

Background: Oxidative stress leads to increased PRMT1 expression and ADMA accumulation.
Results: PRMT1 activity is increased by reductants and decreased by oxidants.
Conclusion: PRMT1 activity is regulated in a redox-sensitive manner.
Significance: The role of PRMT1 activity in the oxidative stress response may be more complex than previously thought.
Keywords: endothelial dysfunction, hydrogen peroxide, posttranslational modification (PTM), redox regulation, oxidative stress, protein arginine methyltransferase, sulfenic acid, ADMA, PRMT1, arginine methylation
Abstract
Elevated levels of asymmetric dimethylarginine (ADMA) correlate with risk factors for cardiovascular disease. ADMA is generated by the catabolism of proteins methylated on arginine residues by protein arginine methyltransferases (PRMTs) and is degraded by dimethylarginine dimethylaminohydrolase. Reports have shown that dimethylarginine dimethylaminohydrolase activity is down-regulated and PRMT1 protein expression is up-regulated under oxidative stress conditions, leading many to conclude that ADMA accumulation occurs via increased synthesis by PRMTs and decreased degradation. However, we now report that the methyltransferase activity of PRMT1, the major PRMT isoform in humans, is impaired under oxidative conditions. Oxidized PRMT1 displays decreased activity, which can be rescued by reduction. This oxidation event involves one or more cysteine residues that become oxidized to sulfenic acid (-SOH). We demonstrate a hydrogen peroxide concentration-dependent inhibition of PRMT1 activity that is readily reversed under physiological H2O2 concentrations. Our results challenge the unilateral view that increased PRMT1 expression necessarily results in increased ADMA synthesis and demonstrate that enzymatic activity can be regulated in a redox-sensitive manner.
Introduction
Endothelial dysfunction plays a major role in cardiovascular disease, the leading cause of death in the United States (1). Several factors have been suggested to contribute to endothelial dysfunction, such as decreased activity and/or expression of endothelial nitric oxide synthase and/or increased vascular formation of oxygen-derived free radicals (2, 3). Asymmetric dimethylarginine (ADMA)2 is an endogenously synthesized inhibitor of NOS that has been gaining increased attention in the cardiovascular field (2, 4–9). In the heart, ADMA and other NOS inhibitors cause reduced heart rate and cardiac output (10, 11). Interestingly, in addition to decreasing levels of nitric oxide, evidence suggests that ADMA may also uncouple NOS (conditions under which NOS generates superoxide anion), therefore increasing oxidative stress and inducing additional endothelial dysfunction (7, 12). Furthermore, ADMA has been shown to increase endothelial oxidative stress and up-regulate the expression of redox-sensitive genes that encode endothelial adhesion molecules (13), increasing the propensity for plaque buildup. Taken together, the available data indicate that ADMA levels represent a risk factor for the development of endothelial dysfunction.
ADMA is generated through the degradation of cellular proteins containing asymmetrically dimethylated arginine residues (Fig. 1). Arginine residues in certain proteins can be modified by the addition of one or two methyl groups. This modification is catalyzed by the protein arginine methyltransferase (PRMT) family of enzymes. Nine human PRMT isoforms can be subdivided into three types, determined by their final methylation products. Type 1 PRMTs (such as PRMT1) form monomethylarginine (MMA) and/or ADMA and represent the majority of identified PRMTs. Type 2 PRMTs form MMA and/or symmetric dimethylarginine, and type 3 PRMTs produce only MMA (14). Each of the methylated arginine products (MMA, ADMA, and symmetric dimethylarginine) can induce different biological responses in the cell. However, only MMA and ADMA are inhibitors of NOS activity (9, 14). ADMA in the body is metabolized by dimethylarginine dimethylaminohydrolase (DDAH) to citrulline and dimethylamine (15). Therefore, the amount of free ADMA at any given time is a reflection of PRMT, proteasome, and DDAH activities.
FIGURE 1.
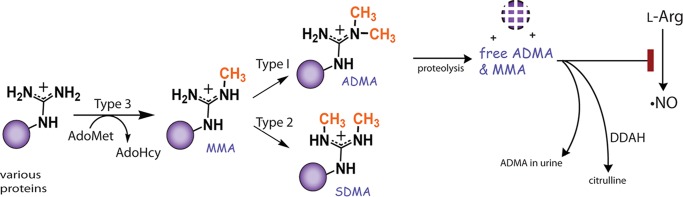
ADMA formation and degradation. PRMTs transfer a methyl group from donor AdoMet to arginine residues in substrate proteins. Type 1 PRMTs can transfer one or two methyl groups to the same-terminal guanidino nitrogen-producing MMA or ADMA, respectively. Upon degradation of the methylated proteins, free MMA and ADMA inhibit NO synthesis by acting as competitive inhibitors of nitric oxide synthase. Free ADMA is catabolized by DDAH to citrulline and dimethylamine or excreted in urine.
Evidence has shown that, under oxidative stress, a condition linked to a variety of disease states, free ADMA levels are increased. In many instances, these studies also showed that PRMT1 RNA or protein expression is increased and DDAH activity is decreased (11, 16–20). This has led many to conclude that the increased expression of PRMT1 protein results in increased protein methylation, giving rise to a larger pool of the precursors for free ADMA (6, 18, 21). Although it is clear that elevated ADMA levels are connected to oxidative stress, the assumption that increased levels of PRMT1 protein expression are directly responsible for increased free ADMA production has not been validated. In fact, a recent report showed a significant reduction in the production of ADMA-containing polypeptides in both replicative and H2O2-induced premature senescent cells (22). To clarify the role of PRMT1 in free ADMA accumulation under oxidative stress conditions, we set out to investigate whether oxidative conditions affect PRMT1 catalytic activity.
Here we report that PRMT1, the major human PRMT isoform, is susceptible to oxidation. Oxidized rat PRMT1, which differs from human PRMT1 at just one residue, is characterized by impaired enzymatic activity that can be rescued by reduction. We demonstrate a reversible, concentration-dependent inhibition of PRMT1 activity by H2O2. Furthermore, we show that this oxidation event involves at least two cysteines that are oxidized to sulfenic acid (-SOH). Our results provide the first direct evidence that PRMT1 enzymatic activity can be regulated in a redox-sensitive manner.
Experimental Procedures
Reagents
AdoMet was purchased from Sigma as a chloride salt (≥80%, from yeast). [3H]AdoMet (83.1 Ci/mmol) was purchased from PerkinElmer Life Sciences. R3 peptide (acGGRGGFGGRGGFGGRGGRG) was synthesized by the Keck Institute (Yale University) and purified to ≥95%. The lyophilized peptide was dissolved in water, and its concentration was determined by mass or by UV spectroscopy (ϵ280 nm = 5690 M−1cm−1). Bulk histones from calf thymus were purchased from Sigma. Histone 4 protein was purchased from New England Biolabs. ZipTip®C4/C18 pipette tips were purchased from Millipore. The DCP-Rho1 sulfenic acid probe was purchased from Kerafast, and the 5IAF thiol-specific probe was purchased from Life Technologies.
Plasmid Generation
The His6-ratPRMT1 plasmid (HisPRMT1) has been generated previously (23). To create a construct with a cleavable His6 tag, a DNA segment coding for a tobacco etch virus (TEV) cleavage site was designed with both N- and C-terminal NdeI restriction sites. The NdeI restriction enzyme was used to cut open the vector at an NdeI site between the His6 tag and the enzyme coding sequence, and the designed TEV insert was ligated using the Quick LigationTM kit (New England Biolabs) to form the His6-TEV-ratPRMT1 plasmid (HisTevPRMT1). The C101S, C342S, C254S, C208S, and C101S/C208S variants were generated using the HisTevPRMT1 plasmid (or the confirmed HisTevPRMT1 C101S plasmid for C101S/C208S) as a PCR template for site-directed mutagenesis using the QuikChange Lightning kit (Stratagene) and sets of complementary oligonucleotide primers spanning the desired site of mutation. To replace all 11 cysteine residues of PRMT1 with serines and create HisTevPRMT1Δcys (or cys-), properly coded DNA with N-terminal NdeI and C-terminal BamHI restriction sites was ordered from GenScript. The PRMT1 sequence in His6-TEV-PRMT1 was excised with NdeI/BamH1 and replaced with the synthetic PRMT1Δcys insert. The cysteineless with Cys101 reintroduced (cys-Cys101), cysteineless with Cys208 reintroduced (cys-Cys208), and cysteineless with both Cys101 and Cys208 reintroduced (cys-Cys101Cys208) variants were generated using the HisTevPRMT1Δcys plasmid as a PCR template for site-directed mutagenesis using the QuikChange Lightning kit (Stratagene) and sets of complementary oligonucleotide primers spanning the desired site of mutation. All plasmids were transformed using the Escherichia coli DH5α cell line. Plasmids were purified (Qiagen plasmid mini kit) and sequenced to confirm the correctness of the open reading frame prior to protein expression.
