDEF was required for the effects of FOXA3. Th2 inflammatory mediators, goblet cell differentiation, and AHR caused by expression of FOXA3 were significantly inhibited in neonatal Spdef–/– as well as in Spdef–/+ mice (Supplemental Figure 4). Increased expression of Muc5ac, Muc5b, Tslp, Il4, and Il13 mRNAs was decreased in FOXA3-expressing Spdef–/– and Spdef+/– mice compared with that observed in Spdef+/+ littermates (Supplemental Figure 4D). Methacholine challenge tests demonstrated that the effects of FOXA3 on AHR were inhibited in Spdef–/– and Spdef+/– mice (Supplemental Figure 4, A and B). Thus, SPDEF plays a central role in both HDM- and FOXA3-induced goblet cell metaplasia and pulmonary inflammation.
FOXA3 enhances pulmonary responses to HDM.
While FOXA3 was not required for the induction of Th2-mediated inflammation by aeroallergen in adult mice (24, 25), the present findings in neonatal mice demonstrated that FOXA3 was sufficient to induce Th2-mediated inflammation. Since the effects of SPDEF and FOXA3 in pulmonary inflammation were similar, we assessed whether FOXA3 further enhanced inflammatory responses during aeroallergen challenge. Scgb1a1-rtTa OTet7-Foxa3 conditional mice were exposed to HDM extract (50 μg) delivered i.n. AHR to HDM extract was increased in FOXA3-expressing mice (Supplemental Figure 5). Likewise, Spdef and other goblet cell–associated Muc5ac and Muc5b mRNA levels were further increased in the FOXA3-expressing mice after exposure to HDM. mRNAs indicating Th2-related DC and lymphocytic activity were increased by expression of FOXA3 after HDM exposure (Supplemental Figure 5C). Consistent with these findings, Spdef haploinsufficiency inhibited the enhanced AHR response to HDM in Foxa3-expressing transgenic mice (Supplemental Figure 4B).
Anti-TSLP ameliorates the inflammatory effects of FOXA3.
Thymic stromal lymphopoetin (TSLP) is expressed by respiratory epithelial cells and is a known mediator of Th2-dependent inflammation in multiple organs, including skin, intestine, and lung (27, 28). We interrogated previous FOXA3-associated ChIP data from our recent studies (24) and identified strong binding of FOXA3 to the promoter region of the human TSLP gene (Supplemental Figure 6). To test the role of TSLP in the induction of FOXA3-generated pulmonary inflammation in neonatal mice, TSLP activity was inhibited by injection of these mice with anti-TSLP Ab on P3 during the induction of FOXA3 (29, 30). Anti-TSLP substantially inhibited pulmonary inflammation and MUC5B staining (Supplemental Figure 7A). TSLP Ab also inhibited Spdef, Muc5ac, Csf2, Ccl11, Ear11, Il4, Tslp, Il5, Ccl24, Il13, and Ccl17, indicating that TSLP is an important mediator of FOXA3-induced inflammation (Supplemental Figure 7B).
Discussion
Pulmonary innate and acquired immunity is rapidly acquired following birth and exposure of pulmonary tissues to commensal and pathogenic microbes, allergens, and toxicants that recruit BM-derived immune cells to the lung. The present findings demonstrate that the selective expression of SPDEF or FOXA3, both transcription factors normally restricted to conducting airways and submucosal gland epithelial cells, was sufficient to induce airway goblet cell differentiation in the neonatal-postnatal period. Either transcription factor was sufficient to cause the spontaneous pulmonary Th2 and eosinophilic inflammation associated with enhanced recruitment and activation of ST2+ group 2 ILCs, DCs, and Th2-expressing lymphocytes, mediated in part by the expression of cytokines and chemokines by respiratory epithelial cells. Remarkably, SPDEF was required for Th2 responses to HDM extract in both neonatal and adult mice and for the inflammatory effects of FOXA3. The present findings demonstrate a critical role for SPDEF and goblet cells in the acquisition of pulmonary Th2 immune responses.
