

Background: Telomere fragility occurs following replication stress.
Results: Leading strand-specific telomere fragility is induced by FEN1 depletion and transcription inhibition and is rescued by ectopic RNase H1 expression.
Conclusion: RNA:DNA hybrids contribute to telomere fragility that is limited by FEN1.
Significance: This is the first explanation for leading strand-specific telomere fragility and is the first leading strand-specific role for FEN1.
Keywords: cancer, DNA replication, genomic instability, telomere, transcription, FEN1
Abstract
The existence of redundant replication and repair systems that ensure genome stability underscores the importance of faithful DNA replication. Nowhere is this complexity more evident than in challenging DNA templates, including highly repetitive or transcribed sequences. Here, we demonstrate that flap endonuclease 1 (FEN1), a canonical lagging strand DNA replication protein, is required for normal, complete leading strand replication at telomeres. We find that the loss of FEN1 nuclease activity, but not DNA repair activities, results in leading strand-specific telomere fragility. Furthermore, we show that FEN1 depletion-induced telomere fragility is increased by RNA polymerase II inhibition and is rescued by ectopic RNase H1 expression. These data suggest that FEN1 limits leading strand-specific telomere fragility by processing RNA:DNA hybrid/flap intermediates that arise from co-directional collisions occurring between the replisome and RNA polymerase. Our data reveal the first molecular mechanism for leading strand-specific telomere fragility and the first known role for FEN1 in leading strand DNA replication. Because FEN1 mutations have been identified in human cancers, our findings raise the possibility that unresolved RNA:DNA hybrid structures contribute to the genomic instability associated with cancer.
Introduction
DNA replication and repair are high fidelity processes that maintain genome stability. Because of the importance of these processes, robust mechanisms have evolved to ensure they are completed even when components of the replication and repair pathways are compromised or absent due to mutation. In some instances, this compensation is inadequate. Indeed, mutations in specific replication or repair proteins give rise to genetic disorders such as ataxia telangiectasia, Bloom syndrome, and Fanconi anemia. Cells from these patients reveal that although gross DNA metabolism continues largely unabated, mild replication defects and sensitivity to DNA-damaging agents or ionizing radiation contribute to genomic instability and increased cancer incidence (1, 2).
Although the redundancy of replication and repair mechanisms ensures faithful replication of the bulk genome, regions with repetitive sequence or an ability to form secondary structures are problematic and thus particularly sensitive to mutations in DNA replication and repair proteins (3). This is best illustrated at common fragile sites, where replication stressors lead to replication defects and genomic instability. Why particular regions of the genome manifest as fragile sites remains obscure, but insufficient replication origins, repetitive sequences, and replication-transcription interference have all been implicated (4–6).
Recently, telomeres have also been described as fragile sites because treatment with aphidicolin, a potent inducer of replication stress, results in reduced replication fork progression and abnormal telomere structures (7, 8). In checkpoint-competent backgrounds, aphidicolin treatment increases telomere fragility by 1.5–4.5-fold (7–9), whereas suppression of the ataxia telangiectasia and Rad3-related (ATR)6 kinase is sufficient to induce a 1.7-fold increase in telomere fragility in murine Seckel cells (9). Telomere fragility is also induced in the absence of telomere-binding proteins that participate in telomere replication. Indeed, knock-out of the Shelterin complex member TRF1, which is required for replication fork progression through the telomere, increases the rate of telomere fragility in murine cells by 3.0–4.5-fold (7, 8, 10); similarly, depletion of the CST complex members CTC1 or STN1, which are important for replication fork restart at the telomere, causes between a 2.0- and 3.0-fold increase in telomere fragility in human cells (11).
DNA replication and repair proteins are also important in maintaining telomere stability by preventing or suppressing telomere fragility. We previously reported that depletion of flap endonuclease 1 (FEN1) results in a 2.0-fold increase in telomere fragility (12). Loss of the DNA glycosylase Nth1, which participates in the repair of oxidative stress-induced lesions, causes a 1.8-fold increase in telomere fragility (13). Helicases and topoisomerases also play roles in reducing telomere fragility. Depletion of TopoIIα causes up to an ∼7-fold increase in telomere fragility, and depletion of the RecQ helicase BLM induces a 1.9-fold increase in telomere fragility (7, 14). Similarly, RTEL1 depletion or deletion induces 2.3- and 4.0-fold increases in telomere fragility, respectively (7, 10). These studies demonstrate the wide range of genetic manipulations that can induce telomere fragility with varying levels of severity.
The mechanism(s) by which telomere fragility occurs is not clear, but the large number of proteins implicated in the phenotype suggests that multiple mechanisms exist. G-quadruplexes may play a role, as telomere fragility induced by RTEL1 deletion is exacerbated by treatment with the G-quadruplex-stabilizing drug TMPyP4 (10). Given these data, if the molecular event inducing telomere fragility occurs after the replication fork has passed, RTEL1-induced telomere fragility would be expected to exhibit lagging strand specificity; however, few studies have examined strand-specific telomere fragility. Sfeir et al. (7) examined TRF1 knock-out mouse cells using chromosome orientation-fluorescent in situ hybridization (CO-FISH), which is capable of distinguishing telomeres replicated by the leading versus lagging strand DNA replication machinery; they found that telomere fragility induced by loss of TRF1 did not exhibit strand specificity. Similarly, Chawla et al. (15) identified UPF1, an ATPase and helicase associated with cytoplasmic RNA quality control, as a telomere-binding protein; in UPF1-depleted cells, telomere fragility increased at both the leading and lagging strands, with a slightly larger increase observed at the leading strand. Most recently, Arora et al. (16) demonstrated that ectopic expression of ribonuclease H1 (RNase H1) reduced fragile telomere formation on the leading strand in alternative lengthening of telomeres (ALT)-positive cells.
Among the stressors the replisome encounters, transcription has a significant impact on DNA replication. Indeed, head-on collisions between the replisome and RNA polymerase (RNAP) are extremely damaging to the replication process (17). In contrast to head-on collisions, co-directional replisome-RNAP collisions in bacteria are more common and better tolerated by the cell (18, 19). This may be due to a mechanism recently elucidated in viral and prokaryotic polymerases: following a co-directional collision with RNAP on the leading strand-replicated DNA, DNA polymerase III is removed from the template, moves forward to the 3′ end of the nascent transcript, displaces RNAP, and restarts DNA synthesis using the transcript as a primer (20). Despite this mechanism, which would seem to permit damage-free replication across a region being transcribed, co-directional collisions between the replisome and RNAP can lead to unresolved RNA:DNA hybrids. If such collisions occur in mammalian cells, the RNA:DNA hybrids left behind would likely lead to DNA double strand breaks, an ataxia telangiectasia-mutated (ATM)-mediated DNA damage response (DDR), and genomic instability (21, 22). Thus, robust mechanisms would need to evolve to remove the RNA:DNA hybrids produced by a collision event.
The known role of FEN1 in limiting telomere fragility (12), as well as the idea that telomere fragility might be the result of replication stress or interference with transcription, led us to explore the mechanism by which FEN1 reduces telomere fragility. We show that treatment with α-amanitin, which reduces the rate of RNAP elongation and thus may increase the rate of stochastic co-directional replisome-RNAP collisions, exacerbates the telomere fragility induced upon FEN1 depletion. Additionally, we find that the telomere fragility phenotype induced by FEN1 depletion and collision induction is RNA:DNA hybrid-dependent by rescuing telomere fragility with ectopic expression of RNase H1. FEN1's role in limiting telomere fragility is distinct from its role in limiting sister telomere loss, as FEN1 depletion-induced telomere fragility is restricted to the leading strand. Neither FEN1's classical replication role mediated by its interaction with proliferating cell nuclear antigen (PCNA) nor FEN1's DNA repair function mediated by its C-terminal interactions with numerous repair proteins are required for its activity in limiting telomere fragility. We find that FEN1's gap endonuclease and exonuclease activities are also dispensable for limiting telomere fragility but that FEN1's flap endonuclease activity is required. Our data support a model in which co-directional replisome-RNAP collisions on the leading strand-replicated telomere produce RNA:DNA hybrid/flap structures that accumulate in the absence of FEN1. We propose that FEN1, a classical lagging strand replication protein, acts on the leading strand during telomere replication to resolve RNA:DNA hybrid/flap structures resembling Okazaki fragment substrates; in the absence of this activity, the subsequent replication stress and DNA damage manifest as telomere fragility. We believe this to be the first report placing an Okazaki fragment-processing protein explicitly on the leading strand during DNA replication.
