

Background: LRP1 plays a major role in foam cell formation from human vascular smooth muscle cells (hVSMCs).
Results: Antibodies generated against the C-terminal half of cluster II CR9 domain (Gly1127–Cys1140) efficiently prevented hVSMC foam cell formation.
Conclusion: CR9 is key for AgLDL binding and internalization.
Significance: Our results open new avenues for treating vascular lipid deposition in atherosclerosis.
Keywords: LRP1, cluster II domains, AgLDL, VSMC, foam cells
Abstract
Low density lipoprotein receptor-related protein (LRP1) mediates the internalization of aggregated LDL (AgLDL), which in turn increases the expression of LRP1 in human vascular smooth muscle cells (hVSMCs). This positive feedback mechanism is thus highly efficient to promote the formation of hVSMC foam cells, a crucial vascular component determining the susceptibility of atherosclerotic plaque to rupture. Here we have determined the LRP1 domains involved in AgLDL recognition with the aim of specifically blocking AgLDL internalization in hVSMCs. The capacity of fluorescently labeled AgLDL to bind to functional LRP1 clusters was tested in a receptor-ligand fluorometric assay made by immobilizing soluble LRP1 “minireceptors” (sLRP1-II, sLRP1-III, and sLRP1-IV) recombinantly expressed in CHO cells. This assay showed that AgLDL binds to cluster II. We predicted three well exposed and potentially immunogenic peptides in the CR7–CR9 domains of this cluster (termed P1 (Cys1051–Glu1066), P2 (Asp1090–Cys1104), and P3 (Gly1127–Cys1140)). AgLDL, but not native LDL, bound specifically and tightly to P3-coated wells. Rabbit polyclonal antibodies raised against P3 prevented AgLDL uptake by hVSMCs and were almost twice as effective as anti-P1 and anti-P2 Abs in reducing intracellular cholesteryl ester accumulation. Moreover, anti-P3 Abs efficiently prevented AgLDL-induced LRP1 up-regulation and counteracted the down-regulatory effect of AgLDL on hVSMC migration. In conclusion, domain CR9 appears to be critical for LRP1-mediated AgLDL binding and internalization in hVSMCs. Our results open new avenues for an innovative anti-VSMC foam cell-based strategy for the treatment of vascular lipid deposition in atherosclerosis.
Introduction
The low density lipoprotein (LDL) receptor-related protein 1 (LRP12; UniProt entry Q07954) is a ubiquitously expressed cell surface endocytic receptor that belongs to the LDL receptor family (1, 2). LRP1 is synthesized in the endoplasmic reticulum as a 600-kDa single-chain polypeptide and subsequently processed into two subunits in the trans-Golgi network. The mature receptor is thus a heterodimer composed of a 515-kDa extracellular α-chain noncovalently associated with an 85-kDa transmembrane/cytoplasmatic β-chain (2, 3). The α-chain contains long modular arrays of acidic cysteine-rich complement-type repeats (CRs), along with epidermal growth factor (EGF)-like domains and β-propeller modules. Complement-type repeats consist of ∼40 amino acid residues with three conserved disulfide linkages and mediate the specific recognition and internalization of more than 40 different extracellular ligands by LRP1. These ligand-binding domains are organized into four clusters termed I–IV, which are composed of 2, 8, 10, and 11 CRs, respectively. Most of the known LRP1 ligands bind to cluster II and/or IV (4). Ligand binding alters the phosphorylation state of the β-chain intracellular tail, which allows recruitment or release of adaptor molecules triggering downstream signaling cascades (1, 5).
Although no structural information has been reported for any of the LRP1 clusters, the structures of some of its individual CR modules have been studied, either by x-ray crystallography or more commonly by NMR in solution. Most of the characterized repeats belong to cluster II, which spans domains CR3–CR10. For example, the crystal structure of domain CR7 has been reported (6), whereas modules CR3 and CR8 have been characterized by NMR in solution (7). Further, the structure of the CR5/CR6 tandem has been solved, and a model of its complex with the chaperone and universal ligand of LDL receptor family members, RAP, has been presented (8). Finally, a recent NMR analysis of a fusion protein between CR17 and the receptor-binding helix from apolipoprotein E (apoE) has unveiled the recognition mechanism of this apolipoprotein (9). On the other hand, the crystal structure of the ectodomain of the founding member of the family, the LDL receptor, has revealed an overall extended arrangement of a cluster of seven CR modules, which form an arch that covers one of the faces of the bulkier β-propeller domain (10).
LRP1 is a multifunctional receptor involved in several biological processes and signaling pathways (11, 12), including lipoprotein metabolism (13), cell proliferation and migration (14–17), neurotransmission (18), and amyloid-β peptide clearance (19). These functions confer on LRP1 an important role in atherosclerosis, cancer, and Alzheimer disease. In the vasculature, LRP1 protects the integrity of the vascular wall through inhibition of the platelet-derived growth factor (PDGF) pathway (14, 20, 21). However, LRP1 overexpression has been associated with atherosclerosis in both animal (22) and human models of atherosclerosis (23–25). In particular, LRP1 efficiently transfers cholesteryl esters (CE) from aggregated LDL (AgLDL) into human coronary VSMCs (26–28), transforming them into foam cells (29, 30). The formation of AgLDL, detected and isolated from human atherosclerotic lesions (31), is promoted through the interaction between retained LDL particles and proteoglycans in the arterial intima (32). Hypercholesterolemia, one of the major cardiovascular risk factors, strongly promotes VSMC foam cell formation by favoring LDL retention and aggregation into the arterial intima (33) but also by up-regulating LRP1 vascular levels (34). Remarkably, it has been recently reported that more than 50% of the foam cells in the vasculature originate from VSMCs (35).
Although accumulated evidence clearly implicates intimal LDL aggregation and vascular LRP1 overexpression in hypercholesterolemia-associated atherosclerosis, a direct physical interaction between LRP1 and AgLDL has not been demonstrated, and the domain(s) of the LRP1 structure that could be involved in specific AgLDL recognition remains to be elucidated. To explore putative LRP1-AgLDL interactions, we produced the LRP1 ligand-binding clusters as soluble proteins (sLRP1-II to -IV, also termed “minireceptors”). Using these definite recombinant fragments, we show that AgLDLs are specifically recognized by LRP1 cluster II. Further, we generated rabbit polyclonal antibodies against three cluster II peptides predicted to be particularly immunogenic and LRP1-specific based on a three-dimensional model and other bioinformatics tools and tested the ability of these antibodies to inhibit AgLDL internalization. Our results show that anti-cluster II antibodies have a high capacity to block AgLDL-cholesteryl ester internalization by human VSMCs. These findings implicate the linker between domains CR7 and CR8 and the N-terminal portion of the latter (Cys1051–Glu1066), the C terminus of CR8 and the CR8-CR9 linker (Asp1090–Cys1104), and mainly the C-terminal half of CR9 (Gly1127–Cys1140) as pivotal for AgLDL binding and internalization.
Experimental Procedures
Production of Soluble LRP1 Minireceptors
Expression Plasmids
A plasmid containing the entire cDNA for human LRP1 (accession number XM_001056970) was kindly provided by Dr. Joachim Herz (University of Texas Southwestern Medical Center, Dallas, TX) and used as the PCR template. We generated cDNAs for three of the soluble ligand-binding clusters of LRP1, sLRP1-II, sLRP1-III, and sLRP1-IV, with cluster boundaries essentially as reported previously (36). Although formally related to these three minireceptors, the significantly smaller cluster I appears to be dispensable for ligand binding, because no ligands have been described so far for this LRP1 region, and it does not bind to the universal chaperone of the family, RAP (36). Therefore, expression of this cluster was pursued no further in the current investigation. An additional construct that codes for the internal CR4–CR8 domains of cluster II (CR4/CR8) was also generated. The domains included in the three sLRP1s are marked in Fig. 1A and span the following residues: sLRP1-II, from Pro787 to Val1244; sLRP1-III, from Gln2462 to Lys3004; sLRP1-IV, from Arg3274 to Gly3843; and CR4/CR8, from His893 to Gly1099. cDNAs were generated by PCR with the oligonucleotides listed in Table 1. Plasmids were isolated, and cDNAs were then subcloned into the BamHI and EcoRV restriction sites of the pSegTag2B vector (Invitrogen). The resulting plasmids, designated pST2B/sLRP1-II, -III -IV, and -CR4/CR8, respectively, were transformed into TOP10 Escherichia coli cells, isolated, and verified by automated sequence analysis.
FIGURE 1.
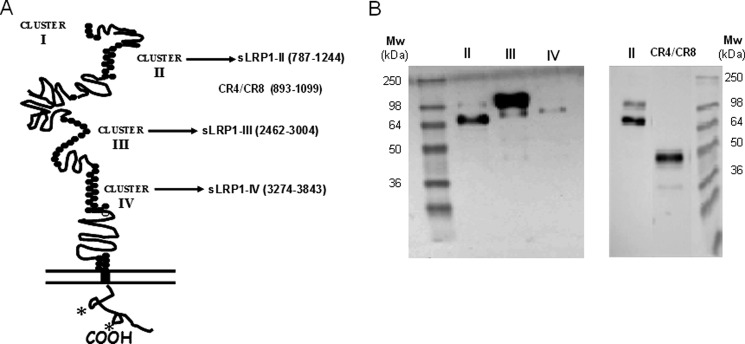
Overexpression of definite human LRP1 fragments for functional assays. A, schematic representation of LRP1 domain structure and fragments cloned for functional analysis of interactions with AgLDL. These fragments were expressed and purified from CHO-K1 cell supernatants. (See “Experimental Procedures” for details.) *, NPXY motifs. B, Western blot analysis of Ni-NTA-purified recombinant proteins (LRP1 clusters II, III, and IV as well as cluster II fragment CR4–CR8) detected using anti-Myc antibody.
