

Background: In glioblastoma, the EGF receptor mutation, EGFRvIII, is a biomarker of tumor aggressiveness even when only a small subpopulation of the cells express EGFRvIII.
Results: EGFRvIII-expressing cells release soluble uPAR (suPAR), which activates cell signaling and promotes migration and invasion of EGFRvIII-negative cells.
Conclusion: suPAR functions as a paracrine cancer-promoting factor in glioblastoma.
Significance: suPAR is biologically active and may contribute to cancer aggressiveness.
Keywords: cell invasion, cell migration, epidermal growth factor receptor (EGFR), glioblastoma, tumor microenvironment, urokinase receptor, EGFRvIII, LRP1, paracrine interaction, paracrine signaling
Abstract
Genomic heterogeneity is characteristic of glioblastoma (GBM). In many GBMs, the EGF receptor gene (EGFR) is amplified and may be truncated to generate a constitutively active form of the receptor called EGFRvIII. EGFR gene amplification and EGFRvIII are associated with GBM progression, even when only a small fraction of the tumor cells express EGFRvIII. In this study, we show that EGFRvIII-positive GBM cells express significantly increased levels of cellular urokinase receptor (uPAR) and release increased amounts of soluble uPAR (suPAR). When mice were xenografted with human EGFRvIII-expressing GBM cells, tumor-derived suPAR was detected in the plasma, and the level was significantly increased compared with that detected in plasma samples from control mice xenografted with EGFRvIII-negative GBM cells. suPAR also was increased in plasma from patients with EGFRvIII-positive GBMs. Purified suPAR was biologically active when added to cultures of EGFRvIII-negative GBM cells, activating cell signaling and promoting cell migration and invasion. suPAR did not significantly stimulate cell signaling or migration of EGFRvIII-positive cells, probably because cell signaling was already substantially activated in these cells. The activities of suPAR were replicated by conditioned medium (CM) from EGFRvIII-positive GBM cells. When the CM was preincubated with uPAR-neutralizing antibody or when uPAR gene expression was silenced in cells used to prepare CM, the activity of the CM was significantly attenuated. These results suggest that suPAR may function as an important paracrine signaling factor in EGFRvIII-positive GBMs, inducing an aggressive phenotype in tumor cells that are EGFRvIII-negative.
Introduction
Urokinase receptor (uPAR)3 is a glycosylphosphatidylinositol-anchored membrane protein and a high affinity receptor for one of the two principal mammalian activators of plasminogen, urokinase-type plasminogen activator (uPA) (1). Binding of uPA to uPAR facilitates activation of proteases at the cell surface and in the extracellular spaces (2), and it also activates cell signaling pathways directed by proximal transmembrane co-receptors such as the EGF receptor (EGFR) (3), integrins (4, 5), and the G-protein-coupled receptor, formyl peptide receptor-like-1 (FPRL1) (6). By activating proteases and by its effects on cell signaling, uPAR promotes cell migration (7).
uPAR exhibits a limited expression pattern in nonmalignant cells (8), with notable exceptions including myeloid cells and their precursors (9), endothelial cells activated in support of angiogenesis (10), and smooth muscle cells that migrate into the neointima of atherosclerotic lesions (11). uPAR expression in fibroblasts is regulated by inflammatory mediators (12). In tumors, uPAR expression may be dramatically increased, either due to expression by the cancer cells themselves or by activated nonmalignant cells in the tumor microenvironment (1). Medical conditions in which uPAR expression is increased have gained considerable interest, because uPAR is released as a soluble receptor (suPAR), which may be detected in the plasma (13).
suPAR has been identified as a plasma biomarker of pneumonia and sepsis (14), HIV (15), rheumatoid arthritis (16), cirrhosis (17), and atherosclerotic disease (18). suPAR also has been identified as a plasma biomarker of multiple forms of cancer (19–26). However, suPAR is more than simply an inert biomarker. A number of studies have demonstrated that suPAR binds directly to membrane proteins, such as FPRL1, and triggers cell signaling (6, 27–29). The nature of the signaling response may be determined by whether suPAR is cleaved between domain-1 and the domain-2–3 region, revealing the epitope, SRSRY, involved in FPRL1 binding (6, 27, 30). suPAR has been implicated in hematopoietic stem cell mobilization (31) and in the development of focal segmental glomerulosclerosis (32).
In this study, we examined the potential of suPAR to function as a paracrine signaling factor in cancer. Glioblastoma (GBM) is a highly malignant primary neoplasia of the brain and an important model of complex processes that arise in many malignancies, including genomic heterogeneity and cross-talk between tumor cells and non-neoplastic cells within the tumor microenvironment (33–36). The gene encoding the EGF receptor (EGFR) is frequently amplified in GBM (37–39). In the context of the EGFR gene amplification, mutations occur, including a common truncation event involving deletion of exons 2–7, which encode the ligand-binding ectodomain (40). The resulting constitutively active mutant is called EGFR variant III (EGFRvIII) (40). Although EGFRvIII may be expressed in only a minority of the tumor cells in a GBM, the resulting malignancy is frequently highly aggressive, leading to the hypothesis that factors secreted by EGFRvIII-expressing GBM cells enhance the aggressiveness of EGFRvIII-negative tumor cells. Factors implicated in paracrine pathways that enhance tumor aggressiveness in GBMs include IL-6 and LIF (41).
We previously demonstrated that membrane-anchored uPAR functions in concert with EGFRvIII to support growth and survival of GBM cells (42). We now show that cellular uPAR is selectively overexpressed and suPAR is selectively released by EGFRvIII-expressing GBM cells. suPAR that is released by EGFRvIII-expressing cells activates cell signaling and promotes cell migration and invasion of EGFRvIII-negative GBM cells. suPAR was detected in the plasma of mice xenografted with EGFRvIII-expressing GBM cells and in plasma samples from patients with EGFRvIII-positive GBMs. We propose that suPAR may be an important paracrine regulator of tumor cell physiology in GBM.
Experimental Procedures
Proteins and Reagents
EGF was from Sigma. Purified suPAR was from R&D Systems. AG1478 was from Sigma, and PD98059 was from Calbiochem. The LDL receptor-related protein-1 (LRP1) antagonist, receptor-associated protein, was expressed as a GST fusion protein (GST-RAP) in Plyss DE3 Rosetta cells from EMD Millipore. In brief, transformed bacteria were cultured at 37 °C with constant shaking until the A600 nm = 0.6. Protein expression was induced with 0.2 mm isopropyl β-d-thiogalactopyranoside for 3 h. GST-RAP was purified by glutathione affinity chromatography using the Profinia System from Bio-Rad.
Antibodies that recognize phospho-EGFR (p-EGFR-Y1068), phospho-ERK1/2 (p-ERK1/2-T202/Y204), total ERK1/2, phospho-Akt (p-Akt-S473), total Akt, and β-actin were from Cell Signaling Technologies. Mab807 against uPAR was from R&D Systems. ATN-658, a mouse monoclonal antibody that also recognizes human uPAR, was kindly provided by Dr. Andrew Mazar (Northwestern University). Antibodies that recognize total EGFR and activated (phospho)STAT5a/b were from EMD-Millipore. Antibody that recognizes α-tubulin was from Sigma.
Cell Culture
Unless otherwise specified, cell lines were obtained from the ATCC and maintained in DMEM high glucose (HyClone) with 10% FBS or tetracycline-approved FBS (HyClone). Parental U87MG cells and U87MG cells that overexpress wild-type EGFR (WT-EGFR) or express EGFRvIII were described previously (43). Parental U373MG cells, U373MG cells that overexpress WT-EGFR, and U373MG cells that express EGFRvIII under the control of a doxycycline-repressible promoter also were previously described (44). These cells were cultured in the presence of puromycin (1 μg/ml) and geneticin (200 μg/ml), with or without doxycycline (1 μg/ml). All cells were studied within 10 passages from the original stock.
To silence uPAR gene expression, cells were transfected with uPAR-specific siRNA (5′-GCCGUUACCUCGAAUGCAU-3′) (50 nm) using Lipofectamine 2000 (Life Technologies, Inc.). Control cells were transfected with nontargeting control (NTC) On-target Plus SMARTpool siRNA (Thermo).