Recombinant Protein Expression and Purification
Full-length His-tagged PRMT1 (residues 1–353) was expressed and purified as described previously (23). Briefly, E. coli BL21 cells were transformed with the above constructs and grown on Luria Broth medium/kanamycin agar plates. Selected colonies were grown in LB broth to A600 = 0.6 and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 24 h at 25 °C. Cell pellets were resuspended in lysis/wash buffer (50 mm sodium phosphate (pH 7.6) and 20 mm imidazole), sonicated, and centrifuged at 47,000 × g for 20 min at 4 °C. The clarified supernatant was incubated with nickel-Sepharose 6 Fast Flow resin (Qiagen), rotating for 2 h at 4 °C. The binding reaction was pelleted at 700 × g; the supernatant was discarded; and the resin was washed four times with lysis/wash buffer, seven times with wash buffer containing 70 mm imidazole, and then eluted in six washes with 250 mm imidazole buffer. The elutions were pooled and dialyzed into storage buffer (50 mm sodium phosphate (pH 7.6) and 10% glycerol), concentrated to more than 1 mg/ml, and beaded in liquid nitrogen for storage at −80 °C. To generate cleaved constructs, half of the His6-TEV enzymes were put into cleavage buffer (50 mm sodium phosphate, 1 mm DTT, 2 mm EDTA, and 5% glycerol). The TEV enzyme was added and allowed to cleave overnight. The cleaved enzyme was dialyzed into storage buffer and reincubated with nickel resin (removal of TEV and remaining His6 tag), and then the subsequent supernatant was dialyzed, concentrated, and stored in the same way as the His6-tagged elutions. Enzyme cleavage and purity (>90%) were assessed during and after purification using SDS-page. pET21a TbPRMT7 and pET28b human PRMT6 were purified as PRMT1. pET28a human PRMT3 (residues 211–531) was grown and purified as described in Wang et al. (24). Protein concentrations were determined by UV spectroscopy (PRMT1 ϵ280 nm = 54,945 M−1cm−1, PRMT3 ϵ280 nm = 37,735 M−1cm−1, PRMT7 ϵ280 nm = 37,150 M−1cm−1, and PRMT6 ϵ280 nm = 59,040 M−1cm−1) and by Bradford assay with bovine serum albumin as a standard.
Kinetic Assays of PRMT1 Constructs
Conditions of methylation reactions were as published previously (23). Briefly, enzyme activity was assessed at 37 °C in assays containing 100 nm PRMT1, 2 μm AdoMet (1 μm [3H]AdoMet), 0.38 μm bovine serum albumin, 100 nm AdoHcy nucleosidase (MTAN, purified as described in Ref. 25) and initiated with 200 μm peptide substrate or 2 μm protein substrate. Reactions in the presence of DTT were performed by preincubation of the reaction with 1 mm DTT for 10 min at 4 °C prior to initiation with substrate. At different time points, 5-μl samples were removed from reactions and quenched with 6 μl of 8 m guanidinium hydrochloride. Samples were processed with ZipTip®C4/C18 pipette tips (Millipore) (for protein or peptide substrates, respectively) to separate the unreacted [3H]AdoMet and the radiolabeled product (26). Time-dependent incorporation of the radiolabel into substrates was quantified using a scintillation counter (Beckman Coulter). Methyltransferase activity for hPRMT3, hPRMT6, and TbPRMT7 was performed as above, except 1 mm bulk histones (Sigma) and 1 mm Histone 4 protein (New England Biolabs) were used as substrates for hPRMT6 and TbPRMT7, respectively.
Conditions for testing the effect of hydrogen peroxide on enzymatic activity have been published previously (27) and were modified as follows. PRMT1 was prereduced in 1 mm DTT for 2 h on ice and then rapidly desalted on a 7-kDa ZebaTM spin desalting column (Thermo Scientific). PRMT1 (2 μm final concentration) was oxidized with 0, 0.4, 4, 40, 200, 400, or 800 μm H2O2 for 10 min at 37 °C and then incubated with catalase (300 units/ml) for 1 min at 37 °C. The mixture was then kept on ice and used for kinetic assays as above. Methyltransferase activity was unaffected by the addition of catalase (data not shown). In all cases, data reported are the average of at least three independent measurements.
Size Exclusion Chromatography
Gel filtration chromatography was performed on a SuperdexTM 200 10/300GL column (GE Healthcare) in 50 mm sodium phosphate (pH 7.6), 150 mm NaCl, and 5% glycerol at a 0.4 ml/min flow rate. Freshly purified PRMT1 variants were allowed to equilibrate overnight into gel filtration buffer with or without the addition of 1 mm DTT, 2 mm EDTA prior to examination. All constructs were analyzed by loading 300 μl of enzyme at a concentration of 10–20 μm and run at 0.4 ml/min for 75 min.
In Vitro DCP-Rho1 Labeling of Sulfenic Acid in Recombinant PRMT1
DCP-Rho1 labeling of purified PRMT1 was modified from the method described in Poole et al. (28). Briefly, 2.5 μm recombinantly expressed, air-oxidized WT PRMT1, cysteine-less PRMT1, cysteine-lessCys101, cysteine-lessCys208, or cysteine-lessCys101Cys208 enzymes were incubated with 1 mm DTT or with buffer in the presence of 6 m urea for 1 h at 22 °C prior to addition of 10 μm DCP-Rho1 for 20 min at 22 °C in a final reaction volume of 25 μl. The labeling reaction was quenched by addition of 4× SDS loading dye and boiling for 5 min. Labeled proteins were separated from unreacted label by 12% SDS-PAGE. Band quantification was performed using the Image Lab software from Bio-Rad and is reported as the average of three independent measurements.
In Vitro 5IAF Labeling of Free Thiol Content in Recombinant PRMT1
5IAF labeling of purified PRMT1 was modified from the method described in Wu et al. (29) with optimization conditions aided by Hansen et al. (30). Briefly, 2.5 μm recombinantly expressed, air-oxidized WT PRMT1, cysteine-less PRMT1, cysteine-lessCys101, cysteine-lessCys208, or cysteine-lessCys101Cys208 enzymes were incubated with 1 mm DTT or with buffer in the presence of 6 m urea for 1 h at 22 °C prior to addition of 2.5 mm 5IAF for 1 h at 22 °C in a final reaction volume of 25 μl. The labeling reaction was quenched by addition of 4× SDS loading dye and boiling for 5 min. Labeled proteins were separated from the unreacted label by 12% SDS-PAGE. Band quantification was performed using the Image Lab software from Bio-Rad and is reported as the average of three independent measurements.
LC-MS/MS Analysis of Sulfenic Acid Content in Recombinant PRMT1
120 μg of WT PRMT1 was treated with 5 mm dimedone in the presence of 6 m urea for 2 h at 22 °C. The sample was then buffer-exchanged twice into 50 mm ammonium bicarbonate to remove excess dimedone and all urea. After reduction and alkylation, the labeled sample was precipitated by stepwise addition of 6 volumes of cold acetone with continuous vortexing and then incubated overnight at −20 °C. After centrifugation at 20,000 × g for 30 min at 4 °C, the supernatant was removed, and the pellet was allowed to air-dry. Two consecutive digestions steps were employed for the on-pellet digestion. In step 1 (digestion-aided pellet dissolution), Tris buffer (50 mm (pH 8.5)) containing trypsin at an enzyme/substrate ratio of 1:40 (w/w) was added and incubated at 37 °C for 6 h with vortexing at 500 rpm in a ThermoMixer shaking incubator (Eppendorf). In step 2 (complete cleavage), another portion of trypsin solution was added at an enzyme/substrate ratio of 1:40 (w/w) in a 50-μl final volume. The mixture was incubated at 37 °C for 12 h to achieve a complete digestion. The digestion was terminated by adding 1% (v/v) formic acid and centrifuged at 20,000 × g for 30 min at 4 °C. The supernatant was used for LC-MS/MS analysis.