Respiratory epithelial cells are increasingly recognized as active mediators of innate host defense and inflammation, playing multiple roles in mucociliary clearance, barrier function, and recruitment and activation of professional immune cells, the latter being mediated by the elaboration of diverse cytokines and chemokines (15–17). In contrast to the Th2 inflammatory responses seen in the present study in neonatal mice after expression of SPDEF or FOXA3, the expression of these transcription factors in adult mice caused reversible goblet cell differentiation from nonciliated airway epithelial cells (Club cells) and increased mucus production, but did not cause acute inflammation (22–24). Consistent with those studies, subsets of goblet cells induced by expression of FOXA3 coexpressed CCSP and MUC5B, albeit at lower levels for CCSP (Supplemental Figure 2). The present findings support the concept that SPDEF and FOXA3, perhaps in part related to goblet cell differentiation, influence the recruitment and activation of professional immune cells during the adaptation to exposures to aeroallergens and microbes after birth. SPDEF and FOXA3, transcription factors expressed selectively in nonciliated airway epithelial cells, activate a number of genes regulating mucus metaplasia, including MUC5AC, MUC5B, AGR2, and other proteins involved in the synthesis, glycosylation, and packaging of mucus (23, 31). While a rare subset of airway epithelial cells in normal mouse lung, goblet cells are relatively abundant in conducting airways of the neonatal mouse, and their numbers increase in the first weeks of life (32). Acute goblet cell metaplasia is associated with bacterial and viral infections and exposure to allergens and toxicants (17, 20, 21, 33, 34). Chronic goblet cell metaplasia is associated with increased SPDEF and FOXA3 expression in the setting of asthma, COPD, CF, and other chronic inflammatory diseases of the lung (24). We speculate that early infectious or other inflammatory exposure inducing goblet cell differentiation via SPDEF may serve to enhance Th2 polarization of pulmonary innate immunity to influence subsequent inflammatory responses to environmental challenges.
While mechanisms by which SPDEF induces Th2 inflammatory responses in neonatal mice are likely complex, expression of cytokines and chemokines, known to be products of epithelial cells, e.g., IL-33, TSLP, IL-25, and granulocyte-macrophage CSF (GM-CSF), were spontaneously induced by expression of SPDEF and FOXA3 in neonatal mice. Professional immune cells are actively recruited to the lung following birth by exposure to the environment and release of chemokines by pulmonary cells; for example, monocyte and macrophage recruitment is mediated by the expression of GM-CSF by pulmonary parenchymal cells (35, 36). In the present studies, SPDEF and FOXA3 induced chemokines and cytokines known to attract and activate group 2 ILCs and DCs, recruiting and sensitizing lymphocytes that together elaborate Th2-associated cytokines such as IL-4, IL-5, and IL-13 (37, 38). Since IL-13 and IL-4 regulate SPDEF in airway epithelial cells through IL-4 receptor signaling and STAT6 (22), their expression by Th2 lymphocytes and ILCs may serve to further enhance SPDEF and FOXA3 expression. Since a number of cytokines and chemokines were induced by SPDEF and FOXA3, the mediators of pulmonary Th2 inflammation are likely complex. TSLP, a Th2 chemokine expressed by epithelial cells that are expressed, for example, in skin, gastrointestinal tract, and lung in response to injury and allergens (38–40), was induced by either SPDEF or FOXA3; anti-TSLP Ab substantially inhibited FOXA3-induced Th2-mediated inflammation. However, anti-TSLP decreased expression of SPDEF, perhaps influencing expression of Th2 cytokines (e.g., IL-4 and IL-13) known to induce SPDEF. Surprisingly, anti-TSLP treatment resulted in increased expression of FOXA3 (Supplemental Figure 7), perhaps indicating lack of a counterregulatory response, although it is unclear in the present experiments whether the increase in Foxa3 mRNA resulted from the endogenous Foxa3 gene or from changes in the activity of the Scgb1a1 transgene expressing the Foxa3 cDNA. Recent in vivo and in vitro studies showed that FOXA3 induced TSLP expression in human airway epithelial cells in vitro (24); ChIP of FOXA3-associated transcriptional targets identified direct binding of FOXA3 to the promoter region of the human TSLP gene, supporting the concept that TSLP represents an important signaling molecule produced by the airway epithelial cells that regulate Th2 inflammatory responses to FOXA3 and SPDEF. FOXA3 binding was also detected in the first intron and in the 3′ region of the CCL17 gene (Supplemental Figure 6).