Immunoblot Analysis
Cells were extracted in RIPA buffer (20 mm sodium phosphate, 150 mm NaCl, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with EDTA-free protease inhibitor mixture (Thermo Scientific) and 1 mm sodium orthovanadate. Cellular protein in extracts was determined by bicinchoninic acid assay. Equal amounts of cellular extract were loaded in each lane. SDS-PAGE was performed. Proteins were electrotransferred to 0.45-μm PVDF membranes and incubated with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch). Signal was developed using SuperSignal West Pico (Pierce) or Femto substrate (Thermo Fisher Scientific). To detect suPAR in conditioned medium (CM), CM samples were concentrated using 10-kDa Amicon filters (Millipore). The entire sample was subjected to SDS-PAGE. Densitometry analysis of immunoblots was performed using ImageJ software (National Institutes of Health). For immunoblots in which relative band intensity is reported, at least three separate blots were analyzed and subjected to statistical analysis as described below.
qPCR
Total RNA was isolated using the NucleoSpin RNA II kit (Macherey-Nagel). cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). TaqMan Fast Universal PCR Mastermix was from Applied Biosystems. TaqMan primers and probes for human uPAR (Hs00959822_m1), human uPA (HS00170182_m1), and human HPRT1 (Hs02800695_m1) were from Life Technologies, Inc. PCR was performed using a System 7300 Applied Biosystems instrument and analyzed using StepOne software (Applied Biosystems). mRNA expression was standardized against HPRT1 mRNA and determined by the ΔCt method.
Cell Migration and Invasion
Cell migration was analyzed using Transwell permeable supports with 8-μm pores (Corning Glass) according to the manufacturer's instructions. Cells were seeded in upper chambers and allowed to migrate for 18 h. Cells that migrated to the lower surface of the membranes were stained with Diff-Quick HEMA 3 (Fisher). To study invasion, BioCoat Matrigel invasion chambers were used (Corning Glass). Again, cells migrating to the underside surfaces of the membranes were counted.
Xenograft Studies
Fox Chase SCID mice (CB17/Icr-Prkdcscid/IcrIcoCrl) (Charles River) were inoculated subcutaneously in the right flank with 3 × 106 parental U373MG cells (n = 4) or with EGFRvIII-expressing U373MG cells (n = 4) suspended in growth factor-reduced Matrigel (Corning Glass) and 20 mm sodium phosphate, 150 mm NaCl, pH. 7.4 (PBS). Tumors were measured every 2 days from the external surface using calipers. The mice were euthanized when the tumors were 2.0 cm in maximum diameter. The tumors were harvested. Portions of each tumor were allocated for immunoblot analysis. Other portions were formalin-fixed and paraffin-embedded for staining with hematoxylin and eosin. Microscopic images were collected using an Olympus microscope and CellSens digital imaging software. All animal research was conducted in accordance with UCSD IACUC-approved protocols.
ELISA Analysis to Detect suPAR in Mouse Plasma
To test whether human GBM cells in xenografts release suPAR, we measured human uPAR in mouse plasma by ELISA. Plasma was collected from heavily anesthetized mice just prior to euthanasia by cardiac puncture using heparin-coated 25-gauge needles and syringes. Whole blood was immediately centrifuged at 2000 × g for 25 min at 4 °C. EDTA was added to the plasma as an additional anticoagulant. suPAR was quantified using the Quantikine uPAR ELISA kit (R&D Systems) according to the manufacturer's protocol. The equivalent ELISA kit was used to determine suPAR levels in human plasma samples.
Detection of EGFRvIII Circulating Tumor Cell DNA
Plasma samples from patients with GBM were obtained from the Moores UCSD Cancer Center. Plasma samples from patients without diagnosed cancer were from the UCSD Center for Advanced Laboratory Medicine. All patient plasma samples were de-identified and obtained following approval by the UCSD Institutional Review Board. Cell-free DNA encoding EGFRvIII was detected in plasma samples by PCR. Phusion HF polymerase (New England Biolabs) was used in amplification reactions. Primers for EGFRvIII and WT-EGFR were described previously (41). Plasma samples were added directly to the amplification reaction mixtures. PCR products were resolved on 2% agarose gels and stained with SYBR Safe (Life Technologies, Inc.).
Statistics
Statistical analysis was performed using GraphPad Prism 5 (GraphPad software). All results reflect at least three independent experiments. Studies that included only two treatment groups were analyzed by Student's t test. When more than two treatment groups were compared, the data were analyzed by one-way analysis of variance with Tukey's post hoc analysis. The following significance parameters were used: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
uPAR Expression Is Increased in EGFRvIII-expressing GBM Cells
To examine the effects of EGFRvIII on cellular uPAR expression and suPAR in GBM cells, EGFRvIII was expressed in U87MG and U373MG cells (Fig. 1A). We chose an ectopic expression strategy because EGFRvIII is typically lost from human GBM cells when tumors are removed from patients and placed into cell culture, probably due to the extrachromosomal location of the mutated gene (41, 45). WT-EGFR was detected in parental cells only when immunoblots were exposed for extended periods of time. The level of EGFRvIII in transfected cells was about 20-fold greater than the amount of WT-EGFR in parental U87MG and U373MG cells, as determined by comparing immunoblot band intensity in blots that were sufficiently developed to visualize WT-EGFR.
FIGURE 1.

EGFR signaling increases expression of cellular uPAR in human GBM cells. A, immunoblot analysis comparing EGFR expression in U87MG and U373MG parental cells and in the same cells that express EGFRvIII. Activation of the EGFR (P-Y1068), ERK1/2, Akt, and STAT5 was assessed after serum-starving cells for 18 h. Mean relative signal intensities for P-ERK1/2 and P-Akt relative to T-ERK1/2 and T-Akt are shown below the blots (n = 3). B, immunoblot analysis comparing cellular uPAR in parental U87MG and U373MG cells and in the same cells that overexpress WT-EGFR or express EGFRvIII (vIII). uPAR immunoblots were performed using the identical samples but separate blots because the antibody requires nonreducing conditions. Blots were re-probed for tubulin as a control for load. C, U373MG cells that were serum-starved for 18 h were pretreated with 50 nm AG1478 for 2 h, as indicated (+), and then with 10 ng/ml EGF for 6 h, as indicated (+). Immunoblot analysis was performed to detect cellular uPAR, EGFR, and actin as a control for load. D, qPCR was performed to detect uPAR mRNA after completing the identical incubations as in C (mean ± S.D. relative to control, n = 3; **, p < 0.01). E, EGFRvIII-expressing U373MG cells were serum-starved for 18 h and subsequently treated (+) or untreated (−) for 18 h with 25 μm PD98059 in serum-free medium. Immunoblotting was performed to assess the indicated antigens. F, parental and WT-EGFR-expressing U87MG cells were treated with 10 ng/ml EGF (+) or with vehicle (−) in serum-free medium for 18 h. Immunoblot analysis was then performed.
EGFR Tyr-1068 was phosphorylated in EGFRvIII-expressing cells, reflecting the constitutive activity of the mutated receptor. Downstream targets of EGFR signaling, including Akt, ERK1/2, and STAT5, also were activated preferentially in EGFRvIII-expressing cells. ERK1/2 phosphorylation was increased 1.8 ± 0.3- and 1.5 ± 0.3-fold in U87MG and U373MG cells, respectively. Akt phosphorylation was increased 2.6 ± 0.6- and 2.0 ± 0.2-fold in U87MG and U373MG cells, respectively. Phosphorylated STAT5 was detected only in EGFRvIII-expressing GBM cells.
In U373MG cells, EGFRvIII expression was associated with a >30-fold increase in the cellular uPAR protein level (Fig. 1B). For comparison, we examined WT-EGFR-overexpressing U373MG cells. The total level of EGFR in WT-EGFR-overexpressing cells and in EGFRvIII-expressing cells was similar; however, the effects of WT-EGFR on cellular uPAR were modest. In U87MG cells, expression of EGFRvIII or overexpression of WT-EGFR increased cellular uPAR. We hypothesized that uPAR expression in GBM cells is increased as a result of activated EGFR signaling. To test this hypothesis, we treated WT-EGFR-overexpressing U373MG cells with EGF. As shown in Fig. 1C, uPAR protein expression increased >30-fold, and the EGFR tyrosine kinase inhibitor, AG1478, inhibited the response by greater than 70%. The effects of EGF and AG1478 on cellular uPAR were confirmed at the mRNA level by qPCR (Fig. 1D). EGF increased uPAR mRNA 4.2 ± 0.6-fold in WT-EGFR-overexpressing U373MG cells, and AG1478 inhibited the response to EGF by nearly 60% (p < 0.01).