4 μl of the digested solution was analyzed by nanospray LC-MS/MS, which constitutes an Orbitrap Fusion Tribrid mass spectrometer with ETD (Thermo Fisher Scientific, San Jose, CA) coupled to an ultra-high-pressure Eksigent Ekspert NanoLC 425 system (Dublin, CA) with a autosampler of Eksigent NLC 400. A nano-LC/nanospray setup was used to obtain a comprehensive separation of the complex peptide mixture and sensitive detection. Mobile phases A and B were 0.1% formic acid in 1% acetonitrile and 0.1% formic acid in 88% acetonitrile, respectively. Samples were loaded onto a large ID trap (300 μm ID × 5 mm, packed with Zorbax 5 μm C18 material) with 1% B at a flow rate of 10 μl/min for 3 min. A series of nanoflow gradients was used to backflush the trapped samples onto the nano-LC column (75 μm ID × 100 cm, packed with Pepmap 3-μm C18 material). The column was heated at 52 °C to improve both chromatographic resolution and reproducibility. The gradient profile was as follows: a linear increase from 3 to 8% B over 5 min, an increase from 8 to 27% B over 117 min, an increase from 27 to 45% B over 10 min, an increase from 45 to 98% B over 20 min, and isocratic at 98% B for 20 min.
The mass spectrometry was operating under data-dependent product ion mode. A 3-s scan cycle included an MS1 scan (Orbitrap) followed by MS2 scans (dual-cell ion trap) by alternating CID and ETD activation, was programmed. The parameters used for MS and MS/MS data acquisition under the CID mode were as follows: top speed mode with 3-s cycle time; Orbitrap: scan range (m/z) = 400–1600; resolution = 120 K; AGC target = 5 × 105; maximum injection time (milliseconds) = 50; filter: precursor selection range = 400–1500; include charge state = 2–7; dynamic exclude after n times = 1, duration time, 60 s; decision: data-dependent mode: top speed, precursor priority = most intense; for CID, isolation mode = quadrupole; isolation window = 1.6; collision energy (percent) = 30; detection type: ion trap; ion trap scan rate: rapid; AGC target = 1 × 104; maximum injection time (milliseconds) = 50; microscan = 1; and for ddMS2 (ETD), ETD reaction time (milliseconds) 200; ETD reagent target 1.0 × 106; maximum ETD reagent injection time (milliseconds): 200; AGC target = 1 × 104; maximum injection time (milliseconds) = 50; microscan = 1.
CID and ETD activation spectra were processed using Peaks 7. Briefly, raw files were searched against the sequence of PRMT1 with a precursor mass tolerance of 20 ppm and peptide fragment mass tolerance of 1 Da. The static side chain modifications of carbamidomethyl (57.021) and dynamic side chain modifications of Oxidation (15.995) and dimedone (138.068) controlled the protein false discovery rates as 0.1%.
Results
PRMT1 Activity Is Reversibly Inhibited by Oxidation
The elevated levels of free ADMA that are observed under oxidative stress conditions (7) could arise from a few distinct mechanisms. Both proteasomal degradation and methyltransferase activities could affect the rate of free ADMA formation. Increased synthesis of ADMA-containing proteins (the precursors to free ADMA) could occur by increasing the expression of PRMT proteins while maintaining normal enzyme activity or by increasing the methyltransferase activity of the current pool of PRMT proteins. Because PRMT1 is responsible for ∼85% of arginine methylation in cells and is the primary source of ADMA (31), altered expression and/or activity of this isoform would be expected to contribute greatly to altering the levels of ADMA. To determine whether oxidative conditions induce any changes in PRMT1 activity, we treated fully reduced recombinant PRMT1 with hydrogen peroxide, a common cellular oxidant, and measured the resulting methyltransferase activity.
Recombinantly expressed PRMT1 was prereduced with DTT, and rapidly desalted to remove residual DTT. The reduced PRMT1 was then incubated with various concentrations of H2O2 (0.4–800 μm final concentration), followed by the addition of catalase to remove any remaining H2O2. Surprisingly, when the peroxide-treated PRMT1 was assayed, methyltransferase activity was found to be significantly inhibited by peroxide in a dose-dependent manner (Fig. 2A). At concentrations of hydrogen peroxide of more than 4 μm, PRMT1 activity was significantly impaired and could not be fully recovered (data not shown). However, at the physiologically relevant concentration of 0.4 μm (physiological levels in mammals range from 0.001–0.7 μm) (32), hydrogen peroxide was also able to inhibit PRMT1 activity, and subsequent reduction with DTT resulted in full recovery of activity (Fig. 2B). These experiments provide the first evidence that PRMT1 enzymatic activity is susceptible to reversible oxidative impairment under physiologically relevant oxidative conditions.
FIGURE 2.
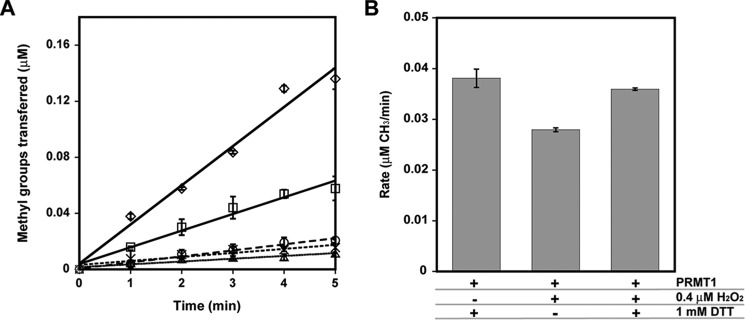
A and B, PRMT1 activity is inhibited by H2O2 in a concentration-dependent manner (A), and activity lost can be recovered by reduction (B). A, reduced PRMT1 was incubated with 0 (♢), 40 (□), 200 (○), 400 (×), or 800 (▵) μm H2O2 for 10 min at 37 °C, followed by the addition of catalase. Methyltransferase activity of the treated PRMT1 was measured with 200 μm R3 peptide as a substrate, as described under “Experimental Procedures.” B, reduced PRMT1 was treated with a physiologically relevant concentration of hydrogen peroxide or PBS and subsequently treated with 1 mm DTT or PBS prior to methyltransferase assays.
PRMT1 Enzymatic Activity Increases after Reduction
To better understand the sensitivity of PRMT1 to oxidation, we purified recombinant PRMT1 in the absence of reductant and measured methyltransferase activity in the absence and presence of a reductant. Preincubation with DTT increased methyltransferase activity more than 10-fold with the R3 peptide substrate (Fig. 3A). We examined the duration of this effect by rapidly desalting PRMT1 following the DTT preincubation step. The activity of the desalted PRMT1 was increased only 2-fold using the R3 peptide (Fig. 3A), indicating that the effect of DTT is transient and that PRMT1 oxidation occurs quickly in the absence of a reductant. We further studied the relationship between enzymatic activity and the concentration of DTT by comparing PRMT1 activity with varied concentrations of DTT ranging from 0.1–10 mm (Fig. 3B). Our results indicate that the effect of DTT is concentration-dependent, achieving a maximal rate enhancement at 1.5–2 mm DTT (Fig. 3B). During the course of this study, more than 50 human and rat PRMT1 proteins were purified in the absence of reductant and showed anywhere from a 1.8- to 70-fold increase in methyltransferase activity when DTT was included in the reaction or the enzyme was preincubated with DTT (data not shown). We note that, in many cases, our purification protocol can be accomplished in as few as ∼8 h. These results demonstrate that PRMT1 is sensitive to oxidation by not only hydrogen peroxide but also cellular conditions and/or the mild conditions used for purification.
FIGURE 3.
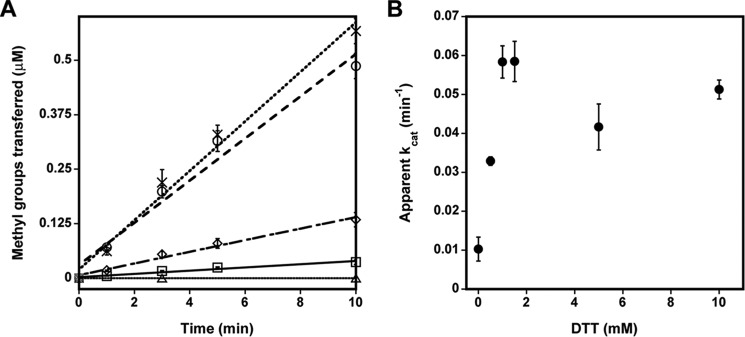
The effect of reducing agents on PRMT1 enzymatic activity. A, PRMT1 methyltransferase activity was measured without substrate (▵) or with 200 μm R3 peptide in the absence (□) and presence of DTT (○) or Tris(2-carboxyethyl)phosphine (×) or after desalting following a 10 min preincubation with DTT (♢). B, methyltransferase activity was measured as a function of DTT concentration.