Experimental Procedures
Cell Culture
Cells were cultured at 37 °C in 5% carbon dioxide and atmospheric oxygen, as reported previously (12, 23, 24). 293T cells and HEK 293 cells were cultured in high glucose Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum (ΔFBS) and 1% penicillin/streptomycin (Sigma). BJ fibroblasts and BJ fibroblasts expressing large T antigen (BJL) were cultured in high glucose Dulbecco's modified Eagle's medium containing 15% medium 199 (HEPES modification), 15% ΔFBS, and 1% penicillin/streptomycin. RPE1 cells were cultured in Dulbecco's modified Eagle's medium (F-12 modification) containing 7.5% ΔFBS and 1% penicillin/streptomycin (Sigma). Treatment with α-amanitin (Sigma) was performed at 10 μg/ml for 12 h prior to collection. All cell cultures were verified free of Mycoplasma contamination by PCR analysis. RPE1 cells were obtained from ATCC; all other cells were obtained from Dr. Robert Weinberg (Massachusetts Institute of Technology).
Virus Production and Infections
Lentiviral production and transductions were carried out as reported previously (25). Briefly, 293T cells were transfected with an 8:1 ratio of pHR′-CMV-8.2ΔR packaging plasmid and pCMV-VSV-G and a pLKO.1-puro plasmid carrying an shRNA using TransIT-LT1 (Mirus Bio, Madison, WI). Supernatant-containing virus was collected 48 h post-transfection and 72 h post-transfection and filtered through a 0.45-μm PVDF membrane. Target cells were infected for 4 h each on 2 consecutive days in the presence of 8 μg/ml protamine sulfate (Sigma). Following infection, transduced BJ and BJL cells were selected with 1 μg/ml puromycin sulfate (Sigma); transduced RPE1 cells were selected with 15 μg/ml puromycin sulfate.
Production of recombinant adenovirus type 5 was carried out using the AdEasy adenoviral vector system (Agilent Technologies, La Jolla, CA) according to the manufacturer's protocol. Following collection of primary adenoviral stock, secondary and tertiary viral stocks were prepared by sequential infection of HEK 293 cells and purification from a cesium gradient. Briefly, infected cells were lysed in 0.5% Nonidet P-40, and cell debris was cleared by centrifugation. Viral particles were precipitated from the lysate with 6.7% PEG 8000, 0.83 m sodium chloride, collected by centrifugation, and washed in PBS. Viral particles were suspended in 1.32 g/ml cesium chloride and centrifuged at 33,000 rpm for 18 h at 4 °C in a swinging-bucket rotor. Intact viral particles were collected from the cesium gradient, dialyzed in PBS, suspended in 33% glycerol, and frozen. Viral stocks were quantified using the AdEasy viral titer kit (Agilent Technologies, La Jolla, CA) according to the manufacturer's instructions.
Adenoviral transduction was carried out following lentiviral transduction. Cells were lifted, combined with concentrated adenovirus, and re-plated in media containing puromycin to select for lentiviral integration. Adenovirus was used at a multiplicity of infection of 20 on RPE1 cells. Following 48 h of simultaneous selection and adenoviral infection, the media were replaced.
Western Blot Analysis
Western blots were conducted as described previously (26). Briefly, cells were washed with PBS and lysed in mammalian cell lysis buffer (100 mm sodium chloride, 50 mm Tris-HCl, pH 8, 5 mm EDTA, 0.5% Nonidet P-40) supplemented with 2 mm dithiothreitol, 1 mm microcystin-LR, 2 mm phenylmethylsulfonyl fluoride, 1 mm sodium fluoride, 1 mm sodium orthovanadate, protease inhibitor mixture (Sigma), and phosphatase inhibitor mixture set I (EMD Millipore, Billerica, MA). Following centrifugation, clarified lysate was quantified using the protein assay from Bio-Rad. Lysates were resolved by SDS-PAGE and transferred to PVDF membranes for western blotting. The following antibodies were used: mouse monoclonal anti-Chk1 (sc8408, Santa Cruz Biotechnology, Santa Cruz, CA); rabbit monoclonal anti-Chk1, phospho-Ser-345 (2348, Cell Signaling Technology, Danvers, MA); rabbit polyclonal anti-FEN1 (A300–255A, Bethyl Laboratories, Montgomery, TX); mouse monoclonal anti-RNase H1 (H00246243-M01, Novus Biologicals, Littleton, CO); rat monoclonal anti-α-tubulin (ab6160, Abcam, Cambridge, UK); mouse monoclonal anti-β-catenin (610154, BD Biosciences); rabbit polyclonal anti-γH2AX (07-164, Millipore, Billerica, MA).
Metaphase Chromosome Preparation
Metaphase chromosome spreads were prepared as described previously (27). Briefly, BJ and BJL fibroblasts were cultured in the presence of 0.1 μg/ml colcemid (Sigma) for 5 h; RPE1 cells were cultured in the presence of 0.3 μg/ml colcemid for 4 h. Following arrest, metaphase cells were collected by mitotic shake-off, swollen in 75 mm potassium chloride, and fixed in 3:1 methanol/acetic acid. Chromosomes were spread by dropping onto glass slides and aged for 18 h at 65 °C. When metaphases were to be analyzed by CO-FISH, 0.3 μg/ml 5-bromo-2′-deoxyuridine (Sigma) and 0.1 μg/ml 5-bromo-2′-deoxycytidine (MP Biomedicals, Santa Ana, CA) were added to the culture media 18 h prior to collection of the cells.
Fluorescent in Situ Hybridization (FISH)
FISH was performed as described previously (27). Metaphase chromosomes were probed with a Cy3-(CCCTAA)3 (telomere) peptide nucleic acid (PNA) probe at 0.03 μg/ml and a FAM-CENPB (centromere) PNA probe at 0.03 μg/ml (PNA Bio, Thousand Oaks, CA) and mounted using ProLong Gold (Life Technologies, Inc.) with 125 ng/ml DAPI (Sigma).
Chromosome Orientation-FISH (CO-FISH)
CO-FISH was conducted as described previously (28) with modifications. Briefly, metaphase chromosomes were rehydrated and treated with 100 μg/ml RNase for 10 min at 37 °C, rinsed, and re-fixed in 4% paraformaldehyde for 10 min at room temperature. Chromosomes were UV-sensitized in 0.5 μg/ml Hoechst 33258 (Sigma) in 2× SSC for 15 min and exposed to 365 nm UV light for 60 min using a UV cross-linker (Vilber-Lourmat, Marne-la-Vallée, France). Chromosomes were then digested with 3 units/μl exonuclease III (Promega, Madison, WI) for 15 min at room temperature, denatured in 70% formamide in 2× SSC at 72 °C for 90 s, and immediately dehydrated in cold ethanol before hybridization. Metaphase chromosomes were probed first with a FAM-(TTAGGG)3 (leading strand telomere) PNA probe at 0.03 μg/ml, then probed with a Cy3-(CCCTAA)3 (lagging strand telomere) PNA probe at 0.03 μg/ml (PNA Bio, Thousand Oaks, CA), and mounted as described for FISH.
Immunofluorescence (IF) and IF-FISH
IF was carried out as described previously (29). For IF-FISH, following the completion of IF, the cells were probed as described for chromosomes above using a Cy3-(CCCTAA)3 (telomere) PNA probe at 0.03 μg/ml (PNA Bio). Antibodies used were as follows: rabbit polyclonal anti-γH2AX (07-164, Millipore, Billerica, MA) and goat anti-rabbit IgG-Alexa Fluor 488 (Life Technologies, Inc.).