TABLE 1.
Oligonucleotides used to generate soluble LRP1 minireceptors
Restriction sites are labeled in boldface type and underlined.
| Oligonucleotide (residues) | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| sLRP1-II (787–1244) | GATCGGATCCAAATGCCGGGTGAACAATGGCGGC | GATCGATATCCCGCTGCGGCAGCTCTCGCCGTCAG |
| sLRP1-III (2462–3004) | GATCGGATCCCCATGCCGAATCAACAACGGTGGC | GATCGATATCCCAGCCTTGCAGCTGTGGGGGTCGC |
| sLRP1-IV (3274–3843) | GATCGGATCCCCCTGCAAGGTCAACAATGGTGGC | GATCGATATCCCGGCCTTGCAGGTGTTGTGCGTCTT |
| CR4/CR8 (893–1099) | GATCGGATCCCACACCTGCCCCTCGGACCGA | GATCGATATCCCTCCCTCACAGCTCTTCTCATC |
Cell Culture and Transfection
CHO-K1 cells were grown in F12-K medium (Gibco) supplemented with 5% fetal bovine serum (FBS), 2 mmol/liter l-glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin at 37 °C in a 5% CO2 atmosphere in a humidified incubator. We transfected CHO-K1 cells with 20 μg of the different pST2B-sLRP1 plasmids using FuGENE 6 reagent (Roche Applied Science) in serum-containing medium according to the manufacturer's instructions. Stably transfected cells were selected using 300 μg/ml zeocin in the culture medium starting 48 h after transfection. CHO-K1 sensitivity to zeocin was determined in preliminary experiments.
Purification of Secreted Minireceptors
Stably transfected CHO-K1 clones were isolated and subcultured in F12-K medium supplemented with 10% FBS, l-glutamine, penicillin/streptomycin, and 300 μg/ml zeocin with the medium changed every 3 days. At near confluence, cells were maintained for 3 days in fresh serum- and protein-free CHO IIIa medium (Gibco) supplemented with 300 μg/ml zeocin, 1× HT supplement (Invitrogen), 2 mmol/liter l-glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin. After this incubation period, the culture medium was collected, and secreted proteins were concentrated with Amicon Ultra-15 centrifugal filters with a 10 kDa nominal molecular mass limit cut-off (Millipore). Secreted polyhistidine-tagged sLRP1s were finally purified with Ni-NTA spin columns (Qiagen). Purified sLRP1s were detected by Western blot with an antibody (Invitrogen) against the N-terminal Myc epitope present in the recombinant minireceptors.
LDL Isolation and Modification
LDL Purification
Human LDL (d1.019–d1.063 g/ml) were obtained from pooled normolipemic plasma by sequential ultracentrifugation in a KBr density gradient. Briefly, VLDLs were first discarded after spinning plasma at 36,000 rpm for 18 h at 4 °C using a fixed angle rotor (50.2 Ti, Beckman) mounted on an Optima L-100 XP ultracentrifuge (Beckman). Subsequently, VLDL-free plasma was layered with 1.063 g/ml KBr solution and centrifuged at 36,000 rpm for 18 h at 4 °C. LDLs were dialyzed against 0.02 m Trizma, 0.15 m NaCl, 1 mm EDTA, pH 7.5, for 18 h and then against normal saline for 2 h. Finally, isolated LDLs were filter-sterilized (0.22-μm Millex-GV filter unit, Millipore). Protein concentration was determined using the BCA protein assay (Thermo Scientific), and the cholesterol concentration was determined with a commercial kit (IL Test Cholesterol, Izasa, Barcelona, Spain).
Fluorescent Labeling of LDL
A stock solution of the fluorescent probe 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanineperchlorate (DiI; Molecular Probes) was prepared by dissolving 30 mg of DiI in 1 ml of DMSO, and then added to the LDL solution to yield a final ratio of 0.5–2 μg of DiI/mg of LDL protein. The mixture was incubated for 18 h at 37 °C. The obtained labeled LDLs (DiI-LDLs), either in their native form or aggregated, were used in fluorometric assays as described below.
Generation of Aggregated LDL
AgLDLs were generated by vortexing 1 mg/ml LDL in PBS for 4 min at room temperature at maximal speed. AgLDLs were then centrifuged at 10,000 × g for 10 min; the precipitable fraction is composed of 97–100% AgLDLs. Of note, the ultrastructure of AgLDL obtained by vortexing is similar to that of LDL modified by versican, one of the main chondroitin sulfate proteoglycans of the arterial intima (28).
Receptor-Ligand Binding Fluorometric Assay
AgLDL binding to sLRP1s was assessed by a fluorometric assay. Anti-c-Myc capture antibody (Sigma) at 5 μg/ml in carbonate/bicarbonate coating buffer (pH 9.6) was immobilized on sterile, tissue culture-treated black polystyrene 96-well plates with lid and clear flat bottom wells (Costar, 3603) overnight at 4 °C. After exhaustive washes with 50 mm Tris, 0.14 m NaCl, 0.05% Tween 20, pH 8.0, the nonspecific protein-binding sites of the coated wells were blocked (blocking buffer: 50 mm Tris, 0.14 m NaCl, 1% BSA, pH 8.0) for 1 h at room temperature. The wells were washed again, and 2 μg of sLRP1s diluted in blocking buffer supplemented with 0.05% Tween 20 were added to the assay and incubated for 2 h at room temperature. After removal of the samples, the plate was washed several times and then incubated with either DiI-labeled native LDL (DiI-nLDL) or DiI-labeled AgLDL (DiI-AgLDL) for 1.5 h at room temperature. Finally, after several washes, the DiI fluorescence was determined in a SpectraMax Gemini EM fluorescence microplate reader (Molecular Devices) with excitation and emission wavelengths set at 551 and 566 nm, respectively.
Polyclonal Antibody Production
Peptide Synthesis
We identified three potentially highly immunogenic peptides in the ligand-binding domain sLRP1-II with the following sequences, using single-letter code: sLRP1-II peptide 1 (P1; Cys1051–Glu1066, CTNQATRPPGGSHTDE); sLRP1-II peptide 2 (P2; Asp1090–Cys1104, DSSDEKSSEGVTHVC); and sLRP1-II peptide 3 (P3; Gly1127–Cys1140, GDNDSEDNSDEENC). These peptides were synthesized as C-terminal amides on a Rink amide MBHA resin (Iris Biotech GmbH, Marktredwitz, Germany) using Fmoc solid-phase protocols in a Prelude instrument (Protein Technologies Inc.) at the Peptide Facility of Pompeu Fabra University (Barcelona, Spain). After deprotection and cleavage with trifluoroacetic acid/water/ethanedithiol/triisopropylsilane (94:2.5:2.5:1, v/v/v/v) for 90 min, the peptides were isolated by precipitation with cold diethyl ether, dissolved in 10% acetic acid, and lyophilized. The crude products were purified to >95% homogeneity by reverse-phase HPLC, and their identity was verified by electrospray mass spectrometry (Applied Biosystems 4700 Proteomics Analyzer). Finally, the peptides were coupled to the carrier proteins, keyhole limpet hemocyanin (for antibody generation), or BSA (for ELISAs), through their N- or C-terminal cysteine residues.
Animal Immunization
Polyclonal antibodies were produced by the Servei de Cultius Cel·lulars, Producció d'Anticossos i Citometria from the Universitat Autònoma de Barcelona (UAB). The animal study was approved by the UAB Animal Research Committee and the Government of Catalonia and conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health. Two rabbits (New Zealand) were used to generate antibodies against each peptide. Rabbits were immunized intradermally with 500 μg of peptide conjugated to keyhole limpet hemocyanin in Freund's complete adjuvant. After the first immunization, rabbits were administered three booster injections (21 days between immunizations) with similar amounts of peptide but using Freund's incomplete adjuvant instead, and a sample of blood was taken to monitor serum-specific antibody levels by ELISA. Total rabbit sera were collected after the final immunization.
ELISA
The presence of specific antibodies in serum samples was determined by ELISA. Briefly, BSA-conjugated cluster II peptides were immobilized on 96-well plates at 1 μg/ml for 60 min in 0.1 m carbonate/bicarbonate buffer, pH 9.6. After blocking with 1% BSA in PBS, the plates were incubated with primary antibodies at several dilutions, followed by 1:1000 diluted horseradish peroxidase-conjugated anti-rabbit IgG (A3673, Sigma-Aldrich). The enzymatic reaction was conducted with SIGMAFAST OPD (o-phenylenediamine dihydrochloride) substrate solution (Sigma-Aldrich). After 30 min, the optical density at 450 nm was measured with a microplate reader (Victor3, PerkinElmer Life Sciences).
Peptide-Ligand Binding Fluorometric Assay
AgLDL binding to cluster II peptides P1-P3 was assessed by a fluorometric assay. Peptide-BSA conjugates were diluted 1:2000 and bound to the wells of a 96-well plate (Costar) overnight at 4 °C. Free BSA was used as a negative control. After exhaustive washes with 50 mm Tris, 0.14 m NaCl, 0.05% Tween 20, pH 8.0, the nonspecific protein-binding sites of the coated wells were blocked for 1 h at room temperature. The wells were washed again and incubated with nLDL or AgLDL for 2 h at room temperature. Finally, after several washes, the concentration of cholesterol was measured using an Amplex® Red cholesterol assay kit (Molecular Probes), following the manufacturer's instructions. Fluorescence intensity was determined in a SpectraMax Gemini EM fluorescence microplate reader (Molecular Devices; excitation and emission wavelengths set at 520 and 578 nm, respectively); the amount of bound cholesterol was calculated by using a standard curve.