Methylation of the sulfhydryl groups in DTT has been observed previously with small-molecule plant O-methyltransferases, where it acted as an intermediate acceptor molecule (33). Even though DTT methylation was not detected in our control reactions lacking peptide substrate (control, Fig. 3A), we replaced DTT in our assay with an alternative thiol-free reductant, Tris(2-carboxyethyl)phosphine. When 1 mm Tris(2-carboxyethyl)phosphine was used to reduce PRMT1, the rate enhancement with the R3 peptide was identical to that observed using 1 mm DTT (Fig. 3A). We conclude that the enhancement of HisPRMT1 methyltransferase activity is due to the reducing power of the DTT or Tris(2-carboxyethyl)phosphine additives.
Several different affinity tags are used for PRMT1 purification, and variable methylation rates have been observed (34–36). We questioned whether our His6 tag was influencing the observed rate changes upon reduction. To address this concern, we expressed a tagless version of rat PRMT1. However, this version of PRMT1 was unstable and lost activity rapidly (data not shown). As an alternative strategy, we created a His6-tagged, TEV-cleavable rat PRMT1 construct (HisTevPRMT1) that allowed us to cleave the tag after an initial purification step and then use the tag-free version (tag-freePRMT1) for kinetic assays. Both tagged and tag-free versions of PRMT1 exhibited rate enhancement upon reduction, although the tag-free version was markedly more active than the tagged version even without DTT in the assay (Fig. 4). This observation is easily explained when the method for acquiring tag-free protein is taken into consideration. The protocol for TEV cleavage includes an overnight dialysis step in a buffer containing 1 mm DTT and 2 mm EDTA, and, therefore, the resulting tag-free enzyme used in the activity assays is already somewhat prereduced. To confirm this theory, we again purified the cleavable (HisTevPRMT1) enzyme by nickel chromatography. Purified enzyme was divided into two separate dialysis bags. TEV was added to one, and the other was left uncleaved. However, both tagged and cleaved samples were dialyzed overnight in the same 1 mm DTT, 2 mm EDTA-containing buffer. Methyltransferase activity of these newly purified tagged and tag-free PRMT1 enzymes revealed no significant rate enhancement upon addition of DTT (Fig. 4). The addition of only EDTA had no significant effect on methyltransferase activity (Fig. 4). These results confirm that it is not the tag but, rather, the reducing treatment of the tag-free enzyme that enhanced the enzymatic activity of PRMT1.
FIGURE 4.
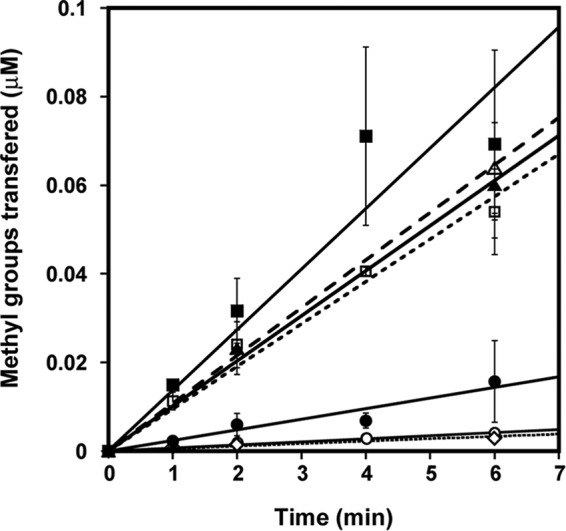
The enhancing effect of DTT on PRMT1 methyltransferase activity is independent of the His6 tag. Enzymatic activity of HisTevPRMT1 (○), cleaved PRMT1 (▵), and dialyzed HisTevPRMT1 (□) measured with 200 μm R3 peptide in the absence (open symbols) and presence (closed symbols) of DTT, respectively, as well as HisTevPRMT1 treated with EDTA only as a control (♢).
Reduction Alters the Oligomeric State of PRMT1
It has been shown that PRMT1 forms at least a homodimer to be catalytically active (36–38). Feng et al. (34) introduced the idea that changing the oligomeric state of PRMT1 affects its enzymatic activity. Because reduction of PRMT1 results in increased activity, we wondered whether reduction had an effect on the oligomeric state of the enzyme. Size exclusion chromatography experiments in the presence or absence of DTT were carried out to determine the oligomeric state of PRMT1 under different redox environments (Fig. 5). Unreduced PRMT1 migrates over a broad range of oligomeric states, with the majority of the protein existing in oligomers that migrate at molecular weights above 660 kDa. As a reference, PRMT1 is thought to be active as an 80-kDa dimer (although a dimer form has not been observed on size exclusion chromatography) (36, 39). Overnight incubation with 1 mm DTT and 2 mm EDTA results in a shift toward a homogeneous oligomeric state migrating between 200 and 450 kDa (Fig. 5). Analytical ultracentrifugation, a more sensitive technique for determining molecular weights, showed the same shift toward a smaller oligomeric species upon reduction, as observed using size exclusion chromatography (data not shown). In conclusion, oxidized PRMT1 forms a large molecular weight functional aggregate, whereas reduction of PRMT1 causes a shift toward a smaller, more uniform oligomeric state that correlated with an increase in enzymatic activity.
FIGURE 5.
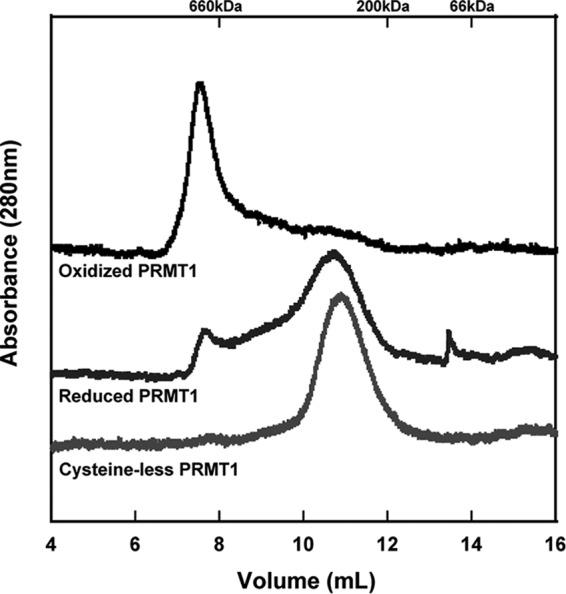
Oligomeric state of PRMT1 proteins assessed by size exclusion chromatography. PRMT1 without DTT treatment (top trace) or incubated overnight with 1 mm DTT and 2 mm EDTA (center trace) compared with cysteineless PRMT1, which migrates at the same position regardless of treatment with DTT or EDTA. Reduction of PRMT1 or removal of all cysteine residues results in a shift toward a smaller oligomeric state.
The Oxidation-dependent Effects on PRMT1 Activity Require one or More Cysteine Residue(s)
One of the most common mechanisms for oxidative damage is the oxidation of cysteine residues (40–43). We took a broad approach to evaluate whether any cysteine residues were responsible for the effects of oxidation/reduction by creating a PRMT1 variant where all 11 rat PRMT1 cysteine residues were mutated to serine (HisTevPRMT1Δcys or cys-). Interestingly, the cysteineless PRMT1 showed no enhancement in methyltransferase activity upon preincubation with DTT (Fig. 6). In addition to activity measurements, the oligomeric state of the cysteineless PRMT1 variant was assessed by size exclusion chromatography (Fig. 5). The cysteineless PRMT1 enzyme exhibited a migration pattern similar to that of the maximally active, reduced PRMT1 regardless of its treatment (data not shown). We conclude from these observations that indeed one (or more) cysteine(s) is/are responsible for the redox switching of PRMT1 activity.
FIGURE 6.