While FOXA3 was not required for goblet cell differentiation and inflammation following antigen sensitization in mice (24, 25), we demonstrated that SPDEF was required for normal Th2 inflammatory responses to HDM allergen in both postnatal and adult mice. Both SPDEF and FOXA3 have antiinflammatory effects on airway epithelial cells, probably functioning by distinct mechanisms. SPDEF bound MyD88 and TRIF, inhibiting NF-κB and interferon responses (41). In contrast, FOXA3 inhibited IFN responses to RV, thereby impairing RV clearance, which was mediated in part by the inhibitory effects of FOXA3 on IRF3 and IFN production (24). Taken together with its ability to enhance expression of Th2-polarizing cytokines and chemokines, we speculate that inhibition of Th1 and IFN responses by SPDEF and FOXA3 may favor Th2 responses to inflammatory stimuli. HDM activates NF-κB through TLR4, the latter being required for normal Th2 responses in the lung (33, 34). While SPDEF and FOXA3 regulate the expression of a number of modulators of Th2 inflammatory responses to HDM, the precise drivers of the spontaneous Th2 inflammatory responses caused by the expression of SPDEF and FOXA3 are unclear. Increased expression of both DC and ILC chemoattractants and activation of Th2 lymphocytes induced by SPDEF or FOXA3 likely serve to enhance Th2-type inflammation in the present models. Alternatively, goblet cells induced by SPDEF may have a direct role in enhancing innate immune responses. Recent findings that goblet cells provide conduits for transepithelial delivery of tolerogenic antigens from the intestinal lumen to CD103+ DCs provide a precedent for a role for goblet cells in communicating with DCs to influence innate immune responses in epithelia (42).
SPDEF and FOXA3 were sufficient to enhance AHR assessed by “enhanced pause” (Penh); however, it is unclear whether these findings represent their effects on goblet cell metaplasia or on bronchial smooth muscle. Acta2 mRNA was increased by expression of Spdef and was decreased in both neonatal and adult Spdef–/– mice after HDM extract exposure. Since Acta2 mRNA is expressed in both vascular and bronchial smooth muscle cells, it is unclear whether these findings are directly related to changes in AHR. Given that increased IL-4 receptor signaling is known to enhance AHR (43, 44), the present findings may be related to increased Th2 cytokine production on airway smooth muscle precursors, to direct effects of IL-4 receptor signaling on airway smooth muscle activity, or to the effects of this signaling on goblet cell differentiation. SPDEF is selectively expressed in airway goblet cells but is also detected in breast, prostate, and intestinal epithelial cells (45, 46). In our past and present studies, SPDEF staining in pulmonary tissues was confined to goblet cells (22–24). While lack of HDM-induced AHR and Th2 inflammation in Spdef–/– mice is likely mediated by the cell-autonomous effects of SPDEF in airway epithelial cells, it remains possible that nonepithelial cells were involved in the present study. Direct activation of Th2 lymphocytes was observed in Spdef–/– mice injected with anti-IgD. These findings and the effects of epithelium-specific expression of both FOXA3 and SPDEF on Th2 inflammatory responses in the present studies support a primary role for lung epithelial SPDEF. The ability of SPDEF to induce FOXA3 likely further enhances the Th2 polarization of inflammatory responses and goblet cell differentiation. In human cells, SPDEF and FOXA3 activate each other, with ChIP-Seq and RNA analyses supporting the concept that FOXA3 binds to and activates the SPDEF promoter to enhance its transcription (24). In those studies, FOXA3-binding sites also were identified in the promoter regions of the genes known to play roles in goblet cell differentiation, including human SPDEF, MUC5B, and AGR2 genes, supporting a direct role for FOXA3 in the regulation of some aspects of goblet cell metaplasia. Actions of FOXA3 on gene expression in human airway epithelial cells in vitro indicated both SPDEF-dependent and -independent roles for FOXA3 in human airway cells in vitro (24). In the present studies, inflammatory responses to HDM were exacerbated by expression of FOXA3 (Supplemental Figure 5).