Fluorescence Imaging
Chromosomes were imaged on a Nikon 90i epifluorescence microscope using a 100× 1.40 NA Plan Apo VC objective (Nikon Instruments, Melville, NY) with Cargille Type FF or Cargille Type LDF immersion oil (Cargille-Sacher Laboratories, Cedar Grove, NJ) at room temperature. Cells were imaged using a 40× 1.0 NA Plan Apo objective (Nikon Instruments) under the same conditions as those for chromosomes. Filter cube sets used were as follows: DAPI-1160B-000-ZERO, FITC-2024B-000-ZERO, and CY3-4040C-000-ZERO (Semrock, Inc., Rochester, NY). Images were captured using a CoolSnap HQ2 CCD camera (Photometrics, Tucson, AZ). Individual channel lookup tables were auto-adjusted nondestructively and linearly, and images were deconvolved with a blind algorithm using NISElements AR (Nikon Instruments) prior to quantification.
RNA Preparation and Northern Hybridization
RNA was prepared using Tri Reagent® (Life Technologies, Inc.). RNA was serially diluted, denatured as described previously (30), and spotted onto a Hybond-XL charged nylon membrane (GE Healthcare) using a Bio-Dot microfiltration apparatus (Bio-Rad) according to the manufacturers' instructions. Samples were also treated with ribonuclease A (Roche Applied Science, Penzberg, Germany) and spotted to identify any DNA contamination in the RNA preparation. Following UV cross-slinking, the membrane was prehybridized in northern hybridization buffer (15% formamide, 1% BSA, 100 mm sodium phosphate, pH 7.7, 1 mm EDTA, 7% SDS) for 1 h at 65 °C. A purified 1.6-kb fragment consisting exclusively of vertebrate telomere repeats was random prime-labeled with [α-32P]dCTP (3000 Ci/mmol) using the High Prime DNA labeling kit (Roche Applied Science) according to the manufacturer's instructions to produce a telomere-specific DNA probe. Similarly, a purified cDNA of the human 5S ribosomal RNA was random prime-labeled to produce a 5S rRNA-specific DNA probe. Probes were purified using Illustra ProbeQuantTM G-50 micro columns (GE Healthcare) and diluted to 1.2 × 106 dpm/ml in 10 ml of northern hybridization buffer. Probes were hybridized to the membrane overnight at 65 °C, after which the membrane was washed and imaged using either autoradiography or a storage phosphor screen and imager. Quantitation was performed in Fiji by first background-subtracting the image and then computing the integrated density for each spot.
Quantitative Reverse Transcription-PCR (qRT-PCR)
For qRT-PCR, cDNA was synthesized using Superscript III reverse transcriptase (Life Technologies, Inc.) according to the manufacturer's instructions. qRT-PCR was conducted using TaqMan Gene Expression Assays (Life Technologies, Inc.) according to the manufacturer's protocol. Target genes used for verification of α-amanitin efficacy were MYC (Hs00153408_m1) and SIAH1 (Hs02339360_m1).
Statistical Analysis
Telomere fragility events were defined as chromatid arms with telomere FISH signal observed as either multiple telomere signals or elongated smears as described previously (7). Fragility was counted in metaphase chromosome spreads; for each experimental condition, a minimum total of 600 chromosomes was counted. The minimum sample size was chosen based on its ability to consistently detect aphidicolin-induced and FEN1 depletion-induced telomere fragility. Chromosomes completely lacking telomere FISH signal or exhibiting no strand specificity in CO-FISH (indicating the technical issue of incomplete brominated strand digestion) were excluded and not quantified. Image groups were blinded prior to quantification. Two or more independent biological replicates were carried out for each experiment.
Where data are shown as representative, the telomere fragility rate was computed for each metaphase chromosome spread (% fragile telomeres), and each experiment was statistically analyzed. Where data are shown as combined, telomere fragility rates were computed for each metaphase chromosome spread, and a normalized value was computed for each metaphase chromosome spread by dividing the raw value by the mean of the control values. The mean of the normalized values from each sample in two independent experiments was computed and graphed with error bars representing the standard error of the mean. For statistical analysis, raw values were centered by computing a t-statistic for each data point: the centered value for each chromosome spread was calculated by dividing the residual of each raw value relative to the control sample's mean by the median absolute deviation of the control values. Centered values from two independent experiments were then combined for statistical analysis. Data are represented either by scatter plots with mean ± standard error of the mean marked by a line and error bars, or by a bar graph with bars indicating the mean and error bars indicating standard error of the mean.
For IF, γH2AX foci were counted in each nucleus. A minimum of 30 nuclei was counted for each condition in an experiment, and two independent biological replicates were combined for data quantification. Data are represented by a box and whiskers plot with the box marking 25th and 75th percentiles, line marking the median, whiskers marking the 5th and 95th percentiles, and dots marking data points outside the 5–95 percentile range.
For all data, p values were computed using a two-tailed Mann-Whitney U test with a 95% confidence interval in Prism 5 (GraphPad Software, La Jolla, CA). The Mann-Whitney U test was chosen because not all samples exhibited normal distributions. All figures except the box and whiskers plot include standard error of the mean as an indicator of variance, and in all cases the variance within samples was similar.
Results
FEN1 Depletion and Transcription Inhibition Induce Replication Stress and a DNA Damage Response
Because telomeres are transcribed to produce telomeric repeat-containing RNA (TERRA) (31, 32), and because interference between replication and transcription is a known cause of genomic instability (5, 33, 34), we turned our attention to the impact that putative collisions between the replication and transcription machinery would have on telomere stability. Unlike in Schizosaccharomyces pombe, where transcription of telomeres and subtelomeres occurs using both strands as templates (35), mammalian telomeres are transcribed exclusively using the C-rich leading strand as a template (31, 32); as such, co-directional collisions are the only type that can occur at the telomere. In bacteria, co-directional collisions are resolved by a mechanism that leaves behind an RNA:DNA hybrid/flap structure (20), which would need to be resolved in a eukaryotic cell to avoid a DDR and genomic instability (21, 22). FEN1 has been previously shown to reduce telomere fragility (12), and the yeast FEN1 homolog Rad27p can hydrolyze RNA flaps (36). We hypothesized that co-directional collisions are a molecular origin of telomere fragility and that FEN1 can prevent post-collision RNA:DNA hybrid/flap structures from accumulating, causing damage, and ultimately leading to fragile telomere formation.
To address this hypothesis, we first examined whether increasing the rate of stochastic collisions between the replisome and RNAP would increase replication stress and trigger a DDR in the context of FEN1 depletion. We treated BJ fibroblasts with the RNA polymerase II (Pol II) elongation inhibitor α-amanitin, a cyclic peptide toxin that reduces the rate of Pol II transcription ∼100-fold, allowing chain elongation to continue without triggering transcript cleavage (37, 38). We expected α-amanitin treatment to increase the frequency of stochastic collisions between the replisome and RNAP and thus increase replication stress and telomere fragility. Following transduction with a validated shRNA targeting the 3′-untranslated region of the FEN1 mRNA (shFEN1) (24) or a control hairpin (shCtrl), we treated BJ fibroblasts with either vehicle or 10 μg/ml α-amanitin for 12 h and collected both RNA and protein lysates from the cells. qRT-PCR analysis confirmed the efficacy of α-amanitin treatment by quantitation of two short lived transcripts, c-Myc and SIAH1. α-amanitin-treated control cells retained 2% and 6% of the c-Myc and SIAH1 mRNAs, respectively, compared with the levels observed in vehicle-treated control cells. Similarly, in FEN1-depleted cells, α-amanitin treatment resulted in 4% and 10% of the levels of c-Myc and SIAH1 mRNAs, respectively, compared with vehicle-treated cells (Fig. 1A). Because transcription inhibition by α-amanitin might reduce steady-state TERRA levels and produce telomere phenotypes as a result of decreased TERRA, we carried out a northern blot analysis of total RNA to detect TERRA. Because TERRA are expressed at low levels in BJ fibroblasts, we utilized a dot blot rather than a gel to maximize signal intensity and hybridized the membrane to a telomere repeat-specific probe; treatment with ribonuclease A was used to show the absence of contaminating DNA, and a 5S rRNA-specific probe was used as a loading control. Northern analysis of vehicle- and α-amanitin-treated cells demonstrated that the α-amanitin treatment conditions subsequently used for western and metaphase analysis did not impact steady-state levels of TERRA in our system, demonstrating that the phenotypes resulting from the treatment were not due to a loss of TERRA, which are known to impact telomere stability (Fig. 1, B and C) (39, 40).