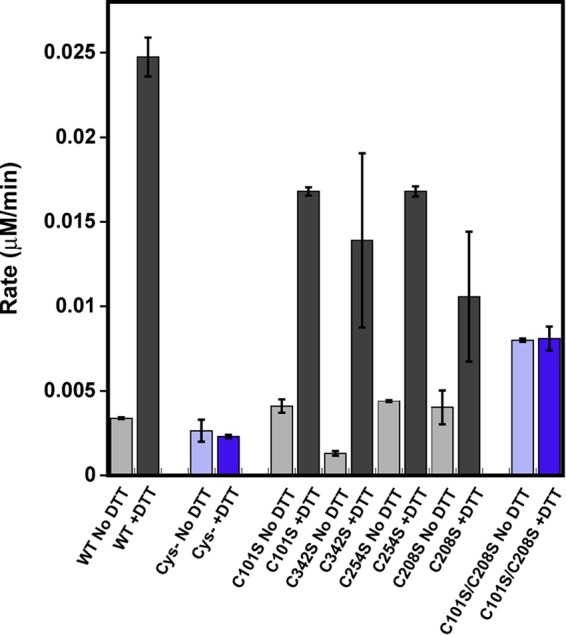
Methyltransferase activity of PRMT1 cysteine variants in the absence or presence of DTT. WT PRMT1, cysteineless PRMT1, C101S PRMT1, C342S PRMT1, C254S PRMT1, C208S PRMT1, and C101S/C208S PRMT1 methylated 200 μm R3 peptide in the absence (light gray/purple) or presence (dark gray/purple) of DTT. The results shown correspond to the average of at least two biological replicates. Removing all cysteine residues from PRMT1 or making Cys101 and Cys208 unavailable for oxidation resulted in abolished redox control over PRMT1 catalytic activity.
Cys101, Cys342, Cys254, and Cys208 Are Not Individually Responsible for Reductive Effects
We examined the rat PRMT1 crystal structures (36) for cysteine residues capable of undergoing oxidation and found that, of the 11 cysteines present in rat PRMT1, cysteines 101, 208, 232, 254, and 342 are all solvent-accessible (Fig. 7). Cysteines 9 and 15 are not visible in the crystal structure and were not considered in this study because a human PRMT1 variant lacking the first 27 amino acids exhibits enhanced activity after reduction (data not shown). A recent quantitative reactivity profiling study identified Cys101 as hyperreactive with 4-hydroxyl-2-nonenal (44), indicating that Cys101 may be susceptible to oxidation. Cysteine 101 is situated on the far edge of the AdoMet binding pocket and directly interacts with the adenine ring structure of AdoMet (Fig. 7) and, therefore, seemed like an excellent target to control enzymatic activity depending on its redox state. Mutation of Cys101 to serine results in a construct mimicking wild-type methyltransferase activity, including the response to DTT (Fig. 6). Closer evaluation of our rat PRMT1 M48L crystal structure (23) showed additional electron density around cysteine 342, suggesting possible oxidation of the thiolate moiety to a reducible sulfenic acid moiety. Although this residue is relatively removed from the active site, it has been suggested that residues distant from the active site can regulate PRMT substrate specificity (45). However, the C342S variant also mimicked wild-type enzymatic activity, including the DTT enhancement (Fig. 6). The PRMT1 crystal structure (36) also shows that Cys254 forms an intermolecular disulfide bond with another PRMT1 dimer and was, therefore, also mutated to determine whether reduction of this disulfide caused the increase in activity and the corresponding shift toward a smaller oligomeric state. However, as we saw with the other two individual cysteine variants, C254S activity was still enhanced upon reduction with DTT (Fig. 6). The C254S PRMT1 variant also behaved as WT PRMT1 when run on size exclusion chromatography (data not shown) (36).
FIGURE 7.

Cysteine residues in rPRMT1. Shown is the PRMT1 dimer (PDB code 1OR8) colored as described in the text (AdoMet binding domain in light gray, β barrel domain in dark gray, dimerization arm in blue, αγ-loop-αZ in orange, AdoHcy in green, and cysteine residues in red). Residues 26–39 were modeled on the basis of the position of this helix in the PRMT3 structure (PDB code 1F3L). A, PRMT1 dimer. B, surface representation showing close active site interactions between AdoHcy, Cys101, and Phe36. C, top view of the dimerization arm in one monomer interacting with the AdoMet binding site in the other monomer. D, back view showing the packing of Cys208 in one monomer with the α helix in the AdoMet binding domain of the other monomer.
As mentioned previously, it is believed that PRMT1 dimerization is necessary for catalytic activity. In fact, all currently available type 1 and type 2 PRMT structures reveal a conserved mode of dimerization between catalytic subunits (36–38, 46, 47). Each subunit contains a dimerization helix-turn-helix dimerization arm (blue, Fig. 7) that extends from the C-terminal β barrel (dark gray, Fig. 7) and rests upon the N-terminal AdoMet binding domain (light gray, Fig. 7) of the other subunit, coming together to form an active dimer with a central cavity and two opposing active sites (Fig. 7).
Zhang et al. (36) reported that removal of the helix-turn-helix dimerization arm of PRMT1 not only eliminated homodimerization but also AdoMet binding and methyltransferase activity. Therefore, it has been suggested that the dimer interaction is required to engage the residues on the other side of the structural elements to interact properly with AdoMet (36). In the analysis of another type 1 PRMT structure, Cheng et al. (37) computed the atomic position fluctuations of the monomer and dimer to determine the impact of dimerization on the motion of atPRMT10, also a type 1 PRMT. They found two regions to have lower atomic position fluctuations in dimeric form than in monomeric form: the αγ-loop-αZ (40–68 in atPRMT10) (orange, Fig. 7) and the dimerization arm (187–235 in atPRMT10) (blue, Fig. 7), suggesting that these regions are stabilized upon dimer formation. The αγ-loop-αZ region is directly involved in AdoMet binding and the formation of the substrate binding groove I (36). It has also been suggested that stabilization of this region by dimerization likely improves the binding of AdoMet and substrate proteins (37). Additionally, Higashimoto et al. (48) demonstrated that, in PRMT4 (CARM1), phosphorylation of Ser229, located on the outer face of the AdoMet binding domain, compromised dimerization, negatively regulated methyltransferase activity, and lowered AdoMet binding. The presence of a charged functional group adjacent to the hydrophobic dimerization arm binding surface likely destabilizes dimerization interactions. Cysteine 208 of PRMT1 is located on the PRMT1 dimer interface (Fig. 7). Given the importance of dimerization for PRMT activity and the predicted susceptibility to oxidation, this residue was also deemed likely to be responsible for the effect of oxidation in impairing methyltransferase activity. Additionally, use of the cysteine oxidation prediction algorithm, which uses distance to the nearest cysteine sulfur, solvent accessibility, and pKa as parameters for thiol oxidation susceptibility, suggested that cysteine 208 is susceptible to oxidation by exposure (49). However, measurement of the enzymatic activity of the individual C208S variant revealed that this construct was also enhanced by DTT (Fig. 6).
If PRMT1 dimerization is indeed stabilizing substrate binding, we rationalized that oxidation of Cys208 in the dimerization arm in conjunction with Cys101 in the AdoMet binding pocket could be responsible for the diminished methyltransferase activity observed under oxidizing conditions. To explore this hypothesis, a C101S/C208S variant of PRMT1 was created, and methyltransferase activity was measured in the presence and absence of DTT. Remarkably, methyltransferase activity of the double variant was the same under both oxidizing and reducing conditions (Fig. 6). We note that the activity of the double variant (C101S/C208S) is lower than the activity of reduced wild-type enzyme, suggesting that these residues may have an additional role in PRMT1 activity. Surprisingly, when both Cys101 and Cys208 are unavailable for oxidation, the protein does not completely shift to a smaller oligomeric state than the cysteine-less construct (data not shown). In conclusion, making Cys101 and Cys208 unavailable for oxidation resulted in abolished redox control over PRMT1 catalytic activity.