Next, we tested the effects of the MEK inhibitor, PD98059, on uPAR protein levels in EGFRvIII-expressing U373MG cells. Fig. 1E shows that PD98059 decreased ERK1/2 phosphorylation in these cells and thus was effective. PD98059 decreased cellular uPAR by greater than 80%. These results suggest that signaling to ERK1/2 is necessary for increased uPAR expression in EGFRvIII-expressing GBM cells but do not rule out a role for other signaling systems as well.
To further test the role of EGFR signaling in the regulation of uPAR expression, we treated WT-EGFR-expressing U87MG cells with EGF. Even though cellular uPAR was already elevated in this cell line, prior to EGF treatment, EGF further increased the uPAR protein level by >3-fold (p < 0.05) (Fig. 1F). EGF also slightly increased cellular uPAR in parental U87MG cells; however, the total abundance of uPAR in the parental cells remained relatively low.
Soluble uPAR Is Generated Selectively by EGFRvIII-expressing GBM Cells
To assess release of suPAR from GBM cells, serum-free CM was concentrated 50× using a standardized procedure and was subjected to immunoblot analysis. As shown in Fig. 2A, suPAR was nearly undetectable in CM from parental U373MG cells, even after the cells were treated with EGF. WT-EGFR-overexpressing cells released low levels of suPAR under standard cell culture conditions; however, EGF induced a 12 ± 2-fold increase in suPAR in CM from these cells (p < 0.01). EGFRvIII-expressing U373MG cells released high levels of suPAR under basal conditions. EGF did not significantly affect the level of suPAR in CM from EGFRvIII-expressing cells, as anticipated because EGF does not bind to EGFRvIII (45). Because the apparent molecular mass of the suPAR detected in CM from EGFRvIII-expressing cells was equivalent to the mass of cellular uPAR in the same cell line (50–60 kDa), the majority of the suPAR in the CM was assumed to be in full-length (uncleaved) form.
FIGURE 2.

EGFRvIII-expressing GBM cells generate increased levels of suPAR. A, parental U373MG cells and U373MG cells that overexpress WT-EGFR (EGFR) or express EGFRvIII (vIII) were serum-starved for 18 h. The cells were then treated with 10 ng/ml EGF (+) or with vehicle (−) for an additional 18 h. CM was recovered, concentrated 50×, and subjected to immunoblot analysis to detect suPAR. The cells from which CM was recovered also were subjected to immunoblot analysis to detect tubulin, as a control to ensure that CM was generated by an equivalent load of cells. Mean relative signal intensities are shown below the blots (n = 3). In each case, the signal intensity is standardized against that observed in the absence of EGF. B, U87MG cells were treated as described in A. CM was subjected to immunoblot analysis to detect suPAR. Cell extracts were analyzed to detect tubulin. C, U373MG cells that express EGFRvIII under the control of a doxycycline (DOX)-repressible promoter were treated (+) or untreated (−) with 1 μg/ml doxycycline for 3 days and subsequently serum-starved for 24 h in the presence or absence of doxycycline. Immunoblot analysis was performed to detect suPAR in CM and uPAR in cell extracts. EGFR and actin in cell extracts also were assessed. D, U87MG cells were cultured in serum-free medium with GST-RAP or GST (150 nm) for 24 or 48 h. CM and cell extracts were subjected to immunoblot analysis to detect uPAR and actin.
Next, we analyzed U87MG cells. In these cells, WT-EGFR overexpression failed to significantly increase suPAR accumulation in CM, even after the cells were treated with EGF (Fig. 2B). By contrast, suPAR was readily detected in CM from EGFRvIII-expressing U87MG cells. These results demonstrate that the effects of EGFRvIII on suPAR release are conserved in two GBM cell culture model systems. Once again, in the U87MG model system, suPAR was not detected in CM from parental cells.
To further test the hypothesis that EGFRvIII expression in GBM cells promotes release of suPAR, we analyzed a model system in which EGFRvIII expression is controlled in U373MG cells by a doxycycline-repressible promoter. Fig. 2C shows that treating these cells with doxycycline for 4 days suppressed EGFRvIII expression by 78 ± 8%. Although doxycycline modestly attenuated expression of cellular uPAR (40–50%), the effects of doxycycline on release of suPAR were more robust (decreased by >90%, p < 0.05).
We and others have shown that neutralizing LRP1 prevents uPAR internalization and degradation, thereby increasing the cell-surface and total cellular abundance of uPAR (46). When EGFRvIII-expressing U373MG cells were cultured in the presence of the LRP1 antagonist, RAP, for 24–48 h, cellular uPAR increased (Fig. 2D), as anticipated (46). This was accompanied by a substantial increase in suPAR accumulation in CM. These results suggest that release of suPAR from GBM cells is at least partially dependent on the abundance of cellular uPAR. Other contributing factors also must be operational, as suggested, for example, by the difference in release of suPAR by WT-EGFR-overexpressing U87MG and U373MG cells, after EGF treatment.
Soluble uPAR Generation by GBM Cells in Vivo
To test whether EGFRvIII-expressing GBM cells release increased amounts of suPAR in vivo, we developed a xenograft model system. EGFRvIII-expressing and parental U373MG cells were inoculated subcutaneously in SCID mice. When the tumors achieved a diameter of 2 cm, as determined by external measurements, the mice were euthanized. Interestingly, the time required for tumors to become 2 cm in diameter was not significantly different when the tumor cells expressed EGFRvIII. Fig. 3A compares the histology of the two tumor types. Parental cells, which were allowed to form tumors in vivo, showed extensive spindle cell morphology, with embedded blood vessels. EGFRvIII-expressing cells were more pleomorphic and epithelioid in shape. Blood vessels were again very prominent.
FIGURE 3.
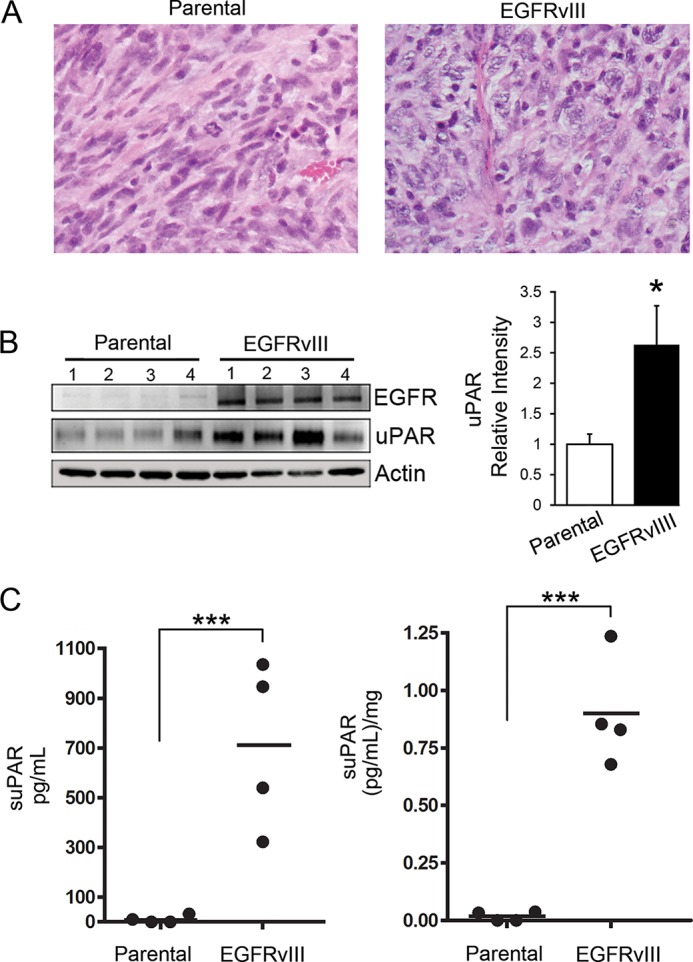
Detection of suPAR in the plasma of mice xenografted with GBM cells. A, representative sections of xenografts recovered from mice when the tumors were 2 cm in maximum diameter. The tissue was formalin-fixed and paraffin-embedded. Sections were stained with hematoxylin and eosin. B, tissue from each xenograft was extracted in RIPA buffer and subjected to SDS-PAGE and immunoblot analysis to detect uPAR and EGFR. Densitometry was performed to quantitate uPAR in each lane, standardized against actin (mean ± S.E.; n = 4; *, p < 0.05). C, plasma was recovered from mice with xenografts at the time of euthanasia. Equivalent samples of plasma were subjected to ELISA to detect human suPAR. Each value represents a different plasma sample tested in duplicate. Results are presented as uncorrected values (left) and relative to tumor weight (right) (n = 4; ***, p < 0.001).