FIGURE 1.

α-Amanitin treatment abrogates expression of mRNAs with short half-lives but does not alter steady-state TERRA levels. A, qPCR analysis of c-Myc and SIAH1 mRNA expression in cells expressing a control hairpin (shCtrl) or FEN1-depleted cells (shFEN1), treated with either vehicle or α-amanitin (α-aman). mRNA levels in α-amanitin-treated cells are shown as a fold change relative to the vehicle-treated cells. Fold changes were calculated using the ΔΔCt method; fold changes from two biological replicates were averaged to produce the graph. Error bars indicate standard error of the mean. B, northern dot blot to detect TERRA. RNA was isolated from cells expressing a control hairpin (shCtrl) or FEN1-depleted cells (shFEN1) that were treated with either vehicle or α-amanitin (α-aman). Serial dilutions of RNA were loaded onto a membrane. Samples treated with RNase A to control for genomic DNA contamination were also loaded (+RNase A). A telomere repeat DNA probe was hybridized to the membrane (telomere probe) to detect TERRA; the membrane was stripped and re-probed with a 5S rRNA DNA probe (5S) as a loading control. The membrane was visualized with autoradiography. C, quantification of TERRA in cells treated with α-amanitin. The northern dot blot in B was imaged with a phosphorimager and analyzed by densitometry using Fiji; TERRA levels in α-amanitin-treated cells are shown as a fold change relative to vehicle-treated cells. Two independent experiments were averaged to produce the graph; error bars represent standard error of the mean.
To determine whether Pol II inhibition induces replication stress and a DDR in the context of FEN1 depletion, we performed western blot analysis to examine phosphorylation of Chk1 at Ser-345 and phosphorylation of histone H2AX at Ser-139 (γH2AX), classical markers for the replication stress response and DDR, respectively. BJ fibroblasts transduced with the control hairpin and treated with vehicle displayed neither Chk1 phosphorylation nor H2AX phosphorylation (Fig. 2A). Treatment with α-amanitin induced a small but detectable increase in Chk1 phosphorylation but did not induce γH2AX, indicating that α-amanitin treatment can induce limited replication stress but is not sufficient to induce a DDR (Fig. 2A). Similarly, BJ fibroblasts depleted of FEN1 and treated with vehicle also displayed a small level of Chk1 phosphorylation and no detectable γH2AX (Fig. 2A). Strikingly, upon treatment with α-amanitin, FEN1-depleted cells showed a robust phosphorylation of Chk1 and strong induction of γH2AX (Fig. 2A).
FIGURE 2.

FEN1 depletion and transcription inhibition induce replication stress, a DNA damage response, and telomere fragility. A, Western analysis of FEN1 expression, Chk1 phosphorylation (pS345), and H2AX phosphorylation (γH2AX) in control (shCtrl) or FEN1-depleted (shFEN1) cells treated with vehicle or α-amanitin (α-aman). β-catenin is shown as a loading control. B, quantification of γH2AX foci per cell. Two independent biological replicates were combined. The box marks the 25th to 75th percentile with the median marked by a horizontal line; whiskers mark the 5th and 95th percentiles, and dots represent values outside the 5–95 percentile range. p values were computed using a two-tailed Mann-Whitney U test (***, p < 0.001 relative to shCtrl). C, representative immunofluorescence images stained with a γH2AX antibody (green) and DAPI (blue) from BJ fibroblasts expressing a control hairpin (shCtrl) or depleted of FEN1 (shFEN1). Cells were treated with vehicle or α-amanitin (α-aman) as indicated. The scale bar (white) represents 25 μm. D, representative quantification of the rate of telomere fragility. p values were computed using a two-tailed Mann-Whitney U test (**, p < 0.01; ***, p < 0.001). Error bars represent standard error of the mean. E, representative metaphase chromosomes processed with FISH from BJ fibroblasts expressing a control hairpin (shCtrl) or depleted of FEN1 (shFEN1). Cells were treated with vehicle or α-amanitin (α-aman) as indicated. Centromeres are green, and telomeres are red. Arrowheads mark fragile telomeres in the magnified images.
We also used IF to examine the formation of γH2AX foci in asynchronous BJ fibroblasts, and IF-FISH to assess the formation of telomere dysfunction-induced foci. Quantification of γH2AX foci demonstrated that although FEN1 depletion induced foci formation (2.14-fold in shFEN1 + vehicle versus shCtrl + vehicle, p < 0.0001), there was no change in γH2AX foci upon treatment with α-amanitin (Fig. 2, B and C). Furthermore, we did not observe an increase in telomere dysfunction-induced foci in response to FEN1 depletion or α-amanitin treatment (data not shown). These results indicate first that the amount of DNA damage induced in conditions that increase collision events causes a response only robust enough to be detected by the more sensitive western analysis. Second, they indicate that FEN1 depletion- and Pol II inhibition-induced replication stress and DNA damage is not restricted to telomeres; rather, DNA damage likely occurs throughout the genome wherever collisions occur. Thus, Pol II inhibition alone induces mild replication stress, and the depletion of FEN1 combined with Pol II inhibition results in a DDR that is not observed when FEN1 is depleted alone.
Inhibition of Transcription Exacerbates the Telomere Fragility Observed upon FEN1 Depletion
We next examined whether the replication stress and DDR phenotypes observed in response to Pol II inhibition and FEN1 depletion manifest as telomere fragility. If failure by FEN1 to resolve the structures induced by collision events between the replisome and RNAP results in fragility, then we anticipated the rate of telomere fragility in α-amanitin-treated and FEN1-depleted cells to mirror the replication stress phenotype. As before, we transduced BJ fibroblasts with either shCtrl (control) or shFEN1 and treated the cells with vehicle or α-amanitin for 12 h prior to collecting metaphase chromosomes. Consistent with our model, cells expressing shCtrl exhibited an increased rate of telomere fragility upon α-amanitin treatment (1.55-fold in shCtrl + α-amanitin versus shCtrl + vehicle, p = 0.0079) (Fig. 2, D and E). When examining only the vehicle-treated cells, we found that as demonstrated previously, FEN1 depletion causes a significant increase in telomere fragility (2.15-fold in shFEN1 + vehicle versus shCtrl + vehicle, p < 0.0001) (Fig. 2, D and E). Strikingly, FEN1-depleted cells treated with α-amanitin displayed a significant 2.76-fold increase in telomere fragility when compared with control and vehicle-treated cells (shFEN1 + α-amanitin versus shCtrl + vehicle, p < 0.0001) and a significant 1.28-fold increase compared with FEN1-depleted, vehicle-treated cells (shFEN1 + α-amanitin versus shFEN1 + vehicle, p = 0.0017) (Fig. 2, D and E). These fragility data mirror the Chk1 phosphorylation phenotype and support a model in which α-amanitin treatment increases co-directional replisome-RNAP collision events that result in structures requiring FEN1 for resolution; without FEN1, the collision events generate replication stress, a DDR, and fragile telomere formation. These experiments suggest that FEN1's role in limiting telomere fragility is dependent upon its ability to resolve structures produced by telomere transcription during DNA replication.