Sulfenic Acid Formation at Cys101 and Cys208 in Vitro
To further analyze the oxidation events occurring at Cys101 and Cys208, we used the sulfenic acid-specific probe DCP-Rho1, which contains a rhodamine attached to the functional core of dimedone. To detect sulfenic acids, air oxidized wild-type PRMT1 (WT), cysteine-less with Cys101 reintroduced (cys-Cys101), cysteineless with Cys208 reintroduced (cys-Cys208), and cysteineless with both Cys101 and Cys208 reintroduced (cys-Cys101Cys208) were incubated with DCP-Rho1 or dimethyl sulfoxide as a negative control in the presence or absence of reductant under denaturing conditions. Cysteine-less PRMT1 (cys-) was also subjected to labeling as a negative control. Analysis of the fluorescent label incorporation (Fig. 8, A and B) clearly shows the presence of sulfenic acid at cysteine 101 and 208 of PRMT1. Interestingly, although both cysteine 101 and 208 are necessary for oxidative impairment of Cys101, we consistently observed more sulfenic acid formation at Cys208, suggesting either that Cys101 is less susceptible to oxidation than Cys208 or that the sulfenic acid form of Cys101 is transient and can be further oxidized to sulfinic or sulfonic acid. Free thiol content analysis using the 5IAF probe (Fig. 8, C and D) and performed in parallel with DCP-Rho1 labeling, shows similar free thiol content for cys-Cys101 and cys-Cys208. When analyzed together, these results are consistent with a higher propensity for irreversible oxidation at Cys101. In addition to the fluorescent probes, WT PRMT1 treated with sulfenic acid-specific dimedone was subjected to LC-MS/MS analysis to further confirm sulfenic acid formation. Using this method, dimedone incorporation was observed at both Cys101 and Cys208 (Fig. 8, E and F), confirming the fluorescent probe results. In conclusion, the reversible activity impairment observed under oxidative conditions is consistent with sulfenic acid formation at Cys101 and Cys208 of PRMT1.
FIGURE 8.

Sulfenic acid detection and free thiol content in PRMT1. A–D, air-oxidized wild-type PRMT1 (WT), cys-, cys-Cys101, cys-Cys208, and cys-Cys101Cys208 were denatured in 6 m urea and incubated with 1 mm DTT or buffer prior to addition of 10 μm DCP-Rho1 or 2.5 mm 5IAF. Labeled samples were resolved by SDS-PAGE. A, representative image of the rhodamine fluorescence signal and the corresponding Coomassie bands. B, graphical representation of triplicate gel analysis. Normalized percent DCP-Rho1 fluorescence represents the percentage of fluorescent signal divided by the amount of protein observed in the Coomassie bands, which is interpreted as the relative amount of sulfenic acid present. C, representative image of the 5IAF fluorescence signal and the corresponding Coomassie bands. D, graphical representation of triplicate 5IAF gel analysis, interpreted as relative amount of free thiols present. E and F, representative MS/MS fragmentation spectra for peptides containing dimedone-modified sulfenic acids in PRMT1. E, the CID fragments of peptide IECSSISDYAVK labeled with dimedone at Cys101. F, ETD fragments of peptide MCSIKDVAIK labeled with dimedone at Cys208. The mass shift by the modification is 138.068 (exact number), as denoted in the peptide sequence.
Discussion
Elevated ADMA concentrations have been proposed to predict cardiovascular mortality in patients with chronic renal insufficiency (50) and acute coronary events (8). Additionally, ADMA is not only a marker but also a mediator of oxidative stress within vascular tissue (7). Given that PRMT1 is the primary source of ADMA in eukaryotes, it is of the utmost importance to understand how PRMT1 activity and ADMA synthesis are regulated under oxidative stress conditions. Here we have shown that PRMT1 activity is decreased in vitro under physiologically relevant oxidative conditions, an effect which is readily reversed upon reduction and is associated with cysteine oxidation to sulfenic acid. Our results complicate the view that the increased PRMT1 protein expression observed under oxidative stress conditions is responsible for increased ADMA levels by supporting a model in which PRMT1 activity can be regulated in a redox-sensitive manner. Although it has long been known that DDAH breakdown of ADMA is under redox control, we provide the first evidence that the activity of enzymes involved in the formation of ADMA precursors is regulated in a redox-sensitive fashion.
Removal of two cysteine residues implicated in PRMT1 dimerization and cofactor binding eliminates the redox-dependent control over PRMT1 methyltransferase activity. Dimerization is strictly conserved in all known PRMTs and seems necessary for methyltransferase activity (36–38). One of the pathways proposed for signal communication between the dimer interface and the catalytic center uses the mainly hydrophobic dimer interactions between the αγ-loop-αZ (orange, Fig. 7) of one monomer and the helix-turn-helix dimerization arm of another (blue, Fig. 7) (36, 37, 46). It has been proposed that this dimer interaction is required to engage residues into the proper conformation for interaction with the AdoMet cofactor (36). It is feasible that oxidation of residues involved in this cooperative effort could disrupt PRMT1 methyltransferase activity by affecting AdoMet binding. Interestingly, cysteine 208 is conserved in PRMT3 and PRMT6, whereas both cysteine 101 and 208 are conserved in PRMT8. Remarkably, the type III PRMT7 from Trypanosoma brucei, which contains only three cysteine residues that do not align with any cysteines in PRMT1 but does contain a cysteine at a different location in its dimerization arm, also shows increased activity upon reduction. Activity measurements using human PRMT3, human PRMT6, and TbPRMT7 demonstrate that these type 1 and type 3 PRMTs are also under redox control (Fig. 9). Ongoing studies will help provide insight as to how these residues affect AdoMet interactions and PRMT methyltransferase activity.
FIGURE 9.
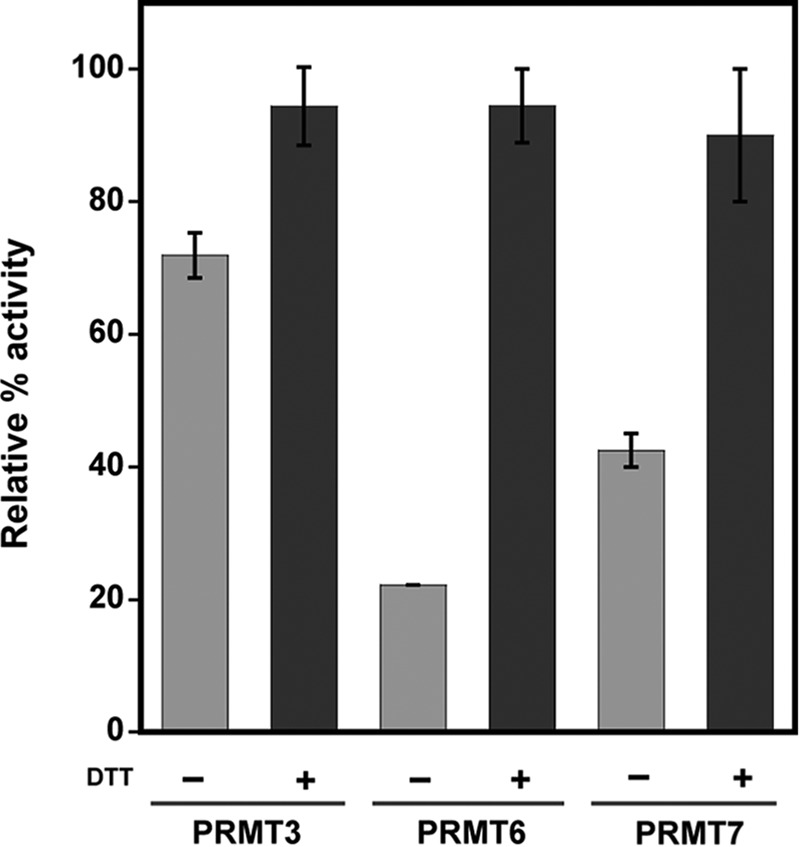
Redox control is conserved among PRMT family members. Human PRMT3 (residues 211–531), human PRMT6, and TbPRMT7 activities were tested with R3 peptide, bulk histones, or Histone 4, respectively, in the absence or presence of DTT. The average of three activity measurements are shown as relative percent activity for each isoform with its corresponding substrate.
Although the active form of PRMT1 is expected to be a dimer, size exclusion chromatography analysis as well as dynamic light scattering have shown that the enzyme typically migrates as a high molecular weight oligomer (34, 36, 38, 51, 52). Feng et al. (34) have suggested that PRMT1 multioligomerization (i.e. greater than a dimer) is concentration-dependent in the range of 0–0.5 μm and that the final PRMT1 multioligomeric complex is the most active form. It is important to note that the study by Feng et al. (34) was done with fully reduced PRMT1 and at significantly lower enzyme concentrations than those used in this study. Our results show that oxidized PRMT1, which displays less activity, shifts from a large oligomeric species to a smaller, but still large oligomeric state under reducing conditions. Although our results seem to be at odds with the study by Feng et al. (34), they only serve to highlight the high degree of complexity that exists in relating PRMT1 oligomerization and activity. Interestingly, although cysteineless PRMT1 remains a smaller oligomer under both reducing and oxidizing conditions, the C101S/C208S PRMT1 variant presents as a mix of both large and smaller oligomers under reducing conditions (data not shown). It is difficult to assess what may be causing PRMT1 to form such large oligomeric aggregates, and the link between PRMT1 cysteine oxidation and protein oligomerization remains to be determined. Although our results add to the larger story, much remains to be discovered regarding the impact of the oligomeric state on PRMT1 activity.