Extracts of recovered tumors were subjected to immunoblot analysis. Fig. 3B shows that EGFRvIII expression was retained in vivo by the U373MG cells (Fig. 3B). uPAR expression was significantly increased in the EGFRvIII-expressing tumors (p < 0.05). Of note, the antibody used for these immunoblots recognizes human uPAR and not mouse uPAR. Thus, the significant increase in uPAR expression in the EGFRvIII-expressing tumors was due to the human tumor cells and not nonmalignant cells that infiltrate the tumors. Next, we performed ELISAs to detect suPAR in the plasma of mice with xenografts at the time of euthanasia. Fig. 3C shows that suPAR was substantially increased in plasma samples from mice with xenografts of EGFRvIII-expressing GBM cells (p < 0.001).
To test whether suPAR is increased in plasma from patients with EGFRvIII-positive GBMs, we obtained plasma samples and applied a PCR assay to detect circulating DNA derived from full-length WT-EGFR and EGFRvIII (47). We used previously described primers (41), which flank the deletion region in EGFRvIII, to specifically detect DNA from this EGFR variant. WT-EGFR was detected using primers that hybridize to exon 2, which is absent in EGFRvIII (Fig. 4A). To test the specificity of our primer sets, we compared cDNA generated from parental U87MG cells and cells transfected to express EGFRvIII. Fig. 4B shows that a band corresponding to the fused segment of the EGFR gene found exclusively in EGFRvIII was observed only in cDNA isolated from EGFRvIII-expressing cells. In four plasma samples from control patients without GBM, WT-EGFR DNA was uniformly detected; however, DNA corresponding to EGFRvIII was totally absent, as anticipated (Fig. 4C). In 12 plasma samples from patients with GBM, WT-EGFR DNA was again uniformly detected, although at differing levels. EGFRvIII-derived DNA was clearly detected in five samples and absent or extremely weak in the other seven samples. Fig. 4D shows six representative plasma samples from the GBM patient cohort. When we performed ELISAs to detect suPAR using the same plasma samples from GBM patients, those that were EGFRvIII DNA-positive demonstrated a statistically significant increase in suPAR compared with the EGFRvIII DNA-negative samples (Fig. 4E).
FIGURE 4.
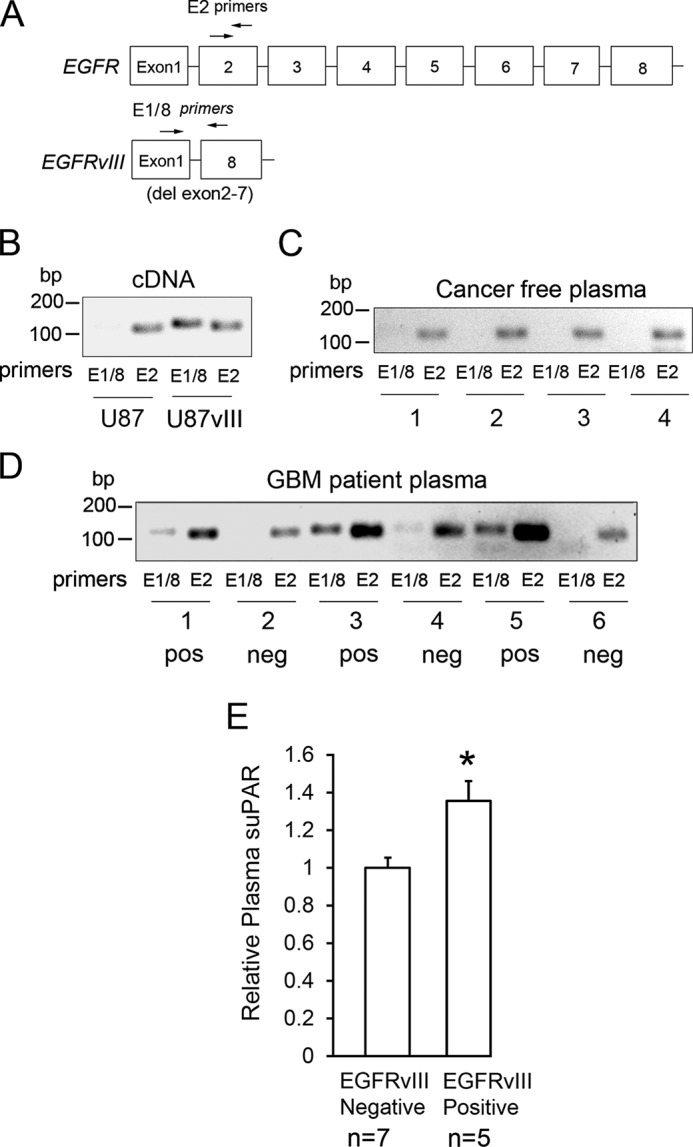
GBM patients with circulating tumor DNA corresponding to EGFRvIII have increased levels of plasma suPAR. A, diagram showing the primers (arrows) used to amplify DNA derived from WT-EGFR for EGFRvIII in plasma samples. B, cDNA was generated from parental U87MG cells and EGFRvIII-expressing U87MG cells. The cDNA was amplified using the primers that detect EGFRvIII (E1/8) and WT-EGFR (E2). The resulting amplicons, which were 119 and 104 bp, respectively, were compared by agarose gel electrophoresis with SYBR Green staining. C, PCR amplification of circulating DNA within cancer-free control plasma samples using the indicated primers. D, PCR amplification of circulating tumor DNA in six representative GBM patient plasma samples. Samples that were judged to be positive for EGFRvIII-derived DNA are marked “pos.” Those judged to be negative are marked “neg.” E, ELISA analysis of suPAR levels in 12 GBM patient plasma samples. Cases were sorted as EGFRvIII-positive (n = 5) or negative (n = 7) based on E1/8 DNA amplification and analysis as shown in D (mean ± S.E.; *, p < 0.05).
Soluble uPAR Activates ERK1/2 and Promotes Migration of GBM Cells
To test whether suPAR may function as a paracrine regulator of GBM cell physiology, first we conducted experiments with purified full-length suPAR. In parental U373MG cells, 10 nm suPAR activated ERK1/2 and Akt, as determined by immunoblot analysis (Fig. 5A). The effects of suPAR on ERK1/2 activation were maximal 5 min after adding the suPAR and sustained for 60 min. In the absence of suPAR, EGFRvIII-expressing U373MG cells demonstrated an increased basal level of ERK1/2 activation, as anticipated because EGFRvIII signals constitutively in these cells. However, suPAR either failed to increase ERK1/2 phosphorylation in EGFRvIII-positive cells or, in some experiments, induced a very minor increase. The increases in ERK1/2 phosphorylation in suPAR-treated EGFRvIII-expressing cells, reported in Fig. 5A, were not statistically significant (n = 3).
FIGURE 5.