Culture of Human Vascular Smooth Muscle Cells (hVSMCs)
hVSMC cultures were obtained from the medium layer isolated from macroscopically healthy coronary artery segments collected from patients undergoing cardiac transplantation at Hospital de la Santa Creu i Sant Pau (Barcelona, Spain). hVSMCs were isolated by a modification of the explant technique, as described previously (25–28). The explants were incubated at 37 °C in a humidified atmosphere of 5% CO2. After 1 week, the cells start to migrate from the explants and proliferate, covering the floor of the culture well. The medium was exchanged every 3 days after the onset of cell outgrowth; a significant outgrowth was reached after 10 days. Tissue fragments were collected with forceps and placed in a new dish with fresh medium. The cells that remained in the dish were cultured until confluence. For characterization, cells were seeded in coverslips and grown to confluence. Cell quiescence was induced by maintaining the cell culture for 24 h in a medium with 0.2% FCS or for 48 h in a medium with 0.4% serum. All experiments used serum-deprived cells between passages 4 and 6; VSMCs at these passages appeared as a relatively homogeneous population, showing a hill-and-valley pattern at confluence. Western blot analysis for specific differentiation markers revealed high levels of α-actin (45 kDa) and calponin (33 kDa). Cell monolayers were grown in medium 199 supplemented with 20% FBS, 2% human serum, 2 mmol/liter l-glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin. The study was approved by the Institutional Ethics Committee at Hospital de la Santa Creu i Sant Pau and conducted in accordance with the Declaration of Helsinki.
Lipid Extraction and Determination of Free and Cholesteryl Ester Content
Serum-deprived human coronary VSMCs were preexposed to anti-sLRP1-II antibodies for 2 h before they were incubated for 18 h with AgLDLs. Following the lipoprotein incubation period, cells were exhaustively washed (twice with PBS, twice with PBS supplemented with 1% BSA, and once with PBS supplemented with both 1% BSA and 100 units/ml heparin) before they were harvested into 1 ml of 0.15 m NaOH. Lipids were extracted using the Bligh and Dyer method (37) with minor modifications (26). The lipid extract was redissolved in dichloromethane, applied to silica gel plates, and separated by thin layer chromatography. Various mixtures of cholesterol, cholesterol palmitate, and mono-, di-, and triglycerides were also run as standards. Heptane or a solvent combination of heptane/diethyl ether/acetic acid (74:21:4, v/v/v) was used as a chromatographic mobile phase. After lipid separation, the plates were dried and stained as reported previously (38). Finally, the spots corresponding to CE and free cholesterol (FC) were quantitated by densitometry against the standard curve using a GS-800 calibrated densitometer (Bio-Rad).
Confocal Microscopy
A stock solution of the fluorescent dye boron-dipyrromethene (BODIPY) was diluted to a final concentration of 1 mg/ml in DMSO. To study lipid droplets, cells were incubated for 30 min with BODIPY (dilution 1:300). Images of immunostained cells were analyzed on a Leica inverted fluorescence confocal microscope (Leica TCS SP2-AOBS; excitation wavelength 480 nm, emission maximum 515 nm). Images were processed with the Leica standard software, TCS-AOBS.
Real-time PCR
Serum-deprived hVSMCs were exposed to lipoproteins in the presence or absence of antibodies. Cells were then washed with PBS, and total RNA was isolated using TriPure reagent (Roche Applied Science) according to the manufacturer's instructions. Reverse transcription was performed using the high capacity cDNA reverse transcription kit (Applied Biosystems), and the reaction mix was subjected to quantitative real-time PCR to detect LRP1 and SREBP-2 using the assays on demand Hs00233899_m1 and Hs00190237_m1 (Applied Biosystems). Human GAPDH from the same provider was used as an endogenous control. Real-time PCR was performed using 1 μl/well of reverse transcription products in 10 μl of TaqMan PCR master mix (PE Applied Biosystems) with primers at 300 nm and probe at 200 nm. PCR was performed in a PCR-7600HT sequence detection system (ABI PRISM, Applied Biosystems) at 95 °C for 10 min for AmpliTaq Gold activation and then run for 40 cycles at 95 °C for 15 s and 60 °C for 1 min. The relative levels of gene expression were quantitated and analyzed using SDS version 2.4 software. The real-time value for each sample was averaged and compared using the CT method, where the amount of target RNA (2−ΔΔCT) was normalized to the endogenous control (ΔCT) and related to the amount of target gene in the cells.
Western Blot Analysis
Serum-deprived cells were exposed to lipoproteins in the presence or absence of antibodies. Cells were then washed with PBS, and total proteins were isolated using TriPure reagent according to the manufacturer's instructions. Equivalent amounts of total protein (25 μg) were electrophoresed under nonreducing conditions on SDS-polyacrylamide gels. The samples were electrotransferred to nitrocellulose membranes, which were then saturated at room temperature for 1 h in TTBS (20 mm Tris-HCl, pH 7.5, 500 mm NaCl, 0.01% Tween 20, and 5% nonfat milk). Western blot analyses were performed with specific monoclonal antibodies against LRP1 β-chain (Research Diagnostics, clone 8B8 RDI 61067, dilution 1:40) and the corresponding secondary antibody (1:10,000 dilution; Dako). Equal protein loading was verified with Ponceau staining and by anti-β-tubulin Western blot (Abcam, ab6046, 1:1000 dilution). Bands were detected using the ECL prime Western blotting detection reagent (Amersham Biosciences) and quantitated by densitometry with the ChemiDoc system and Quantity-One software (Bio-Rad). Results are expressed as arbitrary units of intensity.
Cell Migration and Wound Repair Assays
hVSMCs were seeded on glass bottom sterile culture dishes. Confluent cells were scratched to create a double-sided wound, as described previously (39, 40) and washed with PBS. Human VSMCs were grown in medium 199 supplemented with FBS (10%), penicillin/streptomycin (1%), and l-glutamine (1%). Photographs were taken at various times over an 18-h period, and cell migration and wound repair were analyzed by measuring the injured area covered by cells counted from the wounding borders with the ImageJ software.
Statistical Analysis
Comparisons among groups were performed by parametric (one-factor analysis of variance) or nonparametric (Mann-Whitney U test) analysis as needed. Statistical significance was considered when p was <0.05.
Results
Aggregated but Not Native LDL Binds to LRP1 Cluster II
To investigate AgLDL binding properties of LRP1 clusters II, III, and IV, we recombinantly expressed these soluble minireceptors (hereafter termed sLRP1-II, sLRP1-III, and sLRP1-IV, respectively; Fig. 1A). Plasmids encoding these three clusters, N-terminally extended by His and Myc tags to assist in protein detection and purification, were stably transfected into CHO-K1 cells as described under “Experimental Procedures.” We have excluded the possibility of misfolding or misglycosylation of the receptor fragments by using mammalian cells instead of bacteria for recombinant protein expression. The use of mammalian cells guarantees that post-translational modifications are correctly performed and that only correctly folded proteins are secreted to the extracellular medium. Further, we have performed all minireceptor expression and purification procedures in the presence of CaCl2 to guarantee occupancy of the single Ca2+-binding site present in all CR modules.
Culture media were collected, and secreted LRP1 minireceptors were purified by metal affinity chromatography on Ni-NTA columns. Fig. 1B shows the analysis of Ni-NTA-purified supernatants by immunoblotting with anti-Myc antibodies. The molecular sizes of the three LRP1 clusters expressed in CHO-K1 cells correlated well with those predicted from the amino acid sequence and putative N-linked glycosylation sites. These molecular sizes were also close to those previously found for similar constructs expressed in human glioblastoma U87 cells (36).
Most of the previous studies that aimed to analyze the interaction of LRP1 with different ligands were performed by surface plasmon resonance, using purified receptor fragments immobilized on BIAcore sensor chips. However, the large AgLDL size (diameter of ∼600 nm) precludes the use of surface plasmon resonance for studying its interactions with LRP1. Therefore, to analyze the potential role of each LRP1 cluster in AgLDL binding, we developed a receptor-ligand binding fluorometric assay based on the immobilization of the recombinant LRP1 minireceptors with anti-c-Myc capture antibodies and the use of DiI-labeled AgLDL (Fig. 2A). As a negative control, we used the supernatants from CHO-K1 cells transfected with the empty vector. As shown in Fig. 2B, AgLDLs specifically bind to cluster II at all concentrations tested, although at the highest concentration (250 μg/ml), they also showed a slight binding to clusters III and IV. In contrast, there was no detectable binding of nLDL to any LRP1 cluster even at the maximum concentration tested. After correcting for background binding, we found that AgLDL binding to cluster II increased linearly with the lipoprotein concentration. Finally, we also generated a recombinant construct that spans the five “internal” complement-type repeats of cluster II, CR4/CR8 (Fig. 1B). There was no detectable binding of AgLDL to this receptor fragment (Fig. 2B), suggesting that cluster II domains CR3, CR9, and/or CR10 are critical for AgLDL interaction.
FIGURE 2.