It is also important to point out that much of the work in the PRMT field is carried out exclusively under reducing conditions. Our work emphasizes the importance of PRMT1 as a redox-sensitive enzyme and the need for careful control of its redox environment. Because PRMT1 activity is under redox control, it is possible that experimental conditions might inadvertently alter the activity and/or interaction partners. In the future, it will be highly important to take into account the redox environment of experiments before reaching conclusions. Additionally, although the different affinity tags that were used in this study did not change the increase in activity observed upon PRMT1 reduction, the overall methyltransferase activity was generally lower for HisTevPRMT1 constructs than for HisPRMT1 constructs. Because the tags are located on the N-terminal end of the enzyme, this observation may hint at the importance of the N-terminal PRMT1 sequence in controlling enzymatic activity.
In addition to providing ADMA precursor pools, growing evidence supports the involvement of PRMT1 in the cellular oxidative stress response as a modifier of signal transduction. PRMT1 has been reported recently to be involved in the transcriptional regulation of the human ferritin gene, up-regulation of which is an important cellular defense response to oxidative stress (53). It has also been reported recently that PRMT1 methylation is involved in the transcriptional repression of HIFα (54), a protein needed to activate the cellular adaptive response to hypoxia, another condition linked to high levels of oxidative stress. Finally, we note that oxidative stress in HEK293 cells has been reported to enhance PRMT1-mediated methylation of FOXO1 in the nucleus (55). The observed increase in FOXO1 methylation seems contradictory to our results and highlights the importance of further investigating the effect of oxidation in vivo on PRMT1 substrate specificity, and/or cellular localization. In future studies, it will also be interesting to determine how redox regulation affects the role of PRMT1 in signal transduction.
In summary, this work has demonstrated that PRMT1 is a redox-responsive enzyme. Oxidation at two cysteine residues potentially destabilizes dimerization, leading to diminished methyltransferase activity, whereas reduction readily reverses the effects of oxidation under physiologically relevant conditions, consistent with sulfenic acid formation observed at both cysteine residues. Our results provide the first direct evidence that PRMT1 enzymatic activity can be regulated in a redox-sensitive manner and raise the concern that the current paradigm used to explain free ADMA accumulation in vivo may be incomplete.
Acknowledgments
We thank Dr. Orlando Acevedo and Symon Gathiaka at Auburn University for help with structural modeling, Brooke Siler for the construction of several of the PRMT1 variants, and Jared Hardman for construction of the His-PRMT6 construct. We also thank Dr. Minkui Luo (Memorial Sloan Kettering Cancer Center) for the human PRMT3 plasmid and Dr. Laurie Read (SUNY Buffalo) for the TbPRMT7 construct.
This work was supported by a Utah State University URCO award (to O. M. P.), a Utah State University Dissertation fellowship (to Y. M.), and National Science Foundation Awards 0920776 and 1412405 (to J. M. H.).
- ADMA
- asymmetric dimethylarginine
- PRMT
- protein arginine methyltransferase
- MMA
- monomethylarginine
- DDAH
- dimethylarginine dimethylaminohydrolase
- AdoMet
- S-adenosylmethionine
- TEV
- tobacco etch virus
- NOS
- nitric oxide synthase
- DTT
- dithiothreitol
- EDTA
- ethylenediaminetetraacetic acid
- DCP-Rho1
- rhodamine B [4-[3-(2,4-dioxocyclohexyl)propyl]carbamate]piperazine amide
- 5IAF
- 5-iodoacetamidofluorescein
- AdoHcy
- S-adenosyl homocysteine
- CARM1
- coactivator-associated arginine methyltransferase 1
- HIFα
- hypoxia-inducible factor α
- FOXO
- forkhead box O transcription factor
- CID
- collision-induced dissociation
- ETD
- electron-transfer dissociation
- ID
- inner diameter.
References
- 1. Go A. S., Mozaffarian D., Roger V. L., Benjamin E. J., Berry J. D., Blaha M. J., Dai S., Ford E. S., Fox C. S., Franco S., Fullerton H. J., Gillespie C., Hailpern S. M., Heit J. A., Howard V. J., Huffman M. D., Judd S. E., Kissela B. M., Kittner S. J., Lackland D. T., Lichtman J. H., Lisabeth L. D., Mackey R. H., Magid D. J., Marcus G. M., Marelli A., Matchar D. B., McGuire D. K., Mohler E. R., 3rd, Moy C. S., Mussolino M. E., Neumar R. W., Nichol G., Pandey D. K., Paynter N. P., Reeves M. J., Sorlie P. D., Stein J., Towfighi A., Turan T. N., Virani S. S., Wong N. D., Woo D., Turner M. B., American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2014) Heart Disease and Stroke Statistics-2014 Update A Report From the American Heart Association. Circulation 129, e28–e292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu T. M., Ding Y. A., Charng M. J., Lin S. J. (2003) Asymmetrical dimethylarginine: a novel risk factor for coronary artery disease. Clin. Cardiol. 26, 458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen X., Niroomand F., Liu Z., Zankl A., Katus H. A., Jahn L., Tiefenbacher C. P. (2006) Expression of nitric oxide related enzymes in coronary heart disease. Basic Res. Cardiol. 101, 346–353 [DOI] [PubMed] [Google Scholar]
- 4. Antoniades C., Shirodaria C., Leeson P., Antonopoulos A., Warrick N., Van-Assche T., Cunnington C., Tousoulis D., Pillai R., Ratnatunga C., Stefanadis C., Channon K. M. (2009) Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur. Heart J. 30, 1142–1150 [DOI] [PubMed] [Google Scholar]
- 5. Böger R. H., Bode-Böger S. M., Szuba A., Tsao P. S., Chan J. R., Tangphao O., Blaschke T. F., Cooke J. P. (1998) Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98, 1842–1847 [DOI] [PubMed] [Google Scholar]
- 6. Lüneburg N., Harbaum L., Hennigs J. K. (2014) The endothelial ADMA/NO pathway in hypoxia-related chronic respiratory diseases. BioMed. Res. Int. 2014, 501612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sydow K., Münzel T. (2003) ADMA and oxidative stress. Atheroscler. Suppl. 4, 41–51 [DOI] [PubMed] [Google Scholar]
- 8. Valkonen V. P., Päivä H., Salonen J. T., Lakka T. A., Lehtimäki T., Laakso J., Laaksonen R. (2001) Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet 358, 2127–2128 [DOI] [PubMed] [Google Scholar]
- 9. Vallance P., Leiper J. (2004) Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 24, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 10. Achan V., Broadhead M., Malaki M., Whitley G., Leiper J., MacAllister R., Vallance P. (2003) Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 23, 1455–1459 [DOI] [PubMed] [Google Scholar]
- 11. Kielstein J. T., Impraim B., Simmel S., Bode-Böger S. M., Tsikas D., Frölich J. C., Hoeper M. M., Haller H., Fliser D. (2004) Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation 109, 172–177 [DOI] [PubMed] [Google Scholar]
- 12. Burgoyne J. R., Mongue-Din H., Eaton P., Shah A. M. (2012) Redox signaling in cardiac physiology and pathology. Circ. Res. 111, 1091–1106 [DOI] [PubMed] [Google Scholar]
- 13. Chan J. R., Böger R. H., Bode-Böger S. M., Tangphao O., Tsao P. S., Blaschke T. F., Cooke J. P. (2000) Asymmetric dimethylarginine increases mononuclear cell adhesiveness in hypercholesterolemic humans. Arterioscler. Thromb. Vasc. Biol. 20, 1040–1046 [DOI] [PubMed] [Google Scholar]
- 14. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ogawa T., Kimoto M., Sasaoka K. (1987) Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-l-arginine to l-citrulline in rats. Biochem. Biophys. Res. Commun. 148, 671–677 [DOI] [PubMed] [Google Scholar]
- 16. Böger R. H., Sydow K., Borlak J., Thum T., Lenzen H., Schubert B., Tsikas D., Bode-Böger S. M. (2000) LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res. 87, 99–105 [DOI] [PubMed] [Google Scholar]
- 17. Chobanyan-Jürgens K., Pham V. V., Stichtenoth D. O., Tsikas D. (2011) Asymmetrical dimethylarginine, oxidative stress, and atherosclerosis. Hypertension 58, e184–185; author reply e186 [DOI] [PubMed] [Google Scholar]