While goblet cell differentiation and mucus production are the noisome accompaniments of acute pulmonary infection or injury, enhancement of Th2 inflammation mediated by expression of FOXA3 and SPDEF may influence innate immune responses in chronic conditions associated with mucous metaplasia and may therefore play a role in the sensitization of patients with chronic pulmonary diseases that may be linked to susceptibility to infection and inflammation, as seen in asthma, COPD, and CF. Likewise, persistent mucus hyperproduction, goblet cell metaplasia, and inflammation following acute infection or injury may influence subsequent inflammatory and immune responses to both commensal and pathogenic microbes and aeroallergens.
In summary, we demonstrate that SPDEF and FOXA3 induced goblet cell differentiation and Th2-mediated inflammatory responses after birth, an active time of recruitment and activation of innate and acquired immunity. Exposure to both commensal and pathogenic microbes and antigens influences the acquisition of immune responses after birth that are likely to have long-term effects on the patterning of subsequent immune and inflammatory responses of the lung. Both SPDEF and FOXA3 are induced by aeroallergens and viral exposures that cause goblet cell metaplasia. Induction of goblet cell metaplasia via SPDEF and FOXA3 in airway epithelial cells may play a role in the establishment of Th2 immune polarization, influencing subsequent response patterns to environmental exposures. SPDEF and, by inference, goblet cells are required for normal pulmonary responses to HDM extract.
Methods
Mouse models.
Scgb1a1-rtTA (line 2) mice were either bred with OTet7-TRE2-Spdef mice or OTet7-Foxa3-IRES-EGFP mice, and dams were placed on doxycycline from E16.5 to P15 (22, 24). Spdef–/– mice were obtained from the laboratory of Hans Clevers (Netherlands Institute of Developmental Biology, Utrecht, The Netherlands) and backcrossed onto BALB/c mice until an F9 generation was obtained (46). Similarly, the Spdef+/+ WT mice were also backcrossed onto BALB/c mice to obtain an F9 generation. In some experiments, Scgb1a1-rtTA Foxa3-IRES mice were bred with Spdef–/– mice to obtain Scgb1a1-rtTA Foxa3-IRES Spdef+/– mice, which were subsequently bred again with Spdef–/– mice to obtain Scgb1a1-rtTA Foxa3-IRES Spdef+/– and Scgb1a1-rtTA Foxa3-IRES Spdef–/– mice. During this breeding, the dams were placed on doxycycline from E16.5 to P15. The transgenes were identified by PCR using the primer sets described in Supplemental Table 1.
Bronchoalveolar lavage fluid collection and cell counting.
Mice were euthanized, the lungs were lavaged twice with 0.2 to 0.5 ml ice-cold PBS with 0.1 mM EDTA to collect bronchoalveolar lavage fluid (BALF) cells, and cytospins of BALF cells (30,000–50,000 cells) were prepared and stained with a Diff-Quik Stain Kit (Polysciences Inc.). Differential counts were determined in 300 to 400 cells.
AHR.
AHR was measured in unrestrained mice using whole-body plethysmograph (Buxco Respiratory Products, Data Sciences International). Mice were exposed to increasing concentrations of methacholine (0.1–25 mg/ml). AHR is represented as Penh in response to methacholine. Penh values measured during this period were averaged and expressed as absolute Penh values.
Histology, immunochemistry, and immunofluorescence confocal microscopy.