Leading Strand-specific Telomere Fragility Is Caused by RNA:DNA Hybrids
Our data above suggest a role for telomere transcription in telomere fragility induced by FEN1 depletion. Based on findings in prokaryotes, if co-directional collisions occur between the replisome and an RNAP, a structure resembling an Okazaki fragment with a segment of RNA:DNA hybrid would result (20); we postulate that if not resolved, this structure could give rise to fragile telomeres. Indeed, post-collision structures resemble R-loops, which are semi-stable displacement loops in which a nascent mRNA remains hybridized to its DNA template, while the coding strand DNA remains single-stranded, resulting in replication stress and common fragile site expression (5). At common fragile sites, the enzyme RNase H1 suppresses replication stress phenotypes induced by R-loop formation by hydrolyzing the RNA in RNA:DNA hybrids and thus resolving displacement loops (5). We reasoned that because the post-co-directional collision structure resembles an R-loop, RNA:DNA hybrids might be responsible for telomere fragility, and thus ectopic expression of RNase H1 should resolve the structure and telomere phenotype. Additionally, because our model predicts that the causative structure for fragile telomere formation occurs after the replication fork has passed the locus in question, we wondered whether the telomere fragility observed upon FEN1 depletion manifests only on the leading strand, where collisions could occur. This question was especially prescient given that FEN1 is canonically a lagging strand replication protein and has a previously established role in limiting sister telomere loss at the lagging strand (12).
Following lentiviral transduction with a control hairpin (shCtrl) or FEN1-depleting hairpin (shFEN1), we transduced RPE1 cells with RNase H1 (Ad-RH1) (Fig. 3C) and collected cells for protein analysis and metaphase chromosome preparation. To identify whether telomere fragility exhibited strand specificity, we used CO-FISH, a technique that exploits the fact that the C-rich and G-rich strands of the mammalian telomere are replicated exclusively by the leading and lagging strand machinery, respectively, allowing the use of strand-specific probes to identify which machinery replicated a given telomere on a metaphase chromosome (28). Strikingly, FEN1 depletion significantly increased leading strand-specific telomere fragility (2.30-fold in shFEN1 versus shCtrl, p = 0.0021) (Fig. 3, A and B) with no change observed on lagging strand-replicated telomeres (1.26-fold in shFEN1 versus shCtrl) (Fig. 3, A and B). Additionally, ectopic expression of RNase H1 rescued fragility on the leading strand-replicated telomere, returning fragility levels to those observed in control cells (1.19-fold in shFEN1 + Ad-RH1 versus shCtrl) (Fig. 3, A and B). Given the specificity of RNase H1 for RNA:DNA hybrids, these data indicate that RNA:DNA hybrids lead to telomere fragility and suggest that the hybrid/flap structures that arise from co-directional collisions on the leading strand are responsible for the telomere fragility observed upon FEN1 depletion. Furthermore, given that RPE1 cells are telomerase-positive and telomerase expression rescues the sister telomere loss observed upon FEN1 depletion, these data indicate that FEN1's role in limiting telomere fragility at the leading strand is distinct from its known role in limiting sister telomere loss at the lagging strand (12, 24).
FIGURE 3.

RNA:DNA hybrids are responsible for FEN1 depletion-induced leading strand-specific telomere fragility. A, representative metaphase chromosomes processed with CO-FISH from RPE1 cells expressing a control hairpin (shCtrl) or depleted of FEN1 (shFEN1), with or without ectopically expressed RNase H1 (Ad-RH1). Leading strand-replicated telomeres are green, and lagging strand-replicated telomeres are red. Arrowheads mark fragile telomeres in the magnified images. B, representative quantification of the rate of strand-specific telomere fragility, with leading strand-specific telomere fragility shown in green and lagging strand-specific telomere fragility shown in red. p values were computed using a two-tailed Mann-Whitney U test (*, p < 0.05; **, p < 0.01). Error bars represent standard error of the mean. C, western analysis of FEN1 and RNase H1 expression in control (shCtrl) or FEN1-depleted (shFEN1) cells, with or without ectopically expressed RNase H1 (Ad-RH1). Two exposures of the same RNase H1 blot are shown. α-Tubulin is shown as a loading control.
α-Amanitin is known to slow but not disengage the RNAP from the template strand (37, 38), and its use would be expected to increase replisome-RNAP collisions and RNA:DNA hybrids. Thus, we next wanted to determine whether the fragility we observed upon α-amanitin treatment was also RNA:DNA hybrid-dependent. To address this question, we transduced RPE1 cells with Ad-RH1 (Fig. 4C) and treated the transduced cells with α-amanitin for 12 h prior to metaphase collection. As before, α-amanitin treatment induced an increase in telomere fragility (1.79-fold in α-amanitin versus vehicle, p = 0.0008) (Fig. 4, A and B). As in the case of telomere fragility following FEN1 depletion, ectopic RNase H1 expression protected α-amanitin-treated cells from telomere fragility, resulting in levels similar to those observed in cells treated with vehicle (1.05-fold in Ad-RH1 + α-amanitin versus vehicle) (Fig. 4, A and B). Because α-amanitin treatment exacerbates telomere fragility in the absence of FEN1 (Fig. 2, D and E), the ability of RNase H1 to rescue fragility in both α-amanitin-treated (Fig. 4, A and B) and FEN1-depleted cells (Fig. 3, A and B) suggests that FEN1's role in limiting telomere fragility is to resolve RNA:DNA hybrid/flap structures that are produced following replisome-RNAP collisions.
FIGURE 4.

RNA:DNA hybrids are responsible for α-amanitin-induced telomere fragility. A, representative metaphase chromosomes processed with FISH from RPE1 cells with or without ectopically expressed RNase H1 (Ad-RH1) and treated with either vehicle or α-amanitin (α-aman). Centromeres are green and telomeres are red. Arrowheads mark fragile telomeres in the magnified images. B, representative quantification of the rate of telomere fragility. p values were computed using a two-tailed Mann-Whitney U test (***, p < 0.001). Error bars represent standard error of the mean. C, western analysis of RNase H1 expression in cells with or without ectopically expressed RNase H1 (Ad-RH1) treated with vehicle or α-amanitin (α-aman). Two exposures of the same RNase H1 blot are shown. α-Tubulin is shown as a loading control.
FEN1 Flap Endonuclease Activity Is Required for Limiting Telomere Fragility
Given the unprecedented finding that FEN1 limits leading strand-specific telomere fragility, we sought to identify which of FEN1's known functions were necessary for this activity. FEN1 possesses three unique enzymatic activities as follows: an endonuclease activity on unannealed 5′ flaps consisting of either DNA or RNA; a weak exonuclease activity that cleaves nicks, gaps, or recessed 5′ ends of double-stranded DNA; and a gap endonuclease activity that cleaves double-stranded DNA at the 3′ end of a short single-stranded gap (41–43). FEN1 is also known to interact with PCNA via a PCNA-interacting peptide (PIP) box, directly pertaining to its role in DNA replication, and a number of DNA repair proteins via its C terminus, pertaining to its role in base excision repair (44, 45). We utilized a series of previously described FEN1 mutants that impact FEN1's different roles in replication (D181A, ΔP, and ΔPΔC) versus repair (ΔC, D181A, ΔPΔC, and E160D) in genetic knockdown-rescue experiments (Fig. 5A) (12, 24). To test whether the reduction in telomere fragility mediated by FEN1 requires its DNA repair functions, we used a lentiviral vector to express shCtrl (control) alone, shFEN1 alone, or shFEN1 simultaneously with the wild type (WT), ΔC, or D181A allele of FEN1 (Fig. 5A); following transduction we prepared metaphase chromosomes. As before, FEN1 depletion induced leading strand-specific telomere fragility (2.05-fold in shFEN3 versus shLuc, p < 0.0001) (Fig. 5, B and D). Expression of the WT allele of FEN1 rescued the leading strand-specific induction of telomere fragility upon endogenous FEN1 knockdown, indicating that the phenotype is specific to FEN1 knockdown (1.18-fold in shFEN1 + WT versus shCtrl) (Fig. 5, B and D). Unexpectedly, expression of the ΔC allele also rescued FEN1 depletion-induced telomere fragility on the leading strand (1.02-fold in shFEN1 + ΔC versus shCtrl) (Fig. 5, B and D). In contrast to the WT and ΔC alleles, the D181A nuclease-dead allele, which is deficient in all known nuclease activities (46, 47), failed to rescue the phenotype, instead resulting in an increase in leading strand-specific telomere fragility comparable to the expression of shFEN1 alone (1.83-fold in shFEN1 + D181A versus shCtrl, p < 0.0001) (Fig. 5, B and D). Neither knockdown of FEN1 nor expression of any of the mutant alleles of FEN1 altered the level of telomere fragility on the lagging strand, confirming that FEN1 does not play a role in the phenotype on lagging strand-replicated telomeres (Fig. 5, B and D). These data indicate that FEN1's flap endonuclease activity is required to limit leading strand-specific telomere fragility, but its interactions with several DNA repair proteins, including WRN and BLM (deficient in the ΔC allele), and thus its DNA repair activities, are dispensable for this role. Consequently, FEN1's ability to limit leading strand-specific telomere fragility is distinct from its previously described role in telomere stability, which depends upon FEN1's C-terminally mediated DNA repair activity to suppress sister telomere loss on the lagging strand-replicated telomere (12, 24).
