

Abstract
Dyskeratosis congenita (DC) is an inherited BM failure disorder that is associated with mutations in genes involved with telomere function and maintenance; however, the genetic cause of many instances of DC remains uncharacterized. In this issue of the JCI, Tummala and colleagues identify mutations in the gene encoding the poly(A)-specific ribonuclease (PARN) in individuals with a severe form of DC in three different families. PARN deficiency resulted in decreased expression of genes required for telomere maintenance and an aberrant DNA damage response, including increased levels of p53. Together, the results of this study support PARN as a DC-associated gene and suggest a potential link between p53 and telomere shortening.
Dyskeratosis congenita genes
Dyskeratosis congenita (DC) was first described in 1910 in two brothers who presented with skin pigmentation anomalies, leukoplakia, and nail dystrophy in childhood (1). Anemia later emerged as a common DC feature, and autosomal dominant, recessive, and X-linked recessive forms of inheritance for this disease are now recognized. In the 1990s, the X-linked DC locus was mapped to chromosomal region Xq28 (2, 3), and DKC1, which encodes the 57 kD protein dyskerin, was identified in 1998 as the causative gene within Xq28 (4). Dyskerin is homologous to the well-characterized yeast protein Cbf5p, which is a pseudouridine synthase that acts on ribosomal RNA and spliceosomal small nuclear RNA (snRNA) (5, 6). Following the discovery that dyskerin was also part of the telomerase complex in vertebrates (7) — and the identification of mutations in telomerase RNA (8) and telomerase reverse transcriptase (TERT) (9, 10) as causes of autosomal dominant DC — dysfunctional telomere maintenance was recognized as the basis for the development of DC. Moreover, patients with DC generally have very short telomeres at presentation. In the last decade, seven additional genes have been linked to DC, making a total of ten known DC-causing genes (11), all of which encode products that are involved in telomere maintenance. Some of these products — including TERT, the telomerase RNA component (TERC), dyskerin, and ribonucleoproteins NOP10 and NHP2 — are components of the telomerase complex. TCAB1 is important for telomerase-complex assembly and trafficking. Mutations in TINF2 and ACD — genes that encode components of the shelterin complex, which protects telomeres from degradation or from being recognized by the DNA damage machinery — are also associated with DC (12). Still other DC-associated genes, such as CTS telomere maintenance complex component 1 (CTC1) and regulator of telomere elongation helicase 1 (RTEL1), are necessary for telomere replication. Patients with classical DC and other inherited forms of the disease can present with a wide range of features in addition to the classic mucocutaneous manifestations and BM aplasia. Pulmonary fibrosis, liver cirrhosis, enteropathy, and a variety of cancers — including leukemia — are also commonly observed in these individuals (13).
Poly(A)-specific ribonuclease (PARN) is a new DC gene
In this issue, Tummala et al. identify a DC-associated gene that has no known connection with telomere metabolism (14). Specifically, Tummala and colleagues performed whole exome sequencing on a cohort of patients with severe DC, which is classified as Hoyeraal Hreidarsson syndrome (HHS) and manifests at an early age with immunodeficiency, intrauterine growth deficiency, and cerebellar hypoplasia. Their analysis revealed the presence of mutations in PARN, which encodes a poly(A)-specific ribonuclease (15). In 22 unrelated individuals, biallelic mutations in PARN that were likely to impair function were identified in three families. In these families, the affected children all presented with the HHS phenotype, and statistical analysis suggested that this was extremely unlikely to have occurred by chance, indicating that mutations in PARN underlie these cases of HHS. In one family, a brother and a sister were both homozygous for a missense mutation that results in an A to V substitution at amino acid 393 (PARNA393V), which is in a conserved N-terminal domain of the protein that is essential for nuclease activity. In a second family, the individual with HHS was homozygous for a point mutation that abolishes a donor splicing site in the PARN transcript, and analysis of mRNA in blood from this patient revealed an absence of properly spliced PARN RNA and the presence of two different PARN transcripts, one of which was missing one exon and the other two exons. In a third family, the affected child was a compound heterozygote at the PARN locus and harbored one allele with a single base insertion that resulted in a frameshift and one allele with an insertion in a donor splice site that was predicted to abolish splicing. Moreover, compared to healthy, age-matched controls, affected individuals in two of the families had substantially shorter telomeres. Telomere length was not measured in the affected individual from the third family. Together, the identification of PARN mutations in multiple families with cases of HHS and the observed decrease in telomere length in affected individuals strongly support PARN as a DC-associated gene.
It is not clear how PARN would affect telomere maintenance. PARN is a poly(A)-specific deadenylase that regulates gene expression by shortening poly(A) tails on transcripts, thereby decreasing their stability. Recent studies indicate that PARN activity is differentially regulated under various cellular conditions (16, 17). Tummala and colleagues demonstrated that cells from individuals with PARN mutations had reduced deadenylation activity, which should affect transcript stability. While PARN has been extensively studied at the biochemical level, the range of mRNA substrates and the physiological relevance of PARN-targeted transcripts has not been fully characterized. Tummala and colleagues compared expression of genes involved in telomere maintenance in the blood of healthy controls and individuals with PARN mutations. They found decreased expression of several key regulators of telomere metabolism, including DKC1, TERF1, RTEL, and TERC. The same transcripts were also decreased in HEK293T cells treated with PARN siRNA (14).
p53: guilty by association?