Paraffin-embedded lung tissues from P15 mice were sectioned and subjected to antigen retrieval using citrate buffer. IHC analyses were performed using goat anti-FOXA3 (1:100, SC-5361; Santa Cruz Biotechnology Inc.); guinea pig anti-SPDEF (1:3,000; generated in the Whitsett Laboratory); mouse mAb against MUC5AC (1:500, ab3649; Abcam); and rabbit anti-MUC5B (1:100, H-300; Santa Cruz Biotechnology Inc.). Immunofluorescence confocal microscopic analyses of MUC5B, acetylated tubulin (1:100, T7451; Sigma-Aldrich), and CCSP (guinea pig anti–CCSP-G210, generated in the Whitsett Laboratory at 1:15,000) were performed on 7-μm-thick sections of paraffin-embedded tissue and imaged with a Nikon A1Rsi Confocal Microscope.
Quantitative RT-PCR.
Total RNA was prepared from whole-lung tissues from P15 mice collected in TRIzol (Invitrogen) according to the manufacturer’s protocol, and cDNA was generated by reverse transcription using 500 ng to 1 μg total RNA (iScript cDNA Synthesis Kit; Bio-Rad Laboratories). The cDNAs were amplified in a StepOnePlus Real-time PCR System (Applied Biosystems, Life Technologies) with the TaqMan probes (Applied Biosystems, Life Technologies) described in Supplemental Table 2. The Ct values of the target genes were normalized to that of the endogenous housekeeping gene 18S rRNA to obtain the ΔCt. The average ΔCt of the target genes in transgenic lung tissues was subtracted from the average ΔCt of the target genes in control lung tissues to obtain the ΔΔCt. The change in gene expression relative to that in control lungs was calculated as 2–ΔΔCt and represented graphically (47).
Flow cytometric analysis of lung cells.
Whole-lung cell suspensions were prepared as previously described (48) and used for flow cytometric analysis. Briefly, lung cell suspensions were prepared by enzymatically digesting the lung tissue using 5,000 caseinolytic units of dispase (Discovery Labware Inc., Corning). Single-cell suspensions were then stained with the fluorochrome-labeled Abs described in Supplemental Table 3. For detection of eosinophils in lung cell suspensions, SiglecF and CCR3 were used. For analysis of ILCs, a lineage cocktail with IL7Rα, FLT3, ST2, IL-17RB, and ICOS was used. For analysis of myeloid DCs (mDCs) and plasmacytoid DCs (pDCs), CD45, CD11c, CD11b, B220, and CD103 were used along with CD86 and I-A/E as activation markers. CD45 and CD3 were used for analysis of T cells. For intracellular cytokine detection, whole-lung cell suspensions were cultured in the presence of brefeldin A (BioLegend), followed by surface staining, then permeabilization with Cytofix/Cytoperm solution (BD Biosciences), and staining for IL-4 and IFN-γ. The stained lung cell samples were acquired on a BD FACSCanto III flow cytometer using FACS Diva software.
Anti-TSLP Ab treatment.
The pups obtained after breeding Scgb1a1-rtTA (line 2) and OTet7-Foxa3-IRES-EGFP mice were injected i.p. on P3 with either anti-TSLP Ab (50 μg) (clone eBio28F12; eBioscience) or control Ab (50 μg) (clone eBR2a; eBioscience). On P8, the pups were euthanized and lung specimens collected for total RNA and pathology. Dams were placed on doxycycline from E16.5 to P8.
Pulmonary allergen challenge.
Mice underwent 3 consecutive i.n. challenges with HDM allergen (Dermatophagoides pteronyssinus) or HDM extract (GREER) (50 μg for neonatal mice; 100 μg for adult mice), or with saline alone. After 48 hours of allergen exposure, mice were euthanized and lung tissues collected. The timing of HDM challenges and anti-TSLP treatment is provided in the figure legends.
Total IgE, HDM-specific IgE, and IgG1 ELISA.
Total IgE and HDM-specific IgE and IgG1 were measured in serum by ELISA. Serum was diluted 1:100 for IgE and 1:10,000 for IgG1. Total IgE levels were measured using the appropriate kit (BD Biosciences). For measuring HDM-specific IgG1 and IgE, plates were coated with 5 μg/well HDM in carbonate coating buffer (pH 9.6) overnight at 4°C. The following day, plates were blocked with 10% FCS in PBS for 1 hour and incubated with samples for 2 hours at room temperature. HRP anti-mouse RaM IgG1 (1:1,000) (BD Biosciences) or biotinylated anti-mouse IgE (2 μg/ml, clone R35-118; BD Biosciences) was used to detect IgG1 and IgE Abs, respectively, and the reaction was developed with a tetramethylbenzidine substrate reagent set (1:1; BD Biosciences).