Aggregated LDL is specifically recognized by LRP1 cluster II. A, schematic representation of the fluorometric assay developed to detect AgLDL binding to LRP1 minireceptors. Briefly, recombinantly expressed clusters II–IV or the CR4/CR8 tandem were immobilized on the wells of a microplate using an anti-c-Myc capture antibody and then incubated with various amounts of fluorescent labeled native or aggregated LDL. Supernatants from CHO-K1 cells transfected with the empty vector were used as background control. (See “Experimental Procedures” for details). B, affinity of generated LRP1 fragments for DiI-labeled lipoproteins. Measured fluorescent intensities are given as mean ± S.D. (error bars) of four different experiments performed in duplicate. *, p < 0.05 versus AgLDL binding to background.
Aggregated LDL Specifically Binds to a Peptide Corresponding to the C-terminal Half of Domain CR9
Next, we decided to start exploring in more detail cluster II regions that might be involved in important contacts with AgLDL. Using the structural data summarized in the Introduction and with the help of several bioinformatics tools, we predicted three well exposed and potentially immunogenic peptides in LRP1 cluster II (Fig. 3A). Partial sequence alignments around these regions are given in Fig. 3B. These peptides, corresponding to the Cys1051–Glu1066, Asp1090–Cys1104, and Gly1127–Cys1140 sequences of human LRP1 were synthesized and purified for further characterization. (For simplicity, these peptides are referred to here as P1, P2, and P3, respectively). To analyze whether these peptides possess LDL binding capacity, we coupled them to BSA and exposed these peptide-BSA conjugates to lipoproteins in our predesigned ELISA as explained in the legend to Fig. 4A. The fluorescence (arbitrary units) corresponding to the background control (immobilized BSA) was subtracted from the fluorescence corresponding to P1-, P2-, and P3-coated wells not exposed to LDL or exposed to native or aggregated LDL (100 μg/ml) (Table 2). Then, by using a standard curve of cholesterol, we transformed fluorescence into cholesterol concentrations. As shown in Fig. 4B, P3-coated wells have a strong capacity to specifically bind AgLDL. In contrast, and in line with the lack of AgLDL binding to the CR4/CR8 construct (Fig. 2B), neither P1 nor P2 peptides bound AgLDL or nLDL under these experimental conditions.
FIGURE 3.

Three-dimensional model of LRP1 CR7/CR9 tandem and prediction of immunogenic peptides. A, schematic representation of the three-dimensional model of the CR7/CR9 tandem from human LRP1 cluster II. The three CR-type modules are represented by their secondary structure elements (β-strands, arrows) colored green and marked. Residues that belong to the three well exposed, immunogenic peptides P1–P3 are shown with all of their non-hydrogen atoms, color-coded as follows: green, carbon; blue, nitrogen; red, oxygen; yellow, sulfur. B, partial sequence alignment of the CR7/CR9 tandem from human LRP1 and closely related members of the LDLR family. The sequences of the P1–P3 peptides and topologically equivalent stretches in other receptors are boxed. Notice the large sequence differences, even with the most closely related member of the family, LRP1B.
FIGURE 4.

Demonstration of direct physical interactions between the C-terminal half of domain CR9 and AgLDL. A, schematic representation of the receptor-ligand binding fluorometric assay used to study interactions between immobilized BSA-conjugated P1, P2, and P3 peptides and AgLDL. (See “Experimental Procedures” for details.) B, quantitation of peptide-bound cholesterol according to the results of the fluorometric assay. C, effect of increasing concentrations of anti-P3 Abs on P3-bound cholesterol. Results are shown as mean ± S.D. (error bars) of six different experiments performed in duplicate. *, p < 0.05 versus no Ab.
TABLE 2.
Quantitation of nLDL and AgLDL binding to LRP1 cluster II peptides by receptor-ligand binding fluorometric assay
Peptides P1–P3 correspond to the Cys1051–Glu1066, Asp1090–Cys1104, and Gly1127–Cys1140 sequences of human LRP1. Results are shown as mean ± S.D. of six independent experiments performed in duplicate.
| BSA | P1 | P2 | P3 | |
|---|---|---|---|---|
| nLDL | 7.76 ± 0.32 | 7.16 ± 0.42 | 7.53 ± 0.62 | 7.03 ± 0.36 |
| AgLDL | 16.19 ± 1.72 | 17.15 ± 2.08 | 16.49 ± 2.63 | 50.55 ± 3.13a |
a p < 0.05 versus BSA-bound cholesterol (see “Results” for details).
The C-terminal Half of LRP1 Domain CR9 (Gly1127–Cys1140) Is Crucial for AgLDL Binding and Internalization in hVSMCs
To explore the biological relevance of the AgLDL-LRP1 interactions demonstrated above in vitro, we generated rabbit polyclonal antibodies against each of the studied cluster II peptides, as described under “Experimental Procedures.” (These polyclonal antibodies will be referred to as anti-P1, anti-P2, and anti-P3, respectively.) Notice that the selected sequences are not conserved in any other LDLR family member, which should guarantee specific interference with LRP1-mediated events. As expected, anti-P3 dose-dependently reduced the capacity of the P3 peptide to bind AgLDL, whereas anti-P1 and anti-P2 polyclonal antibodies did not exert any significant effect in this regard (Fig. 4C).
Next, we analyzed whether anti-P1, anti-P2, and/or anti-P3 Abs prevented AgLDL uptake by hVSMCs. To this end, we exposed these cells to AgLDL (75 μg/ml) in the presence or absence of increasing amounts of anti-peptide Abs. hVSMCs unexposed to lipoproteins showed undetectable levels of intracellular CE (41). Therefore, intracellular CE detected in these cells upon exposure to AgLDL derives exclusively from lipoprotein uptake. As expected (26, 41), the uptake of AgLDL (75–100 μg/ml) induced a strong intracellular CE accumulation in hVSMCs. Anti-P1 and anti-P2 Abs (dilution 1:5) slightly decreased AgLDL-derived intracellular CE accumulation to a maximal 45% reduction (Fig. 5A). In contrast, anti-P3 Abs at the same relative concentration were almost twice as effective in reducing CE accumulation, indicating that P3 is pivotally involved in LRP1-mediated AgLDL binding and internalization. As shown in Fig. 5B, further increasing Ab relative concentration (from 5- to 13-fold) did not further reduce hVSMC intracellular CE levels, indicating that 20–30% of Ab is sufficient to saturate receptor binding sites. The combination of the three antibodies was ineffective to further reduce AgLDL-derived intracellular cholesteryl ester accumulation (Fig. 6), suggesting that anti-P1 and/or anti-P2 Abs sterically interfere with the binding of anti-P3 to CR9.
FIGURE 5.
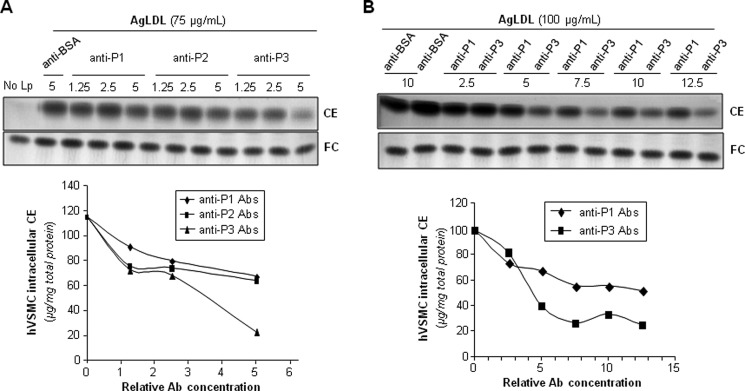
Antibodies raised against the C-terminal half of domain CR9 inhibit AgLDL-derived intracellular CE accumulation in human VSMCs. hVSMCs were preincubated with increasing concentrations of rabbit polyclonal Abs raised against P1–P3 peptides for 2 h and then exposed to AgLDL or nLDL for a further 18 h. Cells were then collected and submitted to lipid extraction. A and B, thin layer chromatography of CE and FC in AgLDL-exposed hVSMCs and bar graphs showing the quantitation of these bands. C, thin layer chromatography of CE and FC in nLDL-exposed hVSMCs.
FIGURE 6.
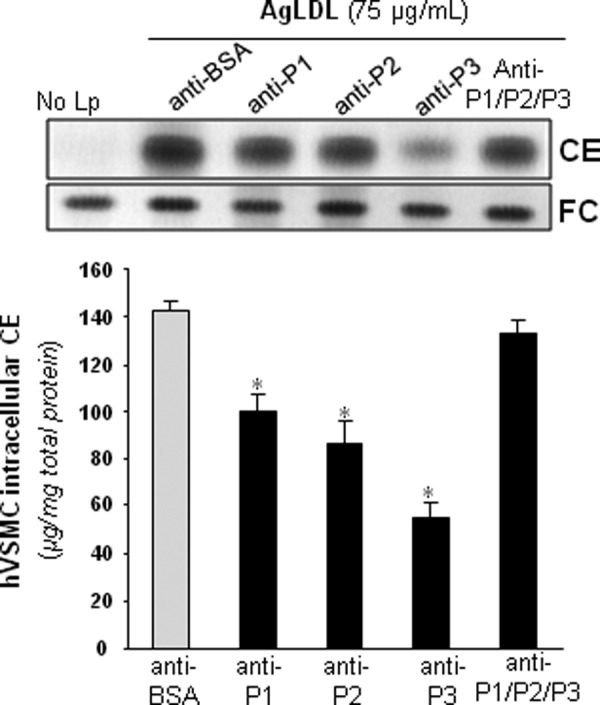
The combination of anti-P1, anti-P2, and anti-P3 antibodies failed to inhibit AgLDL-derived intracellular CE accumulation in human VSMCs. hVSMCs were preincubated with rabbit polyclonal Abs (at 7.5 relative concentration) raised against P1–P3 peptides alone or in combination for 2 h and then exposed to AgLDL for a further 18 h. Cells were then collected and submitted to lipid extraction. Thin layer chromatography showing CE and FC in AgLDL-exposed hVSMCs and bar graphs with the quantitation of these bands. *, p < 0.05 versus anti-BSA Abs. Error bars, S.D.