Purified suPAR activates ERK1/2 and promotes migration of GBM cells. A, purified recombinant uPAR (10 nm) was incubated with parental U373MG cells (parental) or with EGFRvIII-expressing U373MG cells, which had been serum-starved for 18 h, for the indicated time points. Immunoblot analysis was performed to detect phospho-ERK1/2, total ERK1/2, phospho-Akt, and total Akt. Mean relative signal intensities for P-ERK1/2 and P-Akt relative to T-ERK1/2 and T-Akt are shown below the blots (n = 3). B, parental U373MG cells were serum-starved for 18 h, added to Transwells, and allowed to migrate toward 1% serum supplemented with 10 nm suPAR or vehicle (Ctrl) for 18 h. The number of cells that migrated to the underside of the membrane were counted and expressed relative to the control (mean ± S.E.; n = 3, ***, p < 0.001). C, cell migration was studied as in B with parental and EGFRvIII-expressing U373MG cells. The concentration of suPAR was varied as shown (mean ± S.E.; n = 3; *, p < 0.05; **, p < 0.01; ***, p < 0.001). D, parental U373MG cells were allowed to migrate in Transwells for 18 h. The bottom chamber contained serum-free medium and 15 nm suPAR (+) or vehicle (−), as indicated. Other additives to the lower chamber included uPAR-neutralizing antibody (α-uPAR, 50 μg/ml ATN-658 + 15 μg/ml MAB807) or control mouse IgG (65 μg/ml). The number of cells that migrated to the underside of the membranes are expressed relative to the control, which was not suPAR-treated (mean ± S.E.; n = 3; **, p < 0.01). E, parental U373MG cells were allowed to migrate toward 1% serum-containing medium and 15 nm suPAR for 18 h, as indicated (+). 20 μm PD98059 was added to both chambers, as indicated (+). The number of migrated cells were determined and expressed relative to the vehicle-treated control (mean ± S.E.; n = 3; **, p < 0.01; *, p < 0.05). F, qPCR was performed to detect uPA mRNA after treating parental U373MG cells with suPAR for 4 h (mean ± S.E.; n = 3).
Because membrane-anchored uPAR cooperates with EGFR to activate STAT5B (42, 48, 49), we examined STAT5 phosphorylation in parental U373MG cells treated with suPAR for 5–60 min; however, suPAR did not promote STAT5 phosphorylation in these cells (results not shown).
Fig. 5B shows that purified suPAR promoted migration of parental U373MG cells (p < 0.001). The response of the parental cells to suPAR was dose-dependent (Fig. 5C). By contrast, suPAR failed to stimulate migration of EGFRvIII-expressing U373MG cells at each of the concentrations studied.
In the experiments shown in Fig. 5, B and C, suPAR was added to the bottom chamber in low serum medium. When serum was omitted from the Transwell chambers, the effects of suPAR on cell migration were more robust (Fig. 5D). uPAR-specific antibody partially neutralized the effects of purified suPAR on migration of parental U373MG cells. The effect was incomplete suggesting that the antibody may not completely block binding of suPAR to cell-surface receptors. Parental cell migration in response to suPAR also was inhibited by the MEK inhibitor, PD98059 (Fig. 5E), implicating ERK1/2 activation in the pathway by which suPAR promotes GBM cell migration.
We previously demonstrated that uPA promotes GBM cell migration (50). We therefore performed control experiments to test whether suPAR regulates uPA expression in parental U373MG cells. No effect was observed (Fig. 5F), suggesting that the mechanism by which suPAR promotes migration of GBM cells is independent of uPA.
Next, we tested whether CM from EGFRvIII-expressing GBM cells replicates the activity of purified suPAR. Fig. 6A shows that CM activated ERK1/2 in parental U373MG cells (Fig. 6A). Compared with control medium (no exposure to cells), CM from EGFRvIII-expressing cells increased phospho-ERK1/2 6.7 ± 1.2-fold. CM harvested from parental U373MG cells increased phospho-ERK1/2 in separate parental cells by 2.8 ± 0.9-fold. The difference in activity of CM recovered from EGFRvIII-expressing and parental cells was statistically significant (p < 0.05, n = 4).
FIGURE 6.
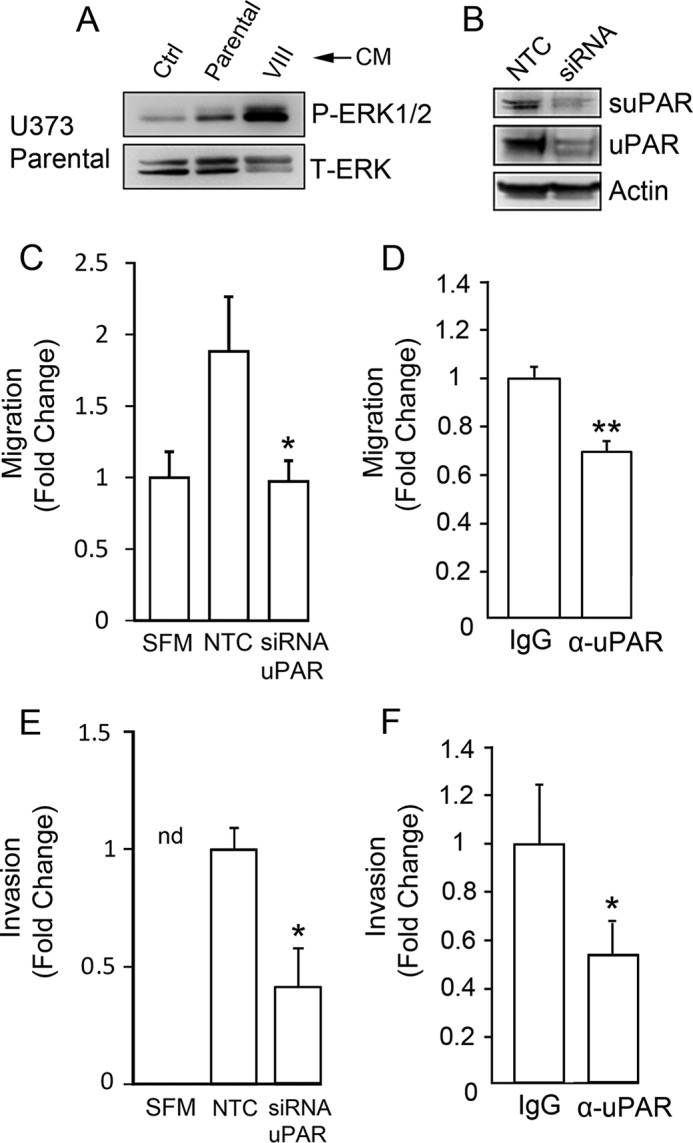
suPAR in conditioned medium from EGFRvIII-expressing GBM cells promotes cell migration. A, serum-free CM was recovered from parental U373MG cells and EGFRvIII-expressing U373MG cells. Nonconditioned medium was used as a control (Ctrl). The CM samples were concentrated 50× and incubated with parental U373MG cells for 10 min. The parental cells were serum-starved for 4 h prior to adding CM or control medium. Immunoblot analysis was performed to detect phospho-ERK1/2 and total ERK1/2. B, uPAR gene expression was silenced in EGFRvIII-expressing cells used to generate CM. Extracts of these cells and cells transfected with NTC siRNA were subjected to immunoblot analysis to detect cellular uPAR. CM was processed to detect suPAR. C, parental U373MG cells were allowed to migrate in Transwells containing serum-free medium (SFM) and coated with 5 μg/ml vitronectin toward CM from EGFRvIII-expressing U373MG cells transfected with NTC or uPAR-specific siRNA (siRNA uPAR). Migration was determined relative to controls in which CM was not added (SFM) (mean ± S.E.; n = 3; *, p < 0.05). D, migration was performed as described in C. CM from EGFRvIII-expressing U373MG cells was added to the lower chamber together with uPAR-specific antibody (α-uPAR, 50 μg/ml ATN-658 + 15 μg/ml MAB807) or nonspecific mouse IgG (65 μg/ml) (mean ± S.E.; n = 3; **, p < 0.01). E, Matrigel invasion assays were performed with parental U373MG cells. The lower chamber contained CM obtained from EGFRvIII-expressing U373MG cells transfected with NTC siRNA (NTC) or uPAR-specific siRNA (siRNA uPAR). Invasion was allowed to proceed for 48 h. No cells invaded toward serum-free medium in the absence of CM (nd). The number of invading cells, detected on the lower membrane surface, was expressed relative to that observed when CM from cells transfected with NTC siRNA was added (mean ± S.E.; n = 3; *, p < 0.05). F, Matrigel invasion was examined as described in E. CM from EGFRvIII-expressing U373MG cells transfected with NTC siRNA was added to the lower chamber. uPAR-neutralizing antibody (α-uPAR) or control mouse IgG was added as specified (mean ± S.E.; n = 3; *, p < 0.05).