In some experiments, the adult Spdef+/+ and Spdef–/– mice were injected i.p. with 200 μl donkey anti-mouse IgD serum, a gift of F. Finkelman (Division of Immunobiology, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, USA). On day 7, serum was collected and ELISA for IgE and IgG1 was performed.
Statistics.
Values are expressed as the mean ± SEM. All statistical analysis was performed using GraphPad Prism 6 (GraphPad Software). Data were analyzed using a 2-tailed, unpaired Student’s t test or 2-way ANOVA, and Bonferroni’s correction was used for multiple comparisons. P values of 0.05 or less were considered statistically significant.
Study approval.
Animals were housed in pathogen-free conditions according to protocols approved by the IACUC of the Cincinnati Children’s Hospital Research Foundation.
Supplementary Material
Acknowledgments
The authors wish to acknowledge Charles Perkins for performing Buxco respiratory studies, Javid Mohammed for performing flow cytometric analysis, Ann Maher for preparation of the manuscript, and Fred Finkelman and Neeru Hershey for helpful discussions. This work was supported by NIH grants HL095580 and HL110964, the Lung Repair and Regeneration Consortium (to J.A. Whitsett).
Footnotes
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(5):2021–2031. doi:10.1172/JCI79422.
Gang Chen’s present address is: CF Center of University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA.
References
- 1.Leeman KT, Fillmore CM, Kim CF. Lung stem and progenitor cells in tissue homeostasis and disease. Curr Top Dev Biol. 2014;107:207–233. doi: 10.1016/B978-0-12-416022-4.00008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogan BL, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15(2):123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambrecht BN, Hammad H. Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Annu Rev Immunol. 2012;30:243–270. doi: 10.1146/annurev-immunol-020711-075021. [DOI] [PubMed] [Google Scholar]
- 4.Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18(5):684–692. doi: 10.1038/nm.2737. [DOI] [PubMed] [Google Scholar]
- 5.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2014;16(1):27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lisciandro JG, van den Biggelaar AH. Neonatal immune function and inflammatory illnesses in later life: lessons to be learnt from the developing world? Clin Exp Allergy. 2010;40(12):1719–1731. doi: 10.1111/j.1365-2222.2010.03629.x. [DOI] [PubMed] [Google Scholar]
- 7.Ege MJ, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364(8):701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 8.Ege MJ, et al. Prenatal exposure to a farm environment modifies atopic sensitization at birth. J Allergy Clin Immunol. 2008;122(2):407–412. doi: 10.1016/j.jaci.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Kusel MM, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119(5):1105–1110. doi: 10.1016/j.jaci.2006.12.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riedler J, et al. Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet. 2001;358(9288):1129–1133. doi: 10.1016/S0140-6736(01)06252-3. [DOI] [PubMed] [Google Scholar]
- 11.Jackson DJ, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178(7):667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abrahamsson TR, Sandberg Abelius M, Forsberg A, Bjorksten B, Jenmalm MC. A Th1/Th2-associated chemokine imbalance during infancy in children developing eczema, wheeze and sensitization. Clin Exp Allergy. 2011;41(12):1729–1739. doi: 10.1111/j.1365-2222.2011.03827.x. [DOI] [PubMed] [Google Scholar]
- 13.Holt PG, Upham JW, Sly PD. Contemporaneous maturation of immunologic and respiratory functions during early childhood: implications for development of asthma prevention strategies. J Allergy Clin Immunol. 2005;116(1):16–24. doi: 10.1016/j.jaci.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336(6080):489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18(5):673–683. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 16.Holtzman MJ, Byers DE, Alexander-Brett J, Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14(10):686–698. doi: 10.1038/nri3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallstrand TS, Hackett TL, Altemeier WA, Matute-Bello G, Hansbro PM, Knight DA. Airway epithelial regulation of pulmonary immune homeostasis and inflammation. Clin Immunol. 2014;151(1):1–15. doi: 10.1016/j.clim.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3(9-10):545–556. doi: 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86(1):245–278. doi: 10.1152/physrev.00010.2005. [DOI] [PubMed] [Google Scholar]