Tummala et al. also observed that PARN-deficient patient cells exhibit an abnormal DNA damage response, including increased cell death and an increase in p53 following UV exposure (14). PARN has recently been shown to participate in positive and negative feedback loops that mediate p53 and mRNA processing (18). In nonstressed conditions, PARN deadenylates and destabilizes p53 mRNA through recognition of a sequence element in the 3′UTR. Under DNA damaging conditions, such as UV exposure, p53 accumulates, associates with, and activates PARN by forming a complex that includes cleavage stimulation factor 1 (CSTF1). Based on this model, a DNA damage–associated increase in p53 would subsequently be kept in check by PARN-dependent destabilization of p53 mRNA; therefore, loss of PARN activity would lead to an increase in p53 mRNA levels. Activation of p53 has been observed in a variety of BM failure syndromes, including Fanconi anemia (19, 20), Diamond Blackfan anemia (21), and even some forms of DC (22, 23). Recently, Simeonova et al. (24) described a strain of mice that is homozygous for a Trp53 allele that results in production of a truncated p53, which lacks 31 C-terminal amino acid residues. In these animals, both the truncated p53 and transcripts of p53 targets Cdkn1a, which encodes p21, and oncogene Mdm2 were increased. Most of these mice died 14–43 days after birth and exhibited DC-associated features, including BM failure, dark skin, cerebellar hypoplasia, and pulmonary fibrosis. Transcript analysis in embryonic fibroblasts from these animals also revealed a decrease in transcripts associated with telomere maintenance, including Dkc1, Rtel1, Tinf2, and Terf1, which overlap with some of the telomere-associated genes Tummala et al. (14) determined to be downregulated in PARN-deficient cells.
Together, these studies suggest a potential mechanism for the development of DC-like phenotypes in the chronic absence of PARN (Figure 1). In rapidly dividing PARN-deficient cells, p53 levels would rise in response to an accumulation of DNA damage. The lack of PARN deadenylase activity would allow accumulation of p53 mRNA and further increase protein levels. Elevated p53 would, in turn, decrease transcription of genes required for telomere maintenance, with a subsequent shortening of telomeres. A novel feature in this model is the regulation of p53 at the mRNA level, whereas p53 is typically regulated at the level of protein stability. Such a mechanism would result in a cycle that enhances telomere shortening and increases p53, thereby inducing failure of tissues that are renewed via stem cell expansion. More work will need to be done to validate the involvement of the p53 pathway in aberrant telomere maintenance in PARN-associated DC; however, if correct, this model would provide an alternative pathway by which aberrant p53 activation contributes to the development of disease manifestations such as BM failure. Further investigation of the relationship between telomere metabolism, p53, and PARN may provide crucial information about BM failure and the prevalence of leukemic transformation and other forms of cancer in these disorders (13).
Figure 1. Possible effects of PARN depletion on telomeres.
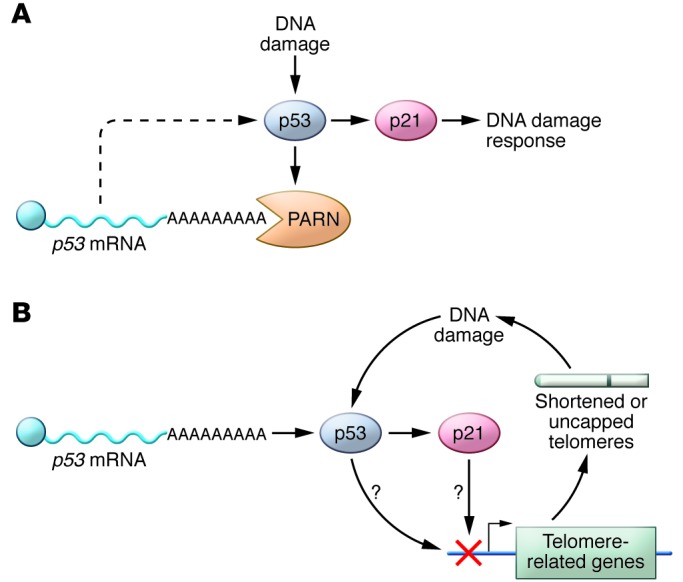
(A) DNA damage in replicating WT cells causes increased levels of the tumor suppressor p53, which in turn induces p21 and the DNA damage response. p53 also activates the PARN deadenylase, which destabilizes p53 mRNA, thereby reducing p53 levels (17). (B) In the absence of PARN, p53 levels can increase unchecked and lead to inhibition of telomere-related genes and short telomeres. Shortened telomeres exacerbate the DNA damage response, further increasing p53 levels, and thereby activating a cycle that leads to short telomeres and cell cycle arrest in rapidly replicating cells — a hallmark of DC pathobiology.