Fig. 6B shows that uPAR gene silencing in EGFRvIII-expressing GBM cells decreased cellular uPAR and the level of suPAR in CM by >60%. Fig. 6C shows that CM from EGFRvIII-expressing U373MG cells, transfected with NTC siRNA, promoted migration of parental cells. CM from cells in which uPAR gene expression was silenced failed to promote parental cell migration beyond the level observed in the absence of CM. Treating CM from EGFRvIII-expressing cells with uPAR-neutralizing antibody also significantly inhibited the activity of the CM in cell migration studies (Fig. 6D).
Next, we tested whether CM from EGFRvIII-expressing U373MG cells promotes the ability of parental U373MG cells to invade Matrigel. In the absence of CM, Matrigel invasion was not observed (Fig. 6E). CM from EGFRvIII-expressing cells, transfected with NTC siRNA, promoted Matrigel invasion. When CM was prepared from cells in which uPAR was silenced, this activity was significantly attenuated (p < 0.05). Similarly, uPAR-neutralizing antibody significantly inhibited the activity of CM in invasion assays (Fig. 6F). These results support a model in which suPAR functions as an important pro-migratory and pro-invasion factor released by EGFRvIII-positive GBM cells.
Discussion
EGFR gene amplification and mutation are common drivers of tumor initiation and progression in GBM; however, many questions remain regarding the function of EGFR in GBM. For example, although EGFRvIII is constitutively active, its activity is highly attenuated compared with EGF-ligated WT-EGFR (40). Nevertheless, tumors in which EGFRvIII is identified tend to be highly aggressive (43, 51). This may reflect novel co-receptor interactions in the cells that express EGFRvIII or activation of downstream signaling pathways that are unique to EGFRvIII (52). We previously showed that in EGFRvIII-expressing GBM cells, membrane-anchored uPAR functions as an important co-receptor promoting activation of STAT5b (42). We now show that expression of cellular uPAR may be increased selectively in EGFRvIII-positive GBM cells. Similarly, EGFR gene amplification or overexpression may increase expression of cellular uPAR when there is sufficient EGFR ligand in the tumor microenvironment.
Using a number of model systems, we demonstrated that in GBM cells, cellular uPAR expression is regulated by EGFR signaling. In U373MG cells that overexpress WT-EGFR, EGF substantially increased uPAR mRNA and protein expression, and the response was blocked by AG1478. In EGFRvIII-expressing cells, inhibiting MEK with PD98059 significantly decreased cellular uPAR. When WT-EGFR was overexpressed, U87MG cells differed from U373MG cells in that the U87MG cells expressed high levels of cellular uPAR even when EGF was not added. This difference may reflect endogenous production of EGFR ligands by U87MG cells. A modest decrease in cellular uPAR was observed when EGFRvIII expression was blocked in U373MG cells with doxycycline. As we have shown previously, this modest decrease in uPAR is accompanied by a major increase in uPA expression, which has the net effect of activating uPAR signaling (42, 53). This shift in GBM cell signaling has been implicated in promoting survival of tumor cells treated with EGFR-targeting drugs and in promoting GBM cell migration (42, 50, 52).
To identify human GBMs in which EGFRvIII is expressed, we amplified circulating DNA. Although this method was effective, it is also indirect and may have detected only those cases in which EGFRvIII was most abundant. Plasma samples from patients who apparently had EGFRvIII-positive GBMs demonstrated a significant increase in suPAR, compared with samples from patients with EGFRvIII-negative tumors. We also examined data from The Cancer Genome Atlas; however, a significant correlation between cellular uPAR expression in the primary tumor and EGFRvIII positivity was not observed (data not shown). This observation probably reflects numerous tumors in this database in which EGFR is amplified or overexpressed in the absence of EGFRvIII.
Single cell genome sequencing has revealed extensive variability in the degree of EGFR gene amplification and expression of mutated forms of the EGFR, among different tumor cells in a single GBM (54). Soluble factors released by one cell may affect properties of neighboring cells, including the capacity for migration, invasion, and survival. We have now demonstrated that GBM cells, which express EGFRvIII, release substantially increased amounts of suPAR. The suPAR was detected in CM from cultured cells, in plasma from mice xenografted with EGFRvIII-expressing GBM cells, and in plasma samples from patients with EGFRvIII-positive tumors.
The mechanism by which suPAR is released from the surfaces of GBM cells remains to be determined. Previously demonstrated pathways for generating suPAR include enzymatic release by glycosylphosphatidylinositol-specific phospholipase D (55) and alternative mRNA splicing (56). In some of our studies, we observed a correlation between the total abundance of cellular uPAR and the amount of suPAR released. However, this pattern was not uniform, and other factors are most likely involved. Enzymes in the phospholipase D gene family have been implicated in cell signaling and cell survival in GBM (57). Drugs that target phospholipase D have shown efficacy in cancer (58).
Using cultures of GBM cells, we have shown that in this form of cancer, purified suPAR may be active, triggering cell signaling and promoting cell migration and invasion. These activities were observed only in studies with parental GBM cells, which express low levels of endogenous WT-EGFR and no EGFRvIII. suPAR was not active against EGFRvIII-positive GBM cells. Thus, suPAR may be viewed as a biologically active paracrine factor, released selectively by GBM cells in which EGFR is activated and promoting an aggressive phenotype in cells that do not overexpress WT-EGFR or express EGFRvIII. Similar factors that also have been implicated in paracrine interactions between genetically distinct cancer cells within a single GBM include IL-6 and LIF (41).
suPAR was identified as a major factor in CM collected from EGFRvIII-expressing GBM cells, which is responsible for the ability of the CM to promote GBM cell migration and invasion. The activity of the CM was attenuated by silencing uPAR gene expression in the EGFRvIII-positive cells or by incubating the CM with uPAR-neutralizing antibody. We propose that similar conditioning of the extracellular tumor microenvironment by EGFRvIII-positive cells may occur in intact tumors.
We previously demonstrated that suPAR may function as a cell signaling agonist or antagonist depending on whether membrane-anchored uPAR in the target cell is actively engaged in cell signaling (27). The basal state of activation of signaling factors such as ERK1/2 in the target cell also is important (27). suPAR may compete with membrane-anchored uPAR for co-receptors such as FPRL-1 (6). Importantly, cleavage of suPAR between domains D1 and D2 is known to fully potentiate the signaling activity of suPAR, converting suPAR from a partial agonist into a full agonist (59–63). In this study, we did not examine cleaved suPAR because our experiments were designed to model the species detected in CM from EGFRvIII-expressing GBM cells. However, it is quite possible that cleaved suPAR may be generated in human tumors.
In conclusion, we have shown that suPAR is a biologically active protein and a potentially important paracrine factor in GBM. Release of suPAR by cells in which EGFR is activated by mutation or amplification may explain why cancer cells within the same tumor that do not have the equivalent genomic changes are more aggressive.
Acknowledgments
We thank Michael S. Lam and Na Du for technical assistance. We also thank Ryan Kim for the GBM patient plasma samples. We thank Alec Saitman for the cancer-free control plasma samples.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA169096 (to S. L. G.) and 2P01CA069246-15A and UH2TR000931 (to B. S. C.).
This article was selected as a Paper of the Week.
- uPAR
- urokinase receptor
- CM
- conditioned medium
- GBM
- glioblastoma
- suPAR
- soluble urokinase receptor
- uPA
- urokinase-type plasminogen activator
- NTC
- nontargeting control
- qPCR
- quantitative PCR.