FIGURE 5.

FEN1 flap endonuclease activity is required to limit leading strand-specific telomere fragility. A, schematic showing FEN1 alleles used in this study. Features indicated include a PIP box (PIP), nuclear localization signal (NLS), C-terminal region (C), and point mutations. The replication competency, repair competency, and ability to rescue telomere fragility (this study) of each allele are shown to the right. B, representative metaphase chromosomes processed with CO-FISH from BJL fibroblasts expressing a control hairpin (shCtrl) or depleted of FEN1 (shFEN1). Leading strand-replicated telomeres are green, and lagging strand-replicated telomeres are red. FEN1 alleles were ectopically expressed where indicated. Arrowheads mark fragile telomeres in the magnified images. C, representative metaphase chromosomes processed with CO-FISH from BJ fibroblasts expressing a control hairpin (shCtrl) or depleted of FEN1 (shFEN1). Leading strand-replicated telomeres are green, and lagging strand-replicated telomeres are red. FEN1 alleles were ectopically expressed where indicated. Arrowheads mark fragile telomeres in the magnified images. D, quantification of strand-specific telomere fragility per chromosome, with leading strand-specific telomere fragility shown in green and lagging strand-specific telomere fragility shown in red. Two independent biological replicates were analyzed, normalized with shCtrl set to 1 for each mutant group, and combined. p values were computed using a two-tailed Mann-Whitney U test (***, p < 0.001 relative to shCtrl). Error bars represent standard error of the mean.
Given that FEN1's repair activity is dispensable for its ability to limit telomere fragility, and telomere fragility is associated with replication stress, we next investigated whether FEN1's interaction with PCNA, and thus its replication activity, might be important in this role. To test this possibility, BJ fibroblasts depleted of FEN1 were transduced with the WT, ΔP, ΔPΔC, or E160D cDNA of FEN1 (Fig. 5A). Analysis of telomere fragility on metaphase chromosomes revealed that as before, the expression of the WT allele rescued the leading strand-specific induction of telomere fragility following FEN1 depletion (1.58-fold in shFEN1 versus shCtrl, p < 0.0001; 0.88-fold in shFEN1 + WT versus shCtrl) (Fig. 5, C and D). Surprisingly, expression of both the ΔP and E160D constructs also rescued the fragility defect (0.77-fold in shFEN1 + ΔP versus shCtrl; 1.20-fold in BJ shFEN1 + E160D versus shCtrl) (Fig. 5, C and D). Only the ΔPΔC allele, a functionally null allele due to its lack of nuclear localization, failed to rescue the leading strand telomere fragility observed upon FEN1 depletion, resulting in an increase similar to that observed upon FEN1 depletion alone (1.61-fold in shFEN1 + ΔPΔC versus shCtrl, p < 0.0001) (Fig. 5, C and D). As in the previous experiment, none of the FEN1 alleles induced lagging strand-specific telomere fragility (Fig. 5, C and D). These data indicate that FEN1 requires neither its interaction with PCNA (deficient in the ΔP allele) nor its gap endonuclease and exonuclease activity (deficient in the E160D allele) to limit leading strand-specific fragility. In combination with the data from expression of the ΔC and D181A mutants, our experiments identify FEN1 flap endonuclease activity as necessary for its role in limiting telomere fragility. These data are consistent with FEN1's known activities, as it has previously been shown to cleave flap structures with numerous modifications, including flaps composed of RNA (36, 43, 48). As such, our data and the literature support a model in which FEN1's flap endonuclease activity could cleave the RNA:DNA hybrid/flap structures produced following a replisome-RNAP collision event (Fig. 6).
FIGURE 6.
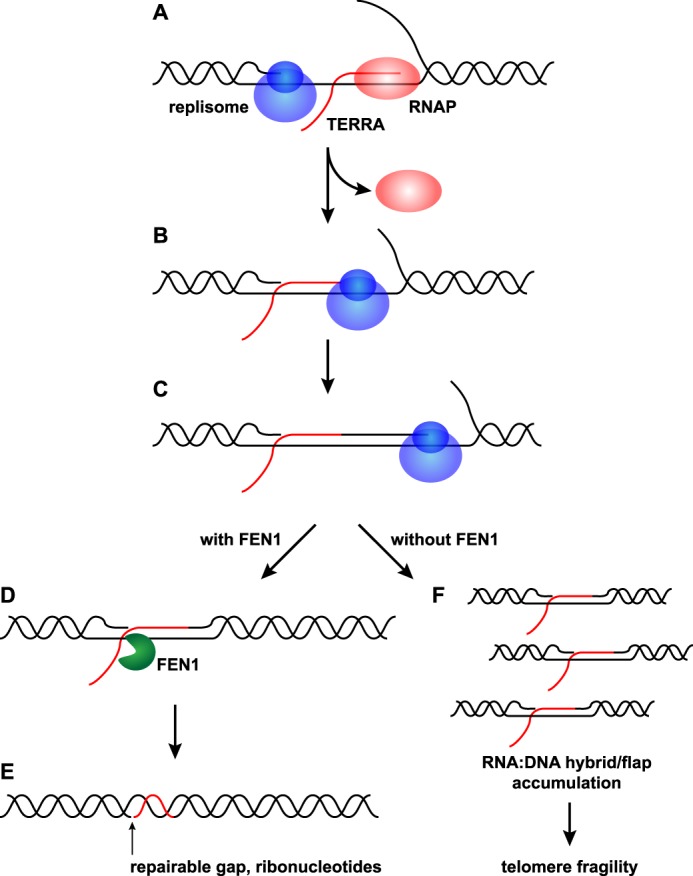
Model of FEN1's role following co-directional replisome-RNAP collisions. A, RNAP transcribes TERRA from the C-rich leading strand. The replisome approaches the transcription complex and a co-directional collision occurs. Pol II dissociates from the nascent TERRA. B, the replisome moves to the 3′ end of the TERRA, leaving a 5′ RNA flap and RNA:DNA hybrid. C, the replisome resumes replication of the leading strand using the 3′ end of the nascent TERRA as a primer. D, FEN1 cleaves the 5′ RNA flap left behind by the collision. E, FEN1's cleavage leaves behind a gap and a stretch of RNA:DNA hybrid that can be repaired. F, in the absence of FEN1, RNA:DNA hybrid/flap structures accumulate and lead to telomere fragility.
Discussion
The role of FEN1 described here provides new insights into the breadth of its functions in maintaining genome stability. In addition to known roles in lagging strand DNA replication, base excision repair, and lagging strand telomere stability, we illustrate for the first time a role for FEN1 in leading strand replication. Furthermore, we have identified transcription as an important contributor to telomere fragility, and we have shown that FEN1 may resolve the RNA:DNA hybrid/flap structures resulting from collisions between the transcription and replication machinery. The strand specificity of telomere fragility observed in the absence of FEN1 shows that it has two independent molecular roles for promoting telomere stability: 1) FEN1 limits sister telomere loss at the lagging strand-replicated telomere by facilitating replication fork reinitiation (12), and 2) FEN1 limits telomere fragility at the leading strand-replicated telomere by resolving RNA:DNA hybrid/flap structures produced by co-directional replisome-RNAP collisions (Fig. 6).