Confocal microscopy analysis of BODIPY-stained cells showed an almost complete absence of AgLDL-induced lipid droplets in anti-P3-treated hVSMCs (Fig. 7). None of the tested Abs at the maxim tested concentration exerted any significant effect on the slight nLDL-derived intracellular CE accumulation (data not shown). Of particular note, anti-P3 antibodies at 7.5 relative concentration showed a similar capacity as RAP (100 μg/ml) to block AgLDL-induced accumulation of intracellular CE in hVSMCs (Fig. 8B). Different from RAP, however, anti-LRP1 Abs were unable to regulate intracellular CE accumulation induced by nLDL in these cells (Fig. 8A). Altogether, these results strongly suggest that domain CR9 in LRP1 cluster II is the most important epitope for AgLDL binding and internalization.
FIGURE 7.
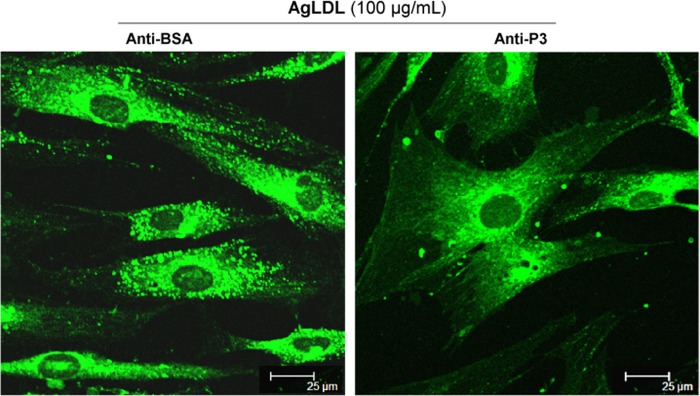
Antibodies raised against the C-terminal half of domain CR9 inhibit AgLDL-induced lipid droplet formation in human VSMCs. hVSMCs were preincubated with anti-P3 Abs (at 7.5 relative concentration) for 2 h and then exposed to AgLDL for a further 4 h. Confocal laser microscopy images showing BODIPY-stained lipid droplets (green dots) in anti-BSA- and anti-P3-treated hVSMCs. Scale bar, 25 μm.
FIGURE 8.

Antibodies against the C-terminal half of domain CR9 interfere with AgLDL-derived intracellular CE accumulation in hVSMCs to a similar extent as the endogenous inhibitor, RAP. hVSMCs were preincubated with Abs (at 7.5 relative concentration) or RAP (100 μg/ml) for 2 h and then exposed to AgLDL or nLDL for a further 18 h. Cells were then collected and submitted to lipid extraction. Thin layer chromatography showing CE and FC bands in nLDL (A) and AgLDL-exposed hVSMCs (B) and the quantitation of these bands are presented graphically below the chromatograms. Results are shown as mean ± S.D. (error bars) of three different experiments performed in duplicate. *, p < 0.05 versus no Ab.
The C-terminal half of LRP1 domain CR9 Is Crucial for AgLDL-induced SREBP-2 Down-regulation and LRP1 Overexpression in hVSMCs
Previous studies from our group have demonstrated that AgLDL positively modulates LRP1 gene expression and protein levels (34, 42, 43), generating a positive feedback loop that efficiently transforms hVSMCs into foam cells (30). Down-regulation of sterol regulatory element-binding protein 2 (SREBP-2) plays a crucial role in the positive modulation of LRP1 by AgLDL (34, 42, 43). In light of these previous findings, we decided to explore the possible impact of anti-peptide antibodies on SREBP-2 down-regulation and LRP1 up-regulation induced by AgLDL. Indeed, anti-P3 Abs strongly prevented SREBP-2 decay induced by AgLDL, whereas anti-P1 and anti-P2 Abs exerted only a slight effect in this regard (Fig. 9A). In line with these findings, and although anti-P1 and anti-P2 Abs diminished the levels of AgLDL-induced LRP1 mRNA expression, a complete reversion of this stimulatory effect was only observed in the presence of anti-P3 Abs (Fig. 9B). Further, and in line with these real-time PCR results, Western blot analysis showed that anti-P3 Abs have a much stronger capacity than anti-P1 and anti-P2 Abs to prevent LRP1 protein up-regulation induced by AgLDL (Fig. 9C). These results are in agreement with the crucial role of domain CR9 in LRP1-mediated AgLDL uptake by human VSMCs.
FIGURE 9.

Anti-P1, anti-P2, and anti-P3 antibodies against the C-terminal half of domain CR9 block the down-regulatory effect of AgLDL on SREBP-2 gene expression and its stimulatory influence on LRP1 gene expression and protein levels in human VSMCs. hVSMCs were preincubated with Abs (at 7.5 relative concentration) for 2 h and then exposed to AgLDL or nLDL for a further 18 h. Cells were then collected in TriPure reagent. A and B, real-time PCR showing SREBP-2 (A) and LRP1 (B) mRNA expression levels. C, Western blot analysis showing LRP1 protein expression. Results are shown as mean ± S.D. (error bars) of three different experiments performed in duplicate. *, p < 0.05 versus anti-BSA Ab.
Anti-P3 Antibodies Reverse the Down-regulatory Effect of AgLDL on hVSMC Migration
Previous studies from our group have demonstrated that AgLDL decreases the migratory capacity of hVSMCs (39, 40). To study the effect of anti-cluster II Abs on this activity, we incubated unexposed and AgLDL-exposed hVSMCs to anti-P3 Abs and measured the capacity of hVSMCs to migrate and cover the scratch after 18 h. In line with all findings presented above, anti-P3 Abs fully reverted the down-regulatory effect of AgLDL on hVSMC migration (Fig. 10).
FIGURE 10.
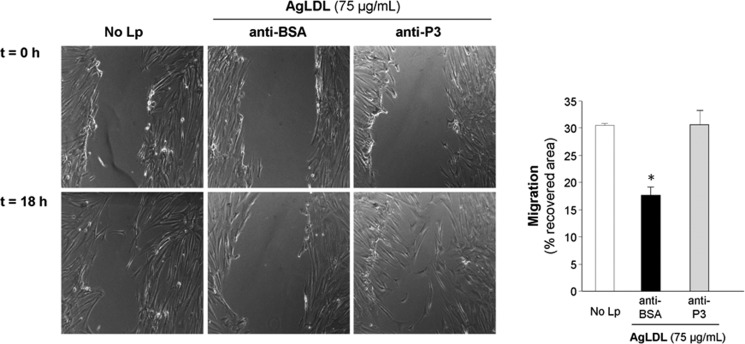
Antibodies raised against the C-terminal half of domain CR9 counteract the down-regulatory effect of AgLDL on the migration of human VSMCs. hVSMCs were preincubated with anti-P3 Abs (at 7.5 relative concentration) for 2 h and then exposed to AgLDL for a further 18 h. Phase-contrast microscopy images show the migratory capacity of hVSMCs exposed or not to AgLDL, treated with anti-BSA or anti-P3 Abs. Bar graphs show the quantitation of cell migration rate over time. Results are shown as mean ± S.D. (error bars) of two experiments performed in triplicate. *, p < 0.05 versus anti-BSA Ab.
Discussion
Our previous studies have consistently demonstrated that LRP1 binds and internalizes AgLDL in human VSMCs, causing a strong intracellular cholesteryl ester accumulation in these cells (26–30). This, in turn, induces LRP1 overexpression (34, 42, 43) and thus a positive feedback loop that efficiently transforms hVSMCs into foam cells (29, 30). These foam cells synthesize and release increased amounts of tissue factor, a key element in the prothrombotic transformation of the vascular wall and thus in the progression of atherosclerosis to thrombosis (44). Remarkably, it has been recently reported that at least 50% of the foam cells previously regarded as monocyte-derived macrophages in human atherosclerotic plaques originate, in fact, from VSMCs (35). Taken together, these findings suggest that generation of hVSMC-derived foam cells through LRP1-mediated AgLDL uptake is a key mechanism for vascular lipid deposition in atherosclerosis.
The LRP1 region(s) that participate in AgLDL binding have remained unknown to date. In the present study, we demonstrate that AgLDL primarily binds to LRP1 cluster II. In particular, the region Gly1127–Cys1140 that spans the C-terminal half of domain CR9 seems to be crucial for AgLDL binding and uptake because antibodies raised against this peptide strongly impaired intracellular cholesteryl ester accumulation caused by AgLDL. Similar to other members of the family, LRP1 cluster II contains eight CRs that are flanked by EGF and β-propeller modules. It is known that CRs in the second and fourth clusters of LRP1 are the most active modules in terms of ligand binding (45), whereas the EGF-like domains and the β-propeller possess a discrete function in ligand binding but are essential for the dissociation of ligands in endosomes (46). The beds-in-a-string arrangement of CR modules in each minireceptor, and in particular in clusters II and IV, seems to be essential for adopting dissimilar quaternary structures and thus creating various ligand binding platforms. This feature, in turn, might explain the wide variety of fully unrelated proteins and protein complexes that are capable of interacting with these clusters, including both free and serpin-bound proteinases (e.g. ADAMTS4 and ADAMTS5 (47) and the uPA·PAI complex (48, 49); the serum pan-proteinase inhibitor, α2-macroglobulin (50, 51); apoE (52); and lipoprotein lipase (53).