References
- 1. Smith H. W., Marshall C. J. (2010) Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 11, 23–36 [DOI] [PubMed] [Google Scholar]
- 2. Collen D. (1999) The plasminogen (fibrinolytic) system. Thromb. Haemost. 82, 259–270 [PubMed] [Google Scholar]
- 3. Liu D., Aguirre Ghiso J., Estrada Y., Ossowski L. (2002) EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 1, 445–457 [DOI] [PubMed] [Google Scholar]
- 4. Wei Y., Lukashev M., Simon D. I., Bodary S. C., Rosenberg S., Doyle M. V., Chapman H. A. (1996) Regulation of integrin function by the urokinase receptor. Science 273, 1551–1555 [DOI] [PubMed] [Google Scholar]
- 5. Tarui T., Mazar A. P., Cines D. B., Takada Y. (2001) Urokinase-type plasminogen activator receptor (CD87) is a ligand for integrins and mediates cell-cell interaction. J. Biol. Chem. 276, 3983–3990 [DOI] [PubMed] [Google Scholar]
- 6. Resnati M., Pallavicini I., Wang J. M., Oppenheim J., Serhan C. N., Romano M., Blasi F. (2002) The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. U.S.A. 99, 1359–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blasi F., Carmeliet P. (2002) uPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 3, 932–943 [DOI] [PubMed] [Google Scholar]
- 8. Solberg H., Ploug M., Høyer-Hansen G., Nielsen B. S., Lund L. R. (2001) The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J. Histochem. Cytochem. 49, 237–246 [DOI] [PubMed] [Google Scholar]
- 9. Plesner T., Ralfkiaer E., Wittrup M., Johnsen H., Pyke C., Pedersen T. L., Hansen N. E., Danø K. (1994) Expression of the receptor for urokinase-type plasminogen activator in normal and neoplastic blood cells and hematopoietic tissue. Am. J. Clin. Pathol. 102, 835–841 [DOI] [PubMed] [Google Scholar]
- 10. Mandriota S. J., Seghezzi G., Vassalli J. D., Ferrara N., Wasi S., Mazzieri R., Mignatti P., Pepper M. S. (1995) Vascular endothelial growth factor increases urokinase receptor expression in vascular endothelial cells. J. Biol. Chem. 270, 9709–9716 [DOI] [PubMed] [Google Scholar]
- 11. Reidy M. A., Irvin C., Lindner V. (1996) Migration of arterial wall cells: expression of plasminogen activators and inhibitors in injured rat arteries. Circ. Res. 78, 405–414 [DOI] [PubMed] [Google Scholar]
- 12. Shetty S., Kumar A., Johnson A. R., Pueblitz S., Holiday D., Raghu G., Idell S. (1996) Differential expression of the urokinase receptor in fibroblasts from normal and fibrotic human lungs. Am. J. Respir. Cell Mol. Biol. 15, 78–87 [DOI] [PubMed] [Google Scholar]
- 13. Holst-Hansen C., Hamers M. J., Johannessen B. E., Brünner N., Stephens R. W. (1999) Soluble urokinase receptor released from human carcinoma cells: a plasma parameter for xenograft tumour studies. Br. J. Cancer 81, 203–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Savva A., Raftogiannis M., Baziaka F., Routsi C., Antonopoulou A., Koutoukas P., Tsaganos T., Kotanidou A., Apostolidou E., Giamarellos-Bourboulis E. J., Dimopoulos G. (2011) Soluble urokinase plasminogen activator receptor (suPAR) for assessment of disease severity in ventilator-associated pneumonia and sepsis. J. Infect. 63, 344–350 [DOI] [PubMed] [Google Scholar]
- 15. Sidenius N., Sier C. F., Ullum H., Pedersen B. K., Lepri A. C., Blasi F., Eugen-Olsen J. (2000) Serum level of soluble urokinase-type plasminogen activator receptor is a strong and independent predictor of survival in human immunodeficiency virus infection. Blood 96, 4091–4095 [PubMed] [Google Scholar]
- 16. Koga T., Okada A., Kawashiri S., Kita J., Suzuki T., Nakashima Y., Tamai M., Satoh K., Origuchi T., Iwamoto N., Yamasaki S., Nakamura H., Migita K., Ida H., Ueki Y., et al. (2011) Soluble urokinase plasminogen activator receptor as a useful biomarker to predict the response to adalimumab in patients with rheumatoid arthritis in a Japanese population. Clin. Exp. Rheumatol. 29, 811–815 [PubMed] [Google Scholar]
- 17. Zimmermann H. W., Koch A., Seidler S., Trautwein C., Tacke F. (2012) Circulating soluble urokinase plasminogen activator is elevated in patients with chronic liver disease, discriminates stage and aetiology of cirrhosis and predicts prognosis. Liver Int. 32, 500–509 [DOI] [PubMed] [Google Scholar]
- 18. Edsfeldt A., Nitulescu M., Grufman H., Grönberg C., Persson A., Nilsson M., Persson M., Björkbacka H., Gonçalves I. (2012) Soluble urokinase plasminogen activator receptor is associated with inflammation in the vulnerable human atherosclerotic plaque. Stroke 43, 3305–3312 [DOI] [PubMed] [Google Scholar]
- 19. Rigolin G. M., Tieghi A., Ciccone M., Bragotti L. Z., Cavazzini F., Della Porta M., Castagnari B., Carroccia R., Guerra G., Cuneo A., Castoldi G. (2003) Soluble urokinase-type plasminogen activator receptor (suPAR) as an independent factor predicting worse prognosis and extra-bone marrow involvement in multiple myeloma patients. Br. J. Haematol. 120, 953–959 [DOI] [PubMed] [Google Scholar]
- 20. Shariat S. F., Roehrborn C. G., McConnell J. D., Park S., Alam N., Wheeler T. M., Slawin K. M. (2007) Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progression, and metastasis. J. Clin. Oncol. 25, 349–355 [DOI] [PubMed] [Google Scholar]
- 21. Henic E., Borgfeldt C., Christensen I. J., Casslén B., Høyer-Hansen G. (2008) Cleaved forms of the urokinase plasminogen activator receptor in plasma have diagnostic potential and predict postoperative survival in patients with ovarian cancer. Clin. Cancer Res. 14, 5785–5793 [DOI] [PubMed] [Google Scholar]
- 22. Lomholt A. F., Christensen I. J., Høyer-Hansen G., Nielsen H. J. (2010) Prognostic value of intact and cleaved forms of the urokinase plasminogen activator receptor in a retrospective study of 518 colorectal cancer patients. Acta Oncol. 49, 805–811 [DOI] [PubMed] [Google Scholar]
- 23. Langkilde A., Hansen T. W., Ladelund S., Linneberg A., Andersen O., Haugaard S. B., Jeppesen J., Eugen-Olsen J. (2011) Increased plasma soluble uPAR level is a risk marker of respiratory cancer in initially cancer-free individuals. Cancer Epidemiol. Biomarkers Prev. 20, 609–618 [DOI] [PubMed] [Google Scholar]
- 24. Jing J., Zheng S., Han C., Du L., Guo Y., Wang P. (2012) Evaluating the value of uPAR of serum and tissue on patients with cervical cancer. J. Clin. Lab. Anal. 26, 16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Almasi C. E., Brasso K., Iversen P., Pappot H., Høyer-Hansen G., Danø K., Christensen I. J. (2011) Prognostic and predictive value of intact and cleaved forms of the urokinase plasminogen activator receptor in metastatic prostate cancer. Prostate 71, 899–907 [DOI] [PubMed] [Google Scholar]
- 26. Riisbro R., Christensen I. J., Piironen T., Greenall M., Larsen B., Stephens R. W., Han C., Høyer-Hansen G., Smith K., Brünner N., Harris A. L. (2002) Prognostic significance of soluble urokinase plasminogen activator receptor in serum and cytosol of tumor tissue from patients with primary breast cancer prognostic significance of soluble urokinase plasminogen activator receptor in serum and cytosol of T. Clin. Cancer Res. 8, 1132–1141 [PubMed] [Google Scholar]
- 27. Jo M., Thomas K. S., Wu L., Gonias S. L. (2003) Soluble urokinase-type plasminogen activator receptor inhibits cancer cell growth and invasion by direct urokinase-independent effects on cell signaling. J. Biol. Chem. 278, 46692–46698 [DOI] [PubMed] [Google Scholar]
- 28. Rao J. S., Gujrati M., Chetty C. (2013) Tumor-associated soluble uPAR-directed endothelial cell motility and tumor angiogenesis. Oncogenesis 2, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sidenius N., Sier C. F., Blasi F. (2000) Shedding and cleavage of the urokinase receptor ( uPAR): Shedding and cleavage of uPA: identification and characterisation of uPAR fragments in vitro and in vivo. FEBS Lett. 475, 52–56 [DOI] [PubMed] [Google Scholar]
- 30. Mazzieri R., D'Alessio S., Kenmoe R. K., Ossowski L., Blasi F. (2006) An uncleavable uPAR mutant allows dissection of signaling pathways in uPA-dependent cell migration. Mol. Biol. Cell 17, 367–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selleri C., Montuori N., Ricci P., Visconte V., Carriero M. V., Sidenius N., Serio B., Blasi F., Rotoli B., Rossi G., Ragno P. (2005) Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood 105, 2198–2205 [DOI] [PubMed] [Google Scholar]