- 18. Jiang J. L., Zhang X. H., Li N. S., Rang W. Q., Feng-Ye, Hu C. P., Li Y. J., Deng H. W. (2006) Probucol decreases asymmetrical dimethylarginine level by alternation of protein arginine methyltransferase I and dimethylarginine dimethylaminohydrolase activity. Cardiovasc. Drugs Ther. 20, 281–294 [DOI] [PubMed] [Google Scholar]
- 19. Chen Y., Xu X., Sheng M., Zhang X., Gu Q., Zheng Z. (2009) PRMT-1 and DDAHs-induced ADMA upregulation is involved in ROS- and RAS-mediated diabetic retinopathy. Exp. Eye Res. 89, 1028–1034 [DOI] [PubMed] [Google Scholar]
- 20. Tyagi N., Sedoris K. C., Steed M., Ovechkin A. V., Moshal K. S., Tyagi S. C. (2005) Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 289, H2649-H2656 [DOI] [PubMed] [Google Scholar]
- 21. Wilcox C. S. (2012) Asymmetric dimethylarginine and reactive oxygen species: unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 59, 375–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim Y., Lee E., Lee J., Oh S., Kim S. (2008) Down-regulation of asymmetric arginine methylation during replicative and H2O2-induced premature senescence in WI-38 human diploid fibroblasts. J. Biochem. 144, 523–529 [DOI] [PubMed] [Google Scholar]
- 23. Gui S., Wooderchak W. L., Daly M. P., Porter P. J., Johnson S. J., Hevel J. M. (2011) Investigation of the molecular origins of protein-arginine methyltransferase I (PRMT1) product specificity reveals a role for two conserved methionine residues. J. Biol. Chem. 286, 29118–29126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang R., Ibáñez G., Islam K., Zheng W., Blum G., Sengelaub C., Luo M. (2011) Formulating a fluorogenic assay to evaluate S-adenosyl-l-methionine analogues as protein methyltransferase cofactors. Mol. BioSyst. 7, 2970–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cornell K. A., Swarts W. E., Barry R. D., Riscoe M. K. (1996) Characterization of recombinant Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase: analysis of enzymatic activity and substrate specificity. Biochem. Biophys. Res. Commun. 228, 724–732 [DOI] [PubMed] [Google Scholar]
- 26. Suh-Lailam B. B., Hevel J. M. (2010) A fast and efficient method for quantitative measurement of S-adenosyl-l-methionine-dependent methyltransferase activity with protein substrates. Anal. Biochem. 398, 218–224 [DOI] [PubMed] [Google Scholar]
- 27. Atmane N., Dairou J., Paul A., Dupret J. M., Rodrigues-Lima F. (2003) Redox regulation of the human xenobiotic metabolizing enzyme arylamine N-acetyltransferase 1 (NAT1). Reversible inactivation by hydrogen peroxide. J. Biol. Chem. 278, 35086–35092 [DOI] [PubMed] [Google Scholar]
- 28. Nelson K. J., Klomsiri C., Codreanu S. G., Soito L., Liebler D. C., Rogers L. C., Daniel L. W., Poole L. B. (2010) Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 473, 95–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu Y., Kwon K. S., Rhee S. G. (1998) Probing cellular protein targets of H2O2 with fluorescein-conjugated iodoacetamide and antibodies to fluorescein. FEBS Lett. 440, 111–115 [DOI] [PubMed] [Google Scholar]
- 30. Hansen R. E., Winther J. R. (2009) An introduction to methods for analyzing thiols and disulfides: reactions, reagents, and practical considerations. Anal. Biochem. 394, 147–158 [DOI] [PubMed] [Google Scholar]
- 31. Tang J., Frankel A., Cook R. J., Kim S., Paik W. K., Williams K. R., Clarke S., Herschman H. R. (2000) PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275, 7723–7730 [DOI] [PubMed] [Google Scholar]
- 32. Stone J. R., Yang S. (2006) Hydrogen peroxide: a signaling messenger. Antioxid. Redox Signal. 8, 243–270 [DOI] [PubMed] [Google Scholar]
- 33. Coiner H., Schröder G., Wehinger E., Liu C. J., Noel J. P., Schwab W., Schröder J. (2006) Methylation of sulfhydryl groups: a new function for a family of small molecule plant O-methyltransferases. Plant J. 46, 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feng Y., Xie N., Jin M., Stahley M. R., Stivers J. T., Zheng Y. G. (2011) A transient kinetic analysis of PRMT1 catalysis. Biochemistry 50, 7033–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kleinschmidt M. A., Streubel G., Samans B., Krause M., Bauer U. M. (2008) The protein arginine methyltransferases CARM1 and PRMT1 cooperate in gene regulation. Nucleic Acids Res. 36, 3202–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang X., Cheng X. (2003) Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11, 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng Y., Frazier M., Lu F., Cao X., Redinbo M. R. (2011) Crystal structure of the plant epigenetic protein arginine methyltransferase 10. J. Mol. Biol. 414, 106–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weiss V. H., McBride A. E., Soriano M. A., Filman D. J., Silver P. A., Hogle J. M. (2000) The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat. Struct. Biol. 7, 1165–1171 [DOI] [PubMed] [Google Scholar]
- 39. Kölbel K., Ihling C., Bellmann-Sickert K., Neundorf I., Beck-Sickinger A. G., Sinz A., Kühn U., Wahle E. (2009) Type I arginine methyltransferases PRMT1 and PRMT-3 act distributively. J. Biol. Chem. 284, 8274–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Simplicio P., Franconi F., Frosalí S., Di Giuseppe D. (2003) Thiolation and nitrosation of cysteines in biological fluids and cells. Amino Acids 25, 323–339 [DOI] [PubMed] [Google Scholar]
- 41. Berlett B. S., Stadtman E. R. (1997) Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316 [DOI] [PubMed] [Google Scholar]
- 42. Stadtman E. R. (1993) Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem. 62, 797–821 [DOI] [PubMed] [Google Scholar]
- 43. Carter E. L., Ragsdale S. W. (2014) Modulation of nuclear receptor function by cellular redox poise. J. Inorg. Biochem. 133, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., Cravatt B. F. (2010) Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Osborne T. C., Obianyo O., Zhang X., Cheng X., Thompson P. R. (2007) Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry 46, 13370–13381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Troffer-Charlier N., Cura V., Hassenboehler P., Moras D., Cavarelli J. (2007) Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J. 26, 4391–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yue W. W., Hassler M., Roe S. M., Thompson-Vale V., Pearl L. H. (2007) Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J. 26, 4402–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Higashimoto K., Kuhn P., Desai D., Cheng X., Xu W. (2007) Phosphorylation-mediated inactivation of coactivator-associated arginine methyltransferase 1. Proc. Natl. Acad. Sci. U.S.A. 104, 12318–12323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanchez R., Riddle M., Woo J., Momand J. (2008) Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 17, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zoccali C., Bode-Böger S., Mallamaci F., Benedetto F., Tripepi G., Malatino L., Cataliotti A., Bellanuova I., Fermo I., Frölich J., Böger R. (2001) Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 358, 2113–2117 [DOI] [PubMed] [Google Scholar]
- 51. Tang J., Gary J. D., Clarke S., Herschman H. R. (1998) PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 273, 16935–16945 [DOI] [PubMed] [Google Scholar]
- 52. Lim Y., Kwon Y. H., Won N. H., Min B. H., Park I. S., Paik W. K., Kim S. (2005) Multimerization of expressed protein-arginine methyltransferases during the growth and differentiation of rat liver. Biochim. Biophys. Acta 1723, 240–247 [DOI] [PubMed] [Google Scholar]
- 53. Huang B. W., Ray P. D., Iwasaki K., Tsuji Y. (2013) Transcriptional regulation of the human ferritin gene by coordinated regulation of Nrf2 and protein arginine methyltransferases PRMT1 and PRMT4. FASEB J. 27, 3763–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lafleur V. N., Richard S., Richard D. E. (2014) Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the protein arginine methyltransferase PRMT1. Mol. Biol. Cell 25, 925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamagata K., Daitoku H., Takahashi Y., Namiki K., Hisatake K., Kako K., Mukai H., Kasuya Y., Fukamizu A. (2008) Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231 [DOI] [PubMed] [Google Scholar]