We have previously shown that native LDL is taken up through the classical LDL receptor in hVSMCs (26) but does not appear to bind LRP1. Why then is AgLDL able to interact with this receptor? Possibly, a modification of the apoB protein during aggregation exposes a cryptic binding site, a “neoepitope,” on the AgLDL surface that is specifically recognized by LRP1 cluster II. This neoepitope might arise due to covalent protein modifications and/or local protein unfolding or through formation of dimers or larger apoB aggregates that are not found in native LDL. It has been reported that LRP1 plays no role in the metabolism of apoB-100-containing lipoproteins (54). In this regard, our previous results indicate that apoB-100 in AgLDL is not degraded in lysosomes, as is the case with the native lipoprotein (29). These results support the interaction of LRP1 with a new AgLDL-specific neoepitope.
In our hands, AgLDL bound specifically to the Gly1127–Cys1140 peptide from LRP1 but neither to the whole CR4/CR8 tandem nor to other exposed epitopes within this region (Cys1051–Glu1066 and Asp1090–Cys1104). These findings indicate that cluster II domain CR9 and in particular its C-terminal stretch are critical for AgLDL binding to LRP1. In line with these biochemical results, functional cellular assays in hVSMCs showed that antibodies directed against the CR9 peptide more efficiently inhibited intracellular AgLDL-derived CE accumulation than did those raised with the CR7/CR8 peptides. We must stress, however, that both anti-P1 and anti-P2 Abs also exerted a partial inhibitory effect on intracellular AgLDL-derived CE accumulation although to a lesser extent. The results obtained using a mixture of the three developed Abs indicate that anti-P1 and anti-P2 antibodies sterically interfere with AgLDL-CR9 interactions and/or “freeze” a receptor conformation that is not competent for lipoprotein binding. Further studies are required to define the specific molecular interactions that are ultimately responsible for AgLDL recognition and the mechanism of internalization. In addition to highly efficient blocking of AgLDL-derived intracellular CE accumulation and lipid droplet formation, the described anti-cluster II Abs also efficiently abolished the down-regulatory effect of AgLDL on SREBP-2 mRNA expression and its stimulatory influence on LRP1 expression. In agreement with our previous studies (34, 42, 43), AgLDL modulated both SREBP-2 and LRP1 mRNA levels in hVSMCs, but this effect was completely reversed by anti-P3 Abs.
Similar to all previously characterized ligands of the LDL receptor family, AgLDL binding was prevented by the universal chaperone, RAP (55–59). Inspection of the structure of the CR5/CR6 tandem bound to RAP domain 1 (8) and sequence comparisons reveal that residues involved in receptor-chaperone interaction are conserved or conservatively replaced in CR9, explaining our experimental findings (Fig. 11). These sequence analyses also reveal a unique cluster of acidic residues in the C-terminal half of module CR9, which may be responsible for the specific recognition of AgLDL. Residues Glu1132, Asp1133, Glu1138, and Glu1141 are thus likely to play a particularly relevant role in this regard.
FIGURE 11.
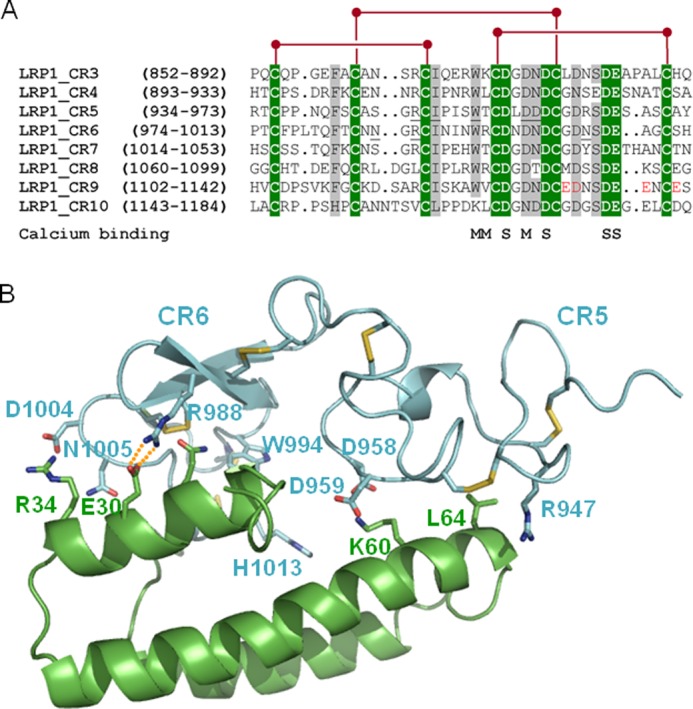
Common and unique sequence features of domain CR9. A, sequence alignment of CR modules from human LRP1 cluster II. Strictly conserved residues are white with green shading, and other well conserved or conservatively replaced residues are shaded gray. Disulfide bridges are shown above, and residues contributing main (M) or side chain oxygen atoms (S) to Ca2+ coordination are indicated below the alignment. Some residues from domains CR5 and CR6 involved in important interactions with RAP are underlined. The residues that form the unique acidic cluster in the C-terminal half of domain CR9 are highlighted in red. B, three-dimensional representation of the complex between the CR5 and CR6 repeats and RAP domain d1. LRP1 domains are given as a blue and RAP as a green schematic. Some interface residues are shown with all of their non-hydrogen atoms, color-coded. Note the presence of exposed salt bridges at the receptor-chaperone interface, in particular between Arg988 (CR6) and the acidic Glu30 in RAP.
Our current results open new avenues for the design of molecules able to specifically inhibit the positive feedback loop that efficiently transforms human VSMCs into foam cells, without or with only minimal side effects (summarized in Fig. 12). VSMCs play a crucial role in the formation of a fibrous cap that stabilizes the atherosclerotic plaque. This is especially important in lipid-enriched atherosclerotic plaques (60, 61). Our results indicate that anti-P3 Abs preserve the migratory capacity of hVSMCs in the presence of AgLDL, suggesting that these antibodies are potentially useful to prevent the rupture of atherosclerotic plaques and thus the progression of atherosclerosis to thrombosis. Remarkably, AgLDL is the first ligand reported to interact with a unique LRP1 domain, CR9, which seems to exclusively recognize aggregated lipoproteins. Along these lines, it is noteworthy that the CR8/CR9 tandem is the only pair of consecutive CR modules from LRP1 cluster II that shows only negligible affinity for serpins PAI-1 and PN1 (62). This makes CR9 an ideal target to counteract direct LRP1-AgLDL interactions without altering other physiologically essential receptor functions. Therefore, the peptides and Abs described in this work may be used to prevent not only VSMC foam cell formation in atherosclerotic lesions but also to retard the evolution of atherosclerosis to thrombosis. In this regard, the inhibition of macrophage foam cell formation has been shown to retard atherosclerosis progression (63–65). We expect beneficial anti-atherosclerotic effects of impairing hVSMC foam cell formation because VSMCs play a major role in the maintenance of vascular integrity and contractility. Interestingly, the administration of a domain from LRP1 cluster IV significantly reduced brain Aβ levels in a mouse model of Alzheimer disease (66). Our results open the possibility of a similar therapeutic approach (e.g. by using P3 peptides or peptidomimetics) for treating atherosclerosis.
FIGURE 12.
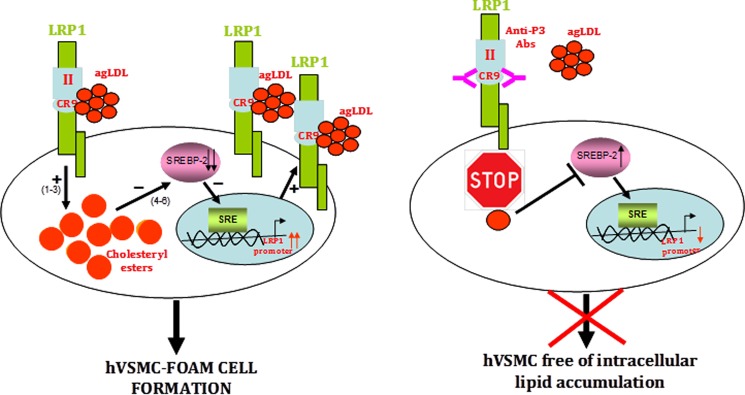
Schematic representation of the mechanism of AgLDL-mediated foam cell formation from human coronary VSMCs and modulation by anti-cluster II antibodies. The figure summarizes our current knowledge on the feedback loop of AgLDL uptake/LRP1 up-regulation/more AgLDL uptake that critically and efficiently contributes to foam cell formation from coronary hVSMCs. To the left, AgLDL are recognized by human VSMCs through LRP1 cluster II, with a particularly relevant role played by domain CR9. Uptake of bound AgLDL results in the accumulation of cholesteryl esters (1–3), which in turn causes the positive modulation of LRP1 promoter through SREBP-2 decay (4–6). To the right, antibodies directed to CR9 are able to interfere with the binding of aggregated LDL, thus effectively inhibiting downstream signaling events.