Although co-directional collisions between the replisome and RNAP are postulated to be less deleterious to DNA replication than head-on collisions, they still necessitate mechanisms to ensure replication fidelity. In bacteria, the primary replicative helicase, DnaB, translocates along the lagging strand template as it unwinds DNA ahead of the replication fork; as such, the helicase can move past an RNAP transcribing from the leading strand, which would result in an inevitable collision between the two polymerases (20). Although accessory helicases such as Rep move along the leading strand template, this activity alone cannot prevent co-directional collisions (20, 49). Bacteria thus can use a mechanism in which replication restarts on the leading strand template following a co-directional collision using the 3′ end of the nascent mRNA as a primer (20). Collisions between the replisome and RNAP also present a problem to the eukaryotic cell, where highly transcribed Pol II and Pol III genes are known to impede replication fork progression (33, 34). Extremely long genes that require more than a single cell cycle to transcribe are also known to induce collision events; these collisions induce common fragile site expression (5). Observations suggest that even though the eukaryotic replicative helicase, a complex of Cdc45, Mcm2–7, and GINS (CMG), translocates along the leading strand (50), its activity is insufficient to prevent collisions from occurring. Indeed, CMG is unable to bypass both biotin-streptavidin and Qdot (20 nm) roadblocks on the leading strand (50). Even though the eukaryotic replicative helicase translocates along the leading strand, our data suggest that it is unable to bypass an RNAP and/or RNA:DNA hybrid on this strand. Together, these observations suggest that eukaryotes require a similar mechanism to that used by bacteria for the resolution of co-directional replisome-RNAP collisions on the leading strand.
Although FEN1 has no known existing roles in leading strand DNA replication, our results provide an explanation consistent with the enzyme's known substrates and activity. The putative RNA:DNA hybrid/flap structure produced following a co-directional replisome-RNAP collision is similar to the Okazaki fragment flaps FEN1 cleaves during lagging strand replication, differing only in that the flap is composed entirely of ribonucleotides. Thus, our model suggests that human FEN1 acts at the leading strand because co-directional collisions at the telomere only happen on the leading strand template. Because FEN1's ability to limit telomere fragility does not require its C-terminal domain, which interacts with the shelterin protein TRF2 to recruit FEN1 to telomeres during S and G2 phases of the cell cycle (24, 51), it is unlikely that FEN1's ability to process post-collision structures is limited to the telomere. However, in other portions of the genome where replication begins from origins to either side of a particular locus, transcription could be more coordinated with replication to prevent head-on collisions from occurring. Wherever co-directional collisions occur, FEN1 is likely able to process the structures produced.
Because the replication fork replicates the telomere in the centromere-to-telomere direction only, and because mammalian telomeres are only transcribed from the C-rich leading-strand template in the same direction (31, 32), replisome-RNAP collisions at the telomere can only occur co-directionally. Our work here, as well as the fact that TERRA depletion induces telomere fragility (39), underscores the role of telomere transcription in fragile telomere formation. Indeed, work in yeast has shown that RNA:DNA hybrids produced by TERRA transcription promote recombination-mediated telomere elongation (40). In ALT-positive cells, RNase H1 has recently been shown to regulate the levels of RNA:DNA hybrids between TERRA and telomeric DNA (16). As in yeast, TERRA RNA:DNA hybrids are hypothesized to promote recombination between ALT telomeres. In the absence of RNase H1, hybrids accumulate and promote excessive replication stress that causes fragile telomere formation and telomere loss; conversely, overexpression of RNase H1 reduces TERRA hybrids such that they cannot promote recombination, leading to progressive telomere shortening (16). Strikingly, the telomere loss that occurs following RNase H1 depletion in ALT cells is leading strand-specific (16). This work, when combined with ours, strongly implicates transcription-associated RNA:DNA hybrid formation at the telomere as a contributor to telomere fragility.
Despite the recency of telomere fragility as a defined phenotype, it has been identified in reports manipulating the expression of many proteins involved in DNA replication and telomere stability. ATR deficiency or depletion, BRCA2 deletion, RAD51 depletion, and RECQL1 depletion all induce elevated rates of telomere fragility (7–9, 52, 53). In addition, CTC1 and STN1, both members of the mammalian CST complex, limit telomere fragility (11). Like FEN1, these proteins participate in replication fork progression, replication fork reinitiation, and telomere stability. To our knowledge, no report has identified any perturbation that induces telomere fragility exclusive to the leading or lagging strand, although RNase H1 overexpression has been shown to reduce telomere fragility at the leading strand (16). Indeed, the lack of strand specificity in the telomere fragility produced by TRF1 deletion (7), as well as the involvement of G-quadruplexes (which form exclusively on the lagging strand) in RTEL1 deletion-induced telomere fragility (10), suggests that there are multiple mechanisms leading to fragile telomere formation. Our work underscores the complexity of DNA replication and, in placing the canonical Okazaki fragment-processing protein FEN1 at the leading strand, reveals the first molecular mechanism for fragile telomere formation on the leading strand.
Acknowledgments
We thank Dr. Peter Burgers, Dr. Susana Gonzalo, Dr. Barry Sleckman, Kevin Flanagan, and Megan Ruhland for critical reading of the manuscript. We thank Jingqin Luo for statistical advice.
This work was supported, in whole or in part, by National Institutes of Health Grant GM95924 (to S. A. S.) and Training Grant GM007067 from NRSA (to D. C. T. and E. A.). This work was also supported by the Siteman Cancer Center at Barnes-Jewish Hospital and Washington University School of Medicine (to S. P. and H. R. M.).
- ATR
- ataxia telangiectasia and Rad3-related
- FEN1
- flap endonuclease 1
- CO-FISH
- chromosome-orientation fluorescent in situ hybridization
- RNase H1
- ribonuclease H1
- RNAP
- RNA polymerase
- DDR
- DNA damage response
- PCNA
- proliferating cell nuclear antigen
- BJL
- BJ fibroblasts expressing large T antigen
- PNA
- peptide nucleic acid
- qRT-PCR
- quantitative RT-PCR
- TERRA
- telomeric repeat-containing RNA
- Pol II
- RNA polymerase II
- shFEN1
- shRNA targeting the FEN1 3′-untranslated region
- shCtrl
- control shRNA
- γH2AX
- histone H2AX phosphorylated at Ser-139
- ALT
- alternative lengthening of telomeres
- IF
- immunofluorescence
- CMG
- Cdc45/Mcm2–7/GINS complex.