Acknowledgments
P.J. Mason’s work is supported in part by NIH/NCI grant R01 CA106995, and M. Bessler’s work is supported in part by NIH/NCI grant R01 CA105312 and by the Buck Family Chair in Hematology.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(5):1796–1798. doi:10.1172/JCI81506.
See the related article beginning on page 2151.
References
- 1.Zinsser F. Atrophia Cutis Reticularis cum Pigmentations, Dystrophia Unguium et Leukoplakis oris (Poikioodermia atrophicans vascularis Jacobi.) Ikonographia Dermatologica. 1910;5:219–223. [Google Scholar]
- 2.Connor JM, Gatherer D, Gray FC, Pirrit LA, Affara NA. Assignment of the gene for dyskeratosis congenita to Xq28. Hum Genet. 1986;72(4):348–351. doi: 10.1007/BF00290963. [DOI] [PubMed] [Google Scholar]
- 3.Knight SW, et al. 1.4 Mb candidate gene region for X linked dyskeratosis congenita defined by combined haplotype and X chromosome inactivation analysis. J Med Genet. 1998;35(12):993–996. doi: 10.1136/jmg.35.12.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heiss NS, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19(1):32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 5.Lafontaine DL, Bousquet-Antonelli C, Henry Y, Caizergues-Ferrer M, Tollervey D. The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 1998;12(4):527–537. doi: 10.1101/gad.12.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins NJ, et al. Cbf5p, a potential pseudouridine synthase, and Nhp2p, a putative RNA-binding protein, are present together with Gar1p in all H BOX/ACA-motif snoRNPs and constitute a common bipartite structure. RNA. 1998;4(12):1549–1568. doi: 10.1017/S1355838298980761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402(6761):551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 8.Vulliamy T, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413(6854):432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 9.Armanios M, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102(44):15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005;34(3):257–263. doi: 10.1016/j.bcmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Gramatges MM, Bertuch AA. Short telomeres: from dyskeratosis congenita to sporadic aplastic anemia and malignancy. Transl Res. 2013;162(6):353–363. doi: 10.1016/j.trsl.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y, et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014;124(18):2767–2774. doi: 10.1182/blood-2014-08-596445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alter BP, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010;150(2):179–188. doi: 10.1111/j.1365-2141.2010.08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tummala H, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151–2160. doi: 10.1172/JCI78963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buiting K, Korner C, Ulrich B, Wahle E, Horsthemke B. The human gene for the poly(A)-specific ribonuclease (PARN) maps to 16p13 and has a truncated copy in the Prader-Willi/Angelman syndrome region on 15q11→q13. Cytogenet Cell Genet. 1999;87(1–2):125–131. doi: 10.1159/000015378. [DOI] [PubMed] [Google Scholar]
- 16.Cevher MA, et al. Nuclear deadenylation/polyadenylation factors regulate 3′ processing in response to DNA damage. EMBO J. 2010;29(10):1674–1687. doi: 10.1038/emboj.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JE, Lee JY, Trembly J, Wilusz J, Tian B, Wilusz CJ. The PARN deadenylase targets a discrete set of mRNAs for decay and regulates cell motility in mouse myoblasts. PLoS Genet. 2012;8(8):e1002901. doi: 10.1371/journal.pgen.1002901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devany E, Zhang X, Park JY, Tian B, Kleiman FE. Positive and negative feedback loops in the p53 and mRNA 3′ processing pathways. Proc Natl Acad Sci U S A. 2013;110(9):3351–3356. doi: 10.1073/pnas.1212533110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceccaldi R, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumitriu B, Young NS. Damage control and its costs: BM failure in Fanconi anemia stems from overactive p53/p21. Cell Stem Cell. 2012;11(1):7–8. doi: 10.1016/j.stem.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boultwood J, Pellagatti A, Wainscoat JS. Haploinsufficiency of ribosomal proteins and p53 activation in anemia: Diamond-Blackfan anemia and the 5q- syndrome. Adv Biol Regul. 2012;52(1):196–203. doi: 10.1016/j.advenzreg.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Ogden GR, Lane DP, Chisholm DM. p53 expression in dyskeratosis congenita: a marker for oral premalignancy? J Clin Pathol. 1993;46(2):169–170. doi: 10.1136/jcp.46.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westin ER, Aykin-Burns N, Buckingham EM, Spitz DR, Goldman FD, Klingelhutz AJ. The p53/p21(WAF/CIP) pathway mediates oxidative stress and senescence in dyskeratosis congenita cells with telomerase insufficiency. Antioxid Redox Signal. 2011;14(6):985–997. doi: 10.1089/ars.2010.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simeonova I, et al. Mutant mice lacking the p53 C-terminal domain model telomere syndromes. Cell Rep. 2013;3(6):2046–2058. doi: 10.1016/j.celrep.2013.05.028. [DOI] [PubMed] [Google Scholar]