This work was supported by FIS PI11/00747 and FIS PI14/01729 from Instituto Salud Carlos III, co-financed by the European Fund for Regional Development, by SAF2010-15668, and by Red de Investigación Cardiovascular (RD RD12/0042/0027).
- LRP1
- low density lipoprotein receptor-related protein 1
- Ab
- antibody
- AgLDL
- aggregated LDL
- CE
- cholesteryl esters
- FC
- free cholesterol
- nLDL
- native LDL
- SREBP-2
- sterol regulatory element-binding protein 2
- VSMC
- vascular smooth muscle cell
- hVSMC
- human VSMC
- DiI
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanineperchlorate
- CR
- complement-type repeat
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Herz J., Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hussain M. M. (2001) Structural, biochemical and signaling properties of the low-density lipoprotein receptor gene family. Front. Biosci. 6, D417–D428 [DOI] [PubMed] [Google Scholar]
- 3. Herz J., Kowal R. C., Goldstein J. L., Brown M. S. (1990) Proteolytic processing of the 600 kd low density lipoprotein receptor-related protein (LRP) occurs in a trans-Golgi compartment. EMBO J. 9, 1769–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Obermoeller-McCormick L. M., Li Y., Osaka H., FitzGerald D. J., Schwartz A. L., Bu G. (2001) Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J. Cell Sci. 114, 899–908 [DOI] [PubMed] [Google Scholar]
- 5. Trommsdorff M., Borg J. P., Margolis B., Herz J. (1998) “Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein.” J. Biol. Chem. 273, 33556–33560 [DOI] [PubMed] [Google Scholar]
- 6. Simonovic M., Dolmer K., Huang W., Strickland D. K., Volz K., Gettins P. G. (2001) Calcium coordination and pH dependence of the calcium affinity of ligand-binding repeat CR7 from the LRP: comparison with related domains from the LRP and the LDL receptor. Biochemistry 40, 15127–15134 [DOI] [PubMed] [Google Scholar]
- 7. Dolmer K., Huang W., Gettins P. G. (2000) NMR solution structure of complement-like repeat CR3 from the low density lipoprotein receptor-related protein. Evidence for specific binding to the receptor binding domain of human α2-macroglobulin. J. Biol. Chem. 275, 3264–3269 [DOI] [PubMed] [Google Scholar]
- 8. Jensen G. A., Andersen O. M., Bonvin A. M., Bjerrum-Bohr I., Etzerodt M., Thøgersen H. C., O'Shea C., Poulsen F. M., Kragelund B. B. (2006) Binding site structure of one LRP-RAP complex: implications for a common ligand-receptor binding motif. J. Mol. Biol. 362, 700–716 [DOI] [PubMed] [Google Scholar]
- 9. Guttman M., Prieto J. H., Handel T. M., Domaille P. J., Komives E. A. (2010) Structure of the minimal interface between ApoE and LRP. J. Mol. Biol. 398, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rudenko G., Henry L., Henderson K., Ichtchenko K., Brown M. S., Goldstein J. L., Deisenhofer J. (2002) Structure of the LDL receptor extracellular domain at endosomal pH. Science 298, 2353–2358 [DOI] [PubMed] [Google Scholar]
- 11. May P., Woldt E., Matz R. L., Boucher P. (2007) The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann. Med. 39, 219–228 [DOI] [PubMed] [Google Scholar]
- 12. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., Strickland D. K. (2008) LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rohlmann A., Gotthardt M., Hammer R. E., Herz J. (1998) Inducible inactivation of hepatic LRP gene by Cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J. Clin. Invest. 101, 689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boucher P., Gotthardt M., Li W. P., Anderson R. G., Herz J. (2003) LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332 [DOI] [PubMed] [Google Scholar]
- 15. Weaver A. M., Hussaini I. M., Mazar A., Henkin J., Gonias S. L. (1997) Embryonic fibroblasts that are genetically deficient in low density lipoprotein receptor-related protein demonstrate increased activity of the urokinase receptor system and accelerated migration on vitronectin. J. Biol. Chem. 272, 14372–14379 [DOI] [PubMed] [Google Scholar]
- 16. Emonard H., Bellon G., de Diesbach P., Mettlen M., Hornebeck W., Courtoy P. J. (2005) Regulation of matrix metalloproteinase (MMP) activity by the low-density lipoprotein receptor-related protein (LRP): a new function for an “old friend”. Biochimie 87, 369–376 [DOI] [PubMed] [Google Scholar]
- 17. Langlois B., Perrot G., Schneider C., Henriet P., Emonard H., Martiny L., Dedieu S. (2010) LRP-1 promotes cancer cell invasion by supporting ERK and inhibiting JNK signaling pathways. PLoS One 5, e11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. May P., Rohlmann A., Bock H. H., Zurhove K., Marth J. D., Schomburg E. D., Noebels J. L., Beffert U., Sweatt J. D., Weeber E. J., Herz J. (2004) Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol. Cell. Biol. 24, 8872–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bu G. (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y., Lu W., Bu G. (2003) Essential role of the low density lipoprotein receptor-related protein in vascular smooth muscle cell migration. FEBS Lett. 555, 346–350 [DOI] [PubMed] [Google Scholar]
- 21. Boucher P., Gotthardt M. (2004) LRP and PDGF signaling: a pathway to atherosclerosis. Trends Cardiovasc. Med. 14, 55–60 [DOI] [PubMed] [Google Scholar]
- 22. Hiltunen T. P., Luoma J. S., Nikkari T., Ylä-Herttuala S. (1998) Expression of LDL receptor, VLDL receptor, LDL receptor-related protein, and scavenger receptor in rabbit atherosclerotic lesions: marked induction of scavenger receptor and VLDL receptor expression during lesion development. Circulation 97, 1079–1086 [DOI] [PubMed] [Google Scholar]
- 23. Moestrup S. K., Gliemann J., Pallesen G. (1992) Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 269, 375–382 [DOI] [PubMed] [Google Scholar]
- 24. Luoma J., Hiltunen T., Särkioja T., Moestrup S. K., Gliemann J., Kodama T., Nikkari T., Ylä-Herttuala S. (1994) Expression of α2-macroglobulin receptor/low density lipoprotein receptor-related protein and scavenger receptor in human atherosclerotic lesions. J. Clin. Invest. 93, 2014–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Llorente-Cortes V., Otero-Viñas M., Berrozpe M., Badimon L. (2004) Intracellular lipid accumulation, low-density lipoprotein receptor-related protein expression, and cell survival in vascular smooth muscle cells derived from normal and atherosclerotic human coronaries. Eur. J. Clin. Invest. 34, 182–190 [DOI] [PubMed] [Google Scholar]
- 26. Llorente-Cortes V., Martínez-González J., Badimon L. (2000) LDL receptor-related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 20, 1572–1579 [DOI] [PubMed] [Google Scholar]
- 27. Llorente-Cortés V., Otero-Viñas M., Badimon L. (2002) Differential role of heparan sulfate proteoglycans on aggregated LDL uptake in human vascular smooth muscle cells and mouse embryonic fibroblasts. Arterioscler. Thromb. Vasc. Biol. 22, 1905–1911 [DOI] [PubMed] [Google Scholar]
- 28. Llorente-Cortés V., Otero-Viñas M., Hurt-Camejo E., Martínez-González J., Badimon L. (2002) Human coronary smooth muscle cells internalize versican-modified LDL through LDL receptor-related protein and LDL receptors. Arterioscler. Thromb. Vasc. Biol. 22, 387–393 [DOI] [PubMed] [Google Scholar]
- 29. Llorente-Cortes V., Otero-Viñas M., Camino-López S., Costales P., Badimon L. (2006) Cholesteryl esters of aggregated LDL are internalized by selective uptake in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 26, 117–123 [DOI] [PubMed] [Google Scholar]
- 30. Llorente-Cortes V., Royo T., Juan-Babot O., Badimon L. (2007) Adipocyte differentiation-related protein is induced by LRP1-mediated aggregated LDL internalization in human vascular smooth muscle cells and macrophages. J. Lipid Res. 48, 2133–2140 [DOI] [PubMed] [Google Scholar]
- 31. Wyler von Ballmoos M., Dubler D., Mirlacher M., Cathomas G., Muser J., Biedermann B. C. (2006) Increased apolipoprotein deposits in early atherosclerotic lesions distinguish symptomatic from asymptomatic patients. Arterioscler. Thromb. Vasc. Biol. 26, 359–364 [DOI] [PubMed] [Google Scholar]
- 32. Oörni K., Pentikainen M. O., Ala-Korpela M., Kovanen P. T. (2000) Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J. Lipid Res. 41, 1703–1714 [PubMed] [Google Scholar]
- 33. Chait A., Wight T. N. (2000) Interaction of native and modified low-density lipoproteins with extracellular matrix. Curr. Opin. Lipidol. 11, 457–463 [DOI] [PubMed] [Google Scholar]
- 34. Llorente-Cortés V., Otero-Viñas M., Sánchez S., Rodríguez C., Badimon L. (2002) Low-density lipoprotein upregulates low-density lipoprotein receptor-related protein expression in vascular smooth muscle cells: possible involvement of sterol regulatory element binding protein-2-dependent mechanism. Circulation 106, 3104–3110 [DOI] [PubMed] [Google Scholar]
- 35. Allahverdian S., Chehroudi A. C., McManus B. M., Abraham T., Francis G. A. (2014) Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129, 1551–1559 [DOI] [PubMed] [Google Scholar]