References
- 1. Sidorova J. M. (2008) Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair 7, 1776–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh D. K., Ahn B., Bohr V. A. (2009) Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology 10, 235–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gilson E., Géli V. (2007) How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 8, 825–838 [DOI] [PubMed] [Google Scholar]
- 4. Burrow A. A., Marullo A., Holder L. R., Wang Y.-H. (2010) Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res. 38, 2865–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Helmrich A., Ballarino M., Tora L. (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977 [DOI] [PubMed] [Google Scholar]
- 6. Letessier A., Millot G. A., Koundrioukoff S., Lachagès A.-M., Vogt N., Hansen R. S., Malfoy B., Brison O., Debatisse M. (2011) Cell type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123 [DOI] [PubMed] [Google Scholar]
- 7. Sfeir A., Kosiyatrakul S. T., Hockemeyer D., MacRae S. L., Karlseder J., Schildkraut C. L., de Lange T. (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martínez P., Thanasoula M., Muñoz P., Liao C., Tejera A., McNees C., Flores J. M., Fernández-Capetillo O., Tarsounas M., Blasco M. A. (2009) Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 23, 2060–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McNees C. J., Tejera A. M., Martínez P., Murga M., Mulero F., Fernandez-Capetillo O., Blasco M. A. (2010) ATR suppresses telomere fragility and recombination but is dispensable for elongation of short telomeres by telomerase. J. Cell Biol. 188, 639–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vannier J.-B., Pavicic-Kaltenbrunner V., Petalcorin M. I., Ding H., Boulton S. J. (2012) RTEL1 Dismantles T Loops and Counteracts Telomeric G4-DNA to Maintain Telomere Integrity. Cell 149, 795–806 [DOI] [PubMed] [Google Scholar]
- 11. Stewart J. A., Wang F., Chaiken M. F., Kasbek C., Chastain P. D., 2nd, Wright W. E., Price C. M. (2012) Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 31, 3537–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saharia A., Teasley D. C., Duxin J. P., Dao B., Chiappinelli K. B., Stewart S. A. (2010) FEN1 ensures telomere stability by facilitating replication fork re-initiation. J. Biol. Chem. 285, 27057–27066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vallabhaneni H., O'Callaghan N., Sidorova J., Liu Y. (2013) Defective repair of oxidative base lesions by the DNA glycosylase Nth1 associates with multiple telomere defects. PLoS Genet. 9, e1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. d'Alcontres M. S., Palacios J. A., Mejias D., Blasco M. A. (2014) TopoIIα prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle 13, 1463–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chawla R., Redon S., Raftopoulou C., Wischnewski H., Gagos S., Azzalin C. M. (2011) Human UPF1 interacts with TPP1 and telomerase and sustains telomere leading-strand replication. EMBO J. 30, 4047–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arora R., Lee Y., Wischnewski H., Brun C. M., Schwarz T., Azzalin C. M. (2014) RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 5, 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rudolph C. J., Dhillon P., Moore T., Lloyd R. G. (2007) Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair 6, 981–993 [DOI] [PubMed] [Google Scholar]
- 18. Liu B., Wong M. L., Tinker R. L., Geiduschek E. P., Alberts B. M. (1993) The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature 366, 33–39 [DOI] [PubMed] [Google Scholar]
- 19. Prado F., Aguilera A. (2005) Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 24, 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pomerantz R. T., O'Donnell M. (2008) The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature 456, 762–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sordet O., Nakamura A. J., Redon C. E., Pommier Y. (2010) DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle 9, 274–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skourti-Stathaki K., Proudfoot N. J. (2014) A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 28, 1384–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stewart S. A., Ben-Porath I., Carey V. J., O'Connor B. F., Hahn W. C., Weinberg R. A. (2003) Erosion of the telomeric single-strand overhang at replicative senescence. Nat. Genet. 33, 492–496 [DOI] [PubMed] [Google Scholar]
- 24. Saharia A., Guittat L., Crocker S., Lim A., Steffen M., Kulkarni S., Stewart S. A. (2008) Flap endonuclease 1 contributes to telomere stability. Curr. Biol. 18, 496–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stewart S. A., Dykxhoorn D. M., Palliser D., Mizuno H., Yu E. Y., An D. S., Sabatini D. M., Chen I. S., Hahn W. C., Sharp P. A., Weinberg R. A., Novina C. D. (2003) Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Honaker Y., Piwnica-Worms H. (2010) Casein kinase 1 functions as both penultimate and ultimate kinase in regulating Cdc25A destruction. Oncogene 29, 3324–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lansdorp P. M., Verwoerd N. P., van de Rijke F. M., Dragowska V., Little M.-T., Dirks R. W., Raap A. K., Tanke H. J. (1996) Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 5, 685–691 [DOI] [PubMed] [Google Scholar]
- 28. Bailey S. M., Cornforth M. N., Kurimasa A., Chen D. J., Goodwin E. H. (2001) Strand-specific postreplicative processing of mammalian telomeres. Science 293, 2462–2465 [DOI] [PubMed] [Google Scholar]
- 29. Duxin J. P., Dao B., Martinsson P., Rajala N., Guittat L., Campbell J. L., Spelbrink J. N., Stewart S. A. (2009) Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 29, 4274–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Chapter 7, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 31. Schoeftner S., Blasco M. A. (2008) Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 10, 228–236 [DOI] [PubMed] [Google Scholar]
- 32. Azzalin C. M., Reichenbach P., Khoriauli L., Giulotto E., Lingner J. (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318, 798–801 [DOI] [PubMed] [Google Scholar]
- 33. Sabouri N., McDonald K. R., Webb C. J., Cristea I. M., Zakian V. A. (2012) DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 26, 581–593 ‘ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Azvolinsky A., Giresi P. G., Lieb J. D., Zakian V. A. (2009) Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 34, 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bah A., Wischnewski H., Shchepachev V., Azzalin C. M. (2012) The telomeric transcriptome of Schizosaccharomyces pombe. Nucleic Acids Res. 40, 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stewart J. A., Campbell J. L., Bambara R. A. (2006) Flap endonuclease disengages Dna2 helicase/nuclease from Okazaki fragment flaps. J. Biol. Chem. 281, 38565–38572 [DOI] [PubMed] [Google Scholar]
- 37. Rudd M. D., Luse D. S. (1996) Amanitin greatly reduces the rate of transcription by RNA polymerase II ternary complexes but fails to inhibit some transcript cleavage modes. J. Biol. Chem. 271, 21549–21558 [DOI] [PubMed] [Google Scholar]
- 38. Bushnell D. A., Cramer P., Kornberg R. D. (2002) Structural basis of transcription: α-amanitin-RNA polymerase II cocrystal at 2.8 A resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 1218–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deng Z., Norseen J., Wiedmer A., Riethman H., Lieberman P. M. (2009) TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 35, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Balk B., Maicher A., Dees M., Klermund J., Luke-Glaser S., Bender K., Luke B. (2013) Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat. Struct. Mol. Biol. 20, 1199–1205 [DOI] [PubMed] [Google Scholar]
- 41. Parrish J. Z., Yang C., Shen B., Xue D. (2003) CRN-1, a Caenorhabditis elegans FEN-1 homologue, cooperates with CPS-6/EndoG to promote apoptotic DNA degradation. EMBO J. 22, 3451–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zheng L., Zhou M., Chai Q., Parrish J., Xue D., Patrick S. M., Turchi J. J., Yannone S. M., Chen D., Shen B. (2005) Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 6, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y., Kao H.-I., Bambara R. A. (2004) Flap endonuclease 1: a central component of DNA metabolism. Annu. Rev. Biochem. 73, 589–615 [DOI] [PubMed] [Google Scholar]
- 44. Li X., Li J., Harrington J., Lieber M. R., Burgers P. M. (1995) Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem. 270, 22109–22112 [DOI] [PubMed] [Google Scholar]
- 45. Guo Z., Chavez V., Singh P., Finger L. D., Hang H., Hegde M. L., Shen B. (2008) Comprehensive mapping of the C-terminus of flap endonuclease-1 reveals distinct interaction sites for five proteins that represent different DNA replication and repair pathways. J. Mol. Biol. 377, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shen B., Nolan J. P., Sklar L. A., Park M. S. (1996) Essential amino acids for substrate binding and catalysis of human flap endonuclease 1. J. Biol. Chem. 271, 9173–9176 [DOI] [PubMed] [Google Scholar]
- 47. Tsutakawa S. E., Classen S., Chapados B. R., Arvai A. S., Finger L. D., Guenther G., Tomlinson C. G., Thompson P., Sarker A. H., Shen B., Cooper P. K., Grasby J. A., Tainer J. A. (2011) Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bornarth C. J., Ranalli T. A., Henricksen L. A., Wahl A. F., Bambara R. A. (1999) Effect of flap modifications on human FEN1 cleavage. Biochemistry 38, 13347–13354 [DOI] [PubMed] [Google Scholar]
- 49. Atkinson J., Gupta M. K., McGlynn P. (2011) Interaction of Rep and DnaB on DNA. Nucleic Acids Res. 39, 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fu Y. V., Yardimci H., Long D. T., Ho T. V., Guainazzi A., Bermudez V. P., Hurwitz J., van Oijen A., Schärer O. D., Walter J. C. (2011) Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146, 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muftuoglu M., Wong H. K., Imam S. Z., Wilson D. M., 3rd, Bohr V. A., Opresko P. L. (2006) Telomere repeat binding factor 2 interacts with base excision repair proteins and stimulates DNA synthesis by DNA polymerase β. Cancer Res. 66, 113–124 [DOI] [PubMed] [Google Scholar]
- 52. Badie S., Escandell J. M., Bouwman P., Carlos A. R., Thanasoula M., Gallardo M. M., Suram A., Jaco I., Benitez J., Herbig U., Blasco M. A., Jonkers J., Tarsounas M. (2010) BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 17, 1461–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Popuri V., Hsu J., Khadka P., Horvath K., Liu Y., Croteau D. L., Bohr V. A. (2014) Human RECQL1 participates in telomere maintenance. Nucleic Acids Res. 42, 5671–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]