- 21.Erle DJ, Sheppard D. The cell biology of asthma. J Cell Biol. 2014;205(5):621–631. doi: 10.1083/jcb.201401050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park KS, et al. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest. 2007;117(4):978–988. doi: 10.1172/JCI29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G, et al. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest. 2009;119(10):2914–2924. doi: 10.1172/JCI39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G, et al. Foxa3 induces goblet cell metaplasia and inhibits innate antiviral immunity. Am J Respir Crit Care Med. 2014;189(3):301–313. doi: 10.1164/rccm.201306-1181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park SW, et al. Distinct roles of FOXA2 and FOXA3 in allergic airway disease and asthma. Am J Respir Crit Care Med. 2009;180(7):603–610. doi: 10.1164/rccm.200811-1768OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris SC, Gause WC, Finkelman FD. IL-4 suppression of in vivo T cell activation and antibody production. J Immunol. 2000;164(4):1734–1740. doi: 10.4049/jimmunol.164.4.1734. [DOI] [PubMed] [Google Scholar]
- 27.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179(2):1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demehri S, Morimoto M, Holtzman MJ, Kopan R. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7(5):e1000067. doi: 10.1371/journal.pbio.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gauvreau GM, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014;370(22):2102–2110. doi: 10.1056/NEJMoa1402895. [DOI] [PubMed] [Google Scholar]
- 30.Taylor BC, et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med. 2009;206(3):655–667. doi: 10.1084/jem.20081499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroeder BW, et al. AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am J Respir Cell Mol Biol. 2012;47(2):178–185. doi: 10.1165/rcmb.2011-0421OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy MG, et al. Mucin production during prenatal and postnatal murine lung development. Am J Respir Cell Mol Biol. 2011;44(6):755–760. doi: 10.1165/rcmb.2010-0020OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15(4):410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poynter ME, et al. NF-κB activation in airways modulates allergic inflammation but not hyperresponsiveness. J Immunol. 2004;173(11):7003–7009. doi: 10.4049/jimmunol.173.11.7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guilliams M, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210(10):1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 2014;15(11):1026–1037. doi: 10.1038/ni.3005. [DOI] [PubMed] [Google Scholar]
- 37.Li BW, Hendriks RW. Group 2 innate lymphoid cells in lung inflammation. Immunology. 2013;140(3):281–287. doi: 10.1111/imm.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol. 2012;130(4):845–852. doi: 10.1016/j.jaci.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allakhverdi Z, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204(2):253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou B, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6(10):1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 41.Korfhagen TR, et al. SAM-pointed domain ETS factor mediates epithelial cell-intrinsic innate immune signaling during airway mucous metaplasia. Proc Natl Acad Sci U S A. 2012;109(41):16630–16635. doi: 10.1073/pnas.1208092109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knoop KA, McDonald KG, McCrate S, McDole JR, Newberry RD. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. 2015;8(1):198–210. doi: 10.1038/mi.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 44.Perkins C, et al. Selective stimulation of IL-4 receptor on smooth muscle induces airway hyperresponsiveness in mice. J Exp Med. 2011;208(4):853–867. doi: 10.1084/jem.20100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sood AK, Kim H, Geradts J. PDEF in prostate cancer. Prostate. 2012;72(6):592–596. doi: 10.1002/pros.21461. [DOI] [PubMed] [Google Scholar]
- 46.Gregorieff A, et al. The ets-domain transcription factor Spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology. 2009;137(4):1333–1345. doi: 10.1053/j.gastro.2009.06.044. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 48.Driscoll B, et al. Isolation and characterization of distal lung progenitor cells. Methods Mol Biol. 2012;879:109–122. doi: 10.1007/978-1-61779-815-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.