- 36. Bu G., Rennke S. (1996) Receptor-associated protein is a folding chaperone for low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 22218–22224 [DOI] [PubMed] [Google Scholar]
- 37. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 38. Huber L. A., Xu Q. B., Jürgens G., Böck G., Bühler E., Gey K. F., Schönitzer D., Traill K. N., Wick G. (1991) Correlation of lymphocyte lipid composition membrane microviscosity and mitogen response in the aged. Eur. J. Immunol. 21, 2761–2765 [DOI] [PubMed] [Google Scholar]
- 39. Otero-Viñas M., Llorente-Cortés V., Peña E., Padró T., Badimon L. (2007) Aggregated low density lipoproteins decrease metalloproteinase-9 expression and activity in human coronary smooth muscle cells. Atherosclerosis 194, 326–333 [DOI] [PubMed] [Google Scholar]
- 40. Revuelta-López E., Castellano J., Roura S., Gálvez-Montón C., Nasarre L., Benitez S., Bayes-Genis A., Badimon L., Llorente-Cortés V. (2013) Hypoxia induces metalloproteinase-9 activation and human vascular smooth muscle cells migration through low-density lipoprotein receptor-related protein 1-mediated Pyk2 phosphorylation. Arterioscler. Thromb. Vasc. Biol. 33, 2877–2887 [DOI] [PubMed] [Google Scholar]
- 41. Llorente-Cortés V., Martínez-González J., Badimon L. (1998) Esterified cholesterol accumulation induced by aggregated LDL uptake in human vascular smooth muscle cells is reduced by HMG-CoA reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 18, 738–746 [DOI] [PubMed] [Google Scholar]
- 42. Llorente-Cortés V., Costales P., Costales P., Bernués J., Camino-Lopez S., Badimon L. (2006) Sterol regulatory element-binding protein-2 negatively regulates low density lipoprotein receptor-related protein transcription. J. Mol. Biol. 359, 950–960 [DOI] [PubMed] [Google Scholar]
- 43. Costales P., Aledo R., Vérnia S., Das A., Shah V. H., Casado M., Badimon L., Llorente-Cortés V. (2010) Selective role of sterol regulatory element binding protein isoforms in aggregated LDL-induced vascular low density lipoprotein receptor-related protein-1 expression. Atherosclerosis 213, 458–468 [DOI] [PubMed] [Google Scholar]
- 44. Llorente-Cortés V., Otero-Viñas M., Camino-López S., Llampayas O., Badimon L. (2004) Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation 110, 452–459 [DOI] [PubMed] [Google Scholar]
- 45. Neels J. G., van Den Berg B. M., Lookene A., Olivecrona G., Pannekoek H., van Zonneveld A. J. (1999) The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J. Biol. Chem. 274, 31305–31311 [DOI] [PubMed] [Google Scholar]
- 46. Davis C. G., Goldstein J. L., Südhof T. C., Anderson R. G., Russell D. W., Brown M. S. (1987) Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature 326, 760–765 [DOI] [PubMed] [Google Scholar]
- 47. Yamamoto K., Owen K., Parker A. E., Scilabra S. D., Dudhia J., Strickland D. K., Troeberg L., Nagase H. (2014) Low density lipoprotein receptor-related protein 1 (LRP1)-mediated endocytic clearance of a disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4): functional differences of non-catalytic domains of ADAMTS-4 and ADAMTS-5 in LRP1 binding. J. Biol. Chem. 289, 6462–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nykjaer A., Kjoller L., Cohen R. L., Lawrence D. A., Garni-Wagner B. A., Todd R. F., 3rd, van Zonneveld A. J., Gliemann J., Andreasen P. A. (1994) Regions involved in binding of urokinase-type-1 inhibitor complex and pro-urokinase to the endocytic α2-macroglobulin receptor/low density lipoprotein receptor-related protein: evidence that the urokinase receptor protects pro-urokinase against binding to the endocytic receptor. J. Biol. Chem. 269, 25668–25676 [PubMed] [Google Scholar]
- 49. Andersen O. M., Petersen H. H., Jacobsen C., Moestrup S. K., Etzerodt M., Andreasen P. A., Thøgersen H. C. (2001) Analysis of a two-domain binding site for the urokinase-type plasminogen activator-plasminogen activator inhibitor-1 complex in low-density-lipoprotein-receptor-related protein. Biochem. J. 357, 289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nielsen K. L., Holtet T. L., Etzerodt M., Moestrup S. K., Gliemann J., Sottrup-Jensen L., Thogersen H. C. (1996) Identification of residues in α-macroglobulins important for binding to the α2-macroglobulin receptor/low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 12909–12912 [DOI] [PubMed] [Google Scholar]
- 51. Andersen O. M., Christensen P. A., Christensen L. L., Jacobsen C., Moestrup S. K., Etzerodt M., Thogersen H. C. (2000) Specific binding of α-macroglobulin to complement-type repeat CR4 of the low-density lipoprotein receptor-related protein. Biochemistry 39, 10627–10633 [DOI] [PubMed] [Google Scholar]
- 52. Küchenhoff A., Harrach-Ruprecht B. (1997) Interaction of apo E-containing lipoproteins with the LDL receptor-related protein LRP. Am. J. Physiol. 272, C369–C382 [DOI] [PubMed] [Google Scholar]
- 53. Williams S. E., Inoue I., Tran H., Fry G. L., Pladet M. W., Iverius P. H., Lalouel J. M., Chappell D. A., Strickland D. K. (1994) The carboxyl-terminal domain of lipoprotein lipase binds to the low density lipoprotein receptor-related protein/α2-macroglobulin receptor (LRP) and mediates binding of normal very low density lipoproteins to LRP. J. Biol. Chem. 269, 8653–8658 [PubMed] [Google Scholar]
- 54. Véniant M. M., Zlot C. H., Walzem R. L., Pierotti V., Driscoll R., Dichek D., Herz J., Young S. G. (1998) Lipoprotein clearance mechanisms in LDL receptor-deficient “Apo-B48-only” and “Apo-B100-only” mice. J. Clin. Invest. 102, 1559–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nykjaer A., Petersen C. M., Møller B., Jensen P. H., Moestrup S. K., Holtet T. L., Etzerodt M., Thøgersen H. C., Munch M., Andreasen P. A. (1992) Purified α2-macroglobulin receptor/LDL receptor-related protein binds urokinase·plasminogen activator inhibitor type-1 complex: evidence that the α2-macroglobulin receptor mediates cellular degradation of urokinase-receptor bound complexes. J. Biol. Chem. 267, 14543–14546 [PubMed] [Google Scholar]
- 56. Mikhailenko I., Battey F. D., Migliorini M., Ruiz J. F., Argraves K., Moayeri M., Strickland D. K. (2001) Recognition of α2-macroglobulin by the low density lipoprotein receptor-related protein requires the cooperation of two ligand binding cluster regions. J. Biol. Chem. 276, 39484–39491 [DOI] [PubMed] [Google Scholar]
- 57. Moestrup S. K., Gliemann J. (1991) Analysis of ligand recognition by the purified α2-macroglobulin receptor (low density lipoprotein receptor-related protein): evidence that high affinity of α2-macroglobulin-proteinase complex is achieved by binding to adjacent receptors. J. Biol. Chem. 266, 14011–14017 [PubMed] [Google Scholar]
- 58. Orth K., Madison E. L., Gething M. J., Sambrook J. F., Herz J. (1992) Complexes of tissue-type plasminogen activator and its serpin inhibitor plasminogen-activator inhibitor type 1 are internalized by means of the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. Proc. Natl. Acad. Sci. U.S.A. 89, 7422–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Warshawsky I., Bu G., Schwartz A. L. (1995) Sites within the 39-kDa protein important for regulating ligand binding to the low-density lipoprotein receptor-related protein. Biochemistry 34, 3404–3415 [DOI] [PubMed] [Google Scholar]
- 60. Newby A. C. (2007) Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 17, 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnson J. L., Dwivedi A., Somerville M., George S. J., Newby A. C. (2011) Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler. Thromb. Vasc. Biol. 31, e35–e44 [DOI] [PubMed] [Google Scholar]
- 62. Jensen J. K., Dolmer K., Gettins P. G. (2009) Specificity of binding of the low density lipoprotein receptor-related protein to different conformational states of the clade E serpins plasminogen activator inhibitor-1 and proteinase nexin-1. J. Biol. Chem. 284, 17989–17997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rousselle A., Qadri F., Leukel L., Yilmaz R., Fontaine J. F., Sihn G., Bader M., Ahluwalia A., Duchene J. (2013) CXCL5 limits macrophage foam cell formation in atherosclerosis. J. Clin. Invest. 123, 1343–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Khan O. M., Akula M. K., Skålen K., Karlsson C., Ståhlman M., Young S. G., Borén J., Bergo M. O. (2013) Targeting GGTase-I activates RHOA, increases macrophage reverse transport, and reduces atherosclerosis in mice. Circulation 127, 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao J. F., Ching L. C., Huang Y. C., Chen C. Y., Chiang A. N., Kou Y. R., Shyue S. K., Lee T. S. (2012) Molecular mechanism of curcumin on the suppression of cholesterol accumulation in macrophage foam cells and atherosclerosis. Mol. Nutr. Food Res. 56, 691–701 [DOI] [PubMed] [Google Scholar]
- 66. Sagare A., Deane R., Bell R. D., Johnson B., Hamm K., Pendu R., Marky A., Lenting P. J., Wu Z., Zarcone T., Goate A., Mayo K., Perlmutter D., Coma M., Zhong Z., Zlokovic B. V. (2007) Clearance of amyloid-β by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]