- 32. Wei C., El Hindi S., Li J., Fornoni A., Goes N., Sageshima J., Maiguel D., Karumanchi S. A., Yap H.-K., Saleem M., Zhang Q., Nikolic B., Chaudhuri A., Daftarian P., Salido E., et al. (2011) Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 17, 952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harada K., Nishizaki T., Ozaki S., Kubota H., Ito H., Sasaki K. (1998) Intratumoral cytogenetic heterogeneity detected by comparative genomic hybridization and laser scanning cytometry in human gliomas. Cancer Res. 58, 4694–4700 [PubMed] [Google Scholar]
- 34. Brennan C. W., Verhaak R. G., McKenna A., Campos B., Noushmehr H., Salama S. R., Zheng S., Chakravarty D., Sanborn J. Z., Berman S. H., Beroukhim R., Bernard B., Wu C.-J., Genovese G., Shmulevich I., et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155, 462–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonavia R., Inda M.-M., Cavenee W. K., Furnari F. B. (2011) Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 71, 4055–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gill B. J., Pisapia D. J., Malone H. R., Goldstein H., Lei L., Sonabend A., Yun J., Samanamud J., Sims J. S., Banu M., Dovas A., Teich A. F., Sheth S. A., McKhann G. M., Sisti M. B., et al. (2014) MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. 111, 12550–12555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanai N., Alvarez-Buylla A., Berger M. S. (2005) Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811–822 [DOI] [PubMed] [Google Scholar]
- 38. Libermann T. A., Razon N., Bartal A. D., Yarden Y., Schlessinger J., Soreq H. (1984) Expression of epidermal growth factor receptors in human brain tumors expression of epidermal growth factor receptors in human brain tumors. Cancer Res. 44, 753–760 [PubMed] [Google Scholar]
- 39. Libermann T. A., Nusbaum H. R., Razon N., Kris R., Lax I., Soreq H., Whittle N., Waterfield M. D., Ullrich A., Schlessinger J. (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313, 144–147 [DOI] [PubMed] [Google Scholar]
- 40. Huang H. S., Nagane M., Klingbeil C. K., Lin H., Nishikawa R., Ji X.-D., Huang C. M., Gill G. N., Wiley H. S., Cavenee W. K., Su Huang H. J. (1997) The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 272, 2927–2935 [DOI] [PubMed] [Google Scholar]
- 41. Inda M.-M., Bonavia R., Mukasa A., Narita Y., Sah D. W., Vandenberg S., Brennan C., Johns T. G., Bachoo R., Hadwiger P., Tan P., Depinho R. A., Cavenee W., Furnari F. (2010) Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24, 1731–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu J., Jo M., Cavenee W. K., Furnari F., VandenBerg S. R., Gonias S. L. (2011) Cross-talk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acad. Sci. 108, 15984–15989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishikawa R., Ji X. D., Harmon R. C., Lazar C. S., Gill G. N., Cavenee W. K., Huang H. J. (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 91, 7727–7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mukasa A., Wykosky J., Ligon K. L., Chin L., Cavenee W. K., Furnari F. (2010) Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc. Natl. Acad. Sci. U.S.A. 107, 2616–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ekstrand A. J., Longo N., Hamid M. L., Olson J. J., Liu L., Collins V. P., James C. D. (1994) Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene. Oncogene 9, 2313–2320 [PubMed] [Google Scholar]
- 46. Gonias S. L., Gaultier A., Jo M. (2011) Regulation of the urokinase receptor (uPAR) by LDL receptor-related protein-1 (LRP1). Curr. Pharm. Des. 17, 1962–1969 [DOI] [PubMed] [Google Scholar]
- 47. Salkeni M. A., Zarzour A., Ansay T. Y., McPherson C. M., Warnick R. E., Rixe O., Bahassi el M. (2013) Detection of EGFRvIII mutant DNA in the peripheral blood of brain tumor patients. J. Neurooncol. 115, 27–35 [DOI] [PubMed] [Google Scholar]
- 48. Jo M., Thomas K. S., Marozkina N., Amin T. J., Silva C. M., Parsons S. J., Gonias S. L. (2005) Dynamic assembly of the urokinase-type plasminogen activator signaling receptor complex determines the mitogenic activity of urokinase-type plasminogen activator. J. Biol. Chem. 280, 17449–17457 [DOI] [PubMed] [Google Scholar]
- 49. Jo M., Thomas K. S., Takimoto S., Gaultier A., Hsieh E. H., Lester R. D., Gonias S. L. (2007) Urokinase receptor primes cells to proliferate in response to epidermal growth factor. Oncogene 26, 2585–2594 [DOI] [PubMed] [Google Scholar]
- 50. Hu J., Muller K. A., Furnari F. B., Cavenee W. K., VandenBerg S. R., Gonias S. L. (2014) Neutralizing the EGF receptor in glioblastoma cells stimulates cell migration by activating uPAR-initiated cell signaling. Oncogene 10.1038/onc.2014.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shinojima N., Tada K., Shiraishi S., Kamiryo T., Kochi M., Nakamura H., Makino K., Saya H., Hirano H., Kuratsu J., Oka K., Ishimaru Y., Ushio Y. (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 63, 6962–6970 [PubMed] [Google Scholar]
- 52. Chumbalkar V., Latha K., Hwang Y., Maywald R., Hawley L., Sawaya R., Diao L., Baggerly K., Cavenee W. K., Furnari F. B., Bogler O. (2011) Analysis of phosphotyrosine signaling in glioblastoma identifies STAT5 as a novel downstream target of Δ EGFR. J. Proteome Res. 10, 1343–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wykosky J., Hu J., Gomez G. G., Taylor T., Villa G. R., Pizzo D., VandenBerg S. R., Thorne A. H., Chen C. C., Mischel P. S., Gonias S. L., Cavenee W. K., Furnari F. B. (2015) A urokinase receptor-bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 75, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Francis J. M., Zhang C.-Z., Maire C. L., Jung J., Manzo V. E., Adalsteinsson V. A., Homer H., Haidar S., Blumenstiel B., Pedamallu C. S., Ligon A. H., Love J. C., Meyerson M., Ligon K. L. (2014) EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 4, 956–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilhelm O. G., Wilhelm S., Escott G. M., Lutz V., Magdolen V., Schmitt M., Rifkin D. B., Wilson E. L., Graeff H., Brunner G. (1999) Cellular glycosylphosphatidylinositol-specific phospholipase D regulates urokinase receptor shedding and cell surface expression. J. Cell. Physiol. 180, 225–235 [DOI] [PubMed] [Google Scholar]
- 56. Pyke C., Eriksen J., Solberg H., Nielsen B. S., Kristensen P., Lund L. R., Danø K. (1993) An alternatively spliced variant of mRNA for the human receptor for urokinase plasminogen activator. FEBS Lett. 326, 69–74 [DOI] [PubMed] [Google Scholar]
- 57. Bruntz R. C., Taylor H. E., Lindsley C. W., Brown H. A. (2014) Phospholipase D2 mediates survival signaling through direct regulation of Akt in glioblastoma cells. J. Biol. Chem. 289, 600–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scott S. A., Selvy P. E., Buck J. R., Cho H. P., Criswell T. L., Thomas A. L., Armstrong M. D., Arteaga C. L., Lindsley C. W. (2009) Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 5, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Degryse B., Resnati M., Czekay R.-P., Loskutoff D. J., Blasi F. (2005) Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J. Biol. Chem. 280, 24792–24803 [DOI] [PubMed] [Google Scholar]
- 60. Montuori N., Carriero M. V., Salzano S., Rossi G., Ragno P. (2002) The cleavage of the urokinase receptor regulates its multiple functions. J. Biol. Chem. 277, 46932–46939 [DOI] [PubMed] [Google Scholar]
- 61. Montuori N., Ragno P. (2009) Multiple activities of a multifaceted receptor: roles of cleaved and soluble upar. Front. Biosci. 14, 2494–2503 [DOI] [PubMed] [Google Scholar]
- 62. Fazioli F., Resnati M., Sidenius N., Higashimoto Y., Appella E., Blasi F. (1997) A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotactic activity. EMBO J. 16, 7279–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Resnati M., Guttinger M., Valcamonica S., Sidenius N., Blasi F., Fazioli F. (1996) Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO J. 15, 1572–1582 [PMC free article] [PubMed] [Google Scholar]