

Background: Streptozotocin produces diabetes and diabetic neuropathy in experimental animals.
Results: Streptozotocin stimulates TRPA1 through oxidation of cysteine residues, which leads to acute sensory changes in vivo.
Conclusion: Cysteine oxidation is a novel mechanism of action for streptozotocin.
Significance: The diabetogenic agent streptozotocin induces acute sensory changes prior to the onset of diabetes.
Keywords: diabetes, dorsal root ganglia, pain, reactive nitrogen species, reactive oxygen species, transient receptor potential channels (TRP channels), streptozotocin
Abstract
Streptozotocin (STZ)-induced diabetes is the most commonly used animal model of diabetes. Here, we have demonstrated that intraplantar injections of low dose STZ evoked acute polymodal hypersensitivities in mice. These hypersensitivities were inhibited by a TRPA1 antagonist and were absent in TRPA1-null mice. In wild type mice, systemic STZ treatment (180 mg/kg) evoked a loss of cold and mechanical sensitivity within an hour of injection, which lasted for at least 10 days. In contrast, Trpa1−/− mice developed mechanical, cold, and heat hypersensitivity 24 h after STZ. The TRPA1-dependent sensory loss produced by STZ occurs before the onset of diabetes and may thus not be readily distinguished from the similar sensory abnormalities produced by the ensuing diabetic neuropathy. In vitro, STZ activated TRPA1 in isolated sensory neurons, TRPA1 cell lines, and membrane patches. Mass spectrometry studies revealed that STZ oxidizes TRPA1 cysteines to disulfides and sulfenic acids. Furthermore, incubation of tyrosine with STZ resulted in formation of dityrosine, suggesting formation of peroxynitrite. Functional analysis of TRPA1 mutants showed that cysteine residues that were oxidized by STZ were important for TRPA1 responsiveness to STZ. Our results have identified oxidation of TRPA1 cysteine residues, most likely by peroxynitrite, as a novel mechanism of action of STZ. Direct stimulation of TRPA1 complicates the interpretation of results from STZ models of diabetic sensory neuropathy and strongly argues that more refined models of diabetic neuropathy should replace the use of STZ.
Introduction
Diabetes induced by administration of streptozotocin (STZ)3 has long been the most widely used rodent model for studies of diabetes and diabetic complications. Sensory neuropathy is one of the most common and serious long term complications of diabetes, and the prevalence of diabetic neuropathy is rising with the global increase in type 2 diabetes (1, 2). Pain and loss of sensation are major symptoms associated with diabetic neuropathy and constitute an immense burden to patients and society (1, 3). Pain is an early manifestation of neuropathy in both type 1 and type 2 diabetic patients, with about half of patients experiencing pain particularly during the early stages when they suffer from a range of symptoms, including burning and shooting pains and aches initially in the distal limbs (4). Although loss of sensitivity is the most common manifestation of sensory neuropathy, some patients also experience hypersensitivities to mechanical (pressure and light touch) and thermal (cold/heat) stimuli. In rodent studies of STZ-induced diabetic neuropathy, rats typically display mechanical and thermal hyperalgesia, whereas mice develop mechanical and thermal hypoalgesia (5). The lack of understanding of the pathogenesis of diabetic neuropathy and pain has hampered the development of mechanism-based therapies. The major current treatments target the management of pain with anti-convulsant, anti-depressant, and opioid drugs that often have limited efficacy or unacceptable side effects (4).
The ion channel TRPA1 is expressed in a subset of nociceptive sensory neurons where it acts as a promiscuous receptor for oxidants and electrophilic agents, but it is also activated by a number of nonreactive compounds (6). Covalent modification of cysteine residues present in the intracellular N terminus of TRPA1 is thought to underlie the agonist activity of oxidants and electrophilic agonists (7, 8). Several inflammatory mediators and transmitters sensitize or activate TRPA1 indirectly through G-protein-coupled receptors (9, 10). Studies with RNAi knockdown or pharmacological inhibitors have shown that TRPA1 plays an important role in the development of mechanical and cold hyperalgesia in models of inflammatory and neuropathic pain (11–13). TRPA1 has attracted interest for its potential role in painful diabetic neuropathy, because inhibition of TRPA1 prevents development of mechanical hyperalgesia in the STZ rat model of diabetes (14, 15). In addition, the glucose metabolite methylglyoxal and several other electrophilic, harmful metabolites formed during hyperglycemia are endogenous TRPA1 agonists (16–18) and may be responsible for hyperalgesia in diabetic neuropathy (19).
Here, we have examined the sensory effects of topical and systemic STZ treatment before the onset of diabetes and the development of diabetic neuropathy. We noticed that topical STZ produced a rapid proalgesic effect, whereas systemic administration of STZ evoked a loss of mechanical and cold sensitivity within an hour of injection. We have identified TRPA1 as the target for the pronociceptive actions of STZ. Using TRPA1-expressing cell lines and cultured sensory neurons, we demonstrate that STZ stimulates TRPA1 directly. Finally, we show that STZ decomposition generates peroxynitrite, which oxidizes TRPA1 cysteine residues found in the TRPA1 N terminus, in good agreement with our current understanding of how oxidants and electrophilic compounds activate TRPA1.
Materials and Methods
Behavioral Experiments
Animal studies were performed according to the UK Home Office Animal Procedures Act (1986) after in-house ethical review. C57BL/6J mice were obtained from Harlan UK Ltd., and Trpa1−/− mice were bred from heterozygous mice kindly provided by Drs. Kelvin Kwan and David Corey (Harvard Medical School, Boston) (20). The mice were backcrossed onto the C57Bl/6J background for 10 generations prior to this set of experiments. The TRPA1 antagonist AP18 was prepared in PBS containing 1% DMSO and 0.5% Tween 80 and administered by either intraperitoneal (2.5 mg/kg in 0.3 ml) or intraplantar (5 μg in 25 μl) injections. STZ stock solutions were prepared in 0.1 mm citric acid (pH 4.5) and adjusted to pH 7.4 (NaHCO3) just before administration. STZ was administered by intraplantar (1–100 μg in 25 μl) or intraperitoneal (180 mg/kg in 0.2 ml) injections. Blood glucose levels were monitored using a Contour XT blood glucose meter (Bayer Healthcare). Before nociceptive testing, the mice were kept in their holding cages to acclimatize (10–15 min) to the experimental room. Thermal sensitivity was assessed using a commercially available hot and cold plate (Ugo Basile, Milan, Italy), by placing a hind paw of a lightly restrained mouse onto the plate surface and measuring the latency to paw withdrawal as described previously (21). Paw withdrawal latencies were determined with the plate set at a chosen temperature (50 °C for hot plate and 10 °C for cold plate tests). Mechanical sensitivity was assessed by measuring paw withdrawal thresholds to an increasing mechanical force applied to the dorsal surface of the paw using an Analgesymeter (Ugo-Basile). The nociceptive threshold was defined as the force in grams at which the mouse withdrew its paw. To avoid tissue injury, the maximum force applied was set to 150 g.
Cell Culture
DRG neurons were prepared from adult male or female Trpa1+/+ and Trpa1−/− C57Bl/6J mice using methods described previously (22). Isolated neurons were plated on poly-d-lysine-coated coverslips and maintained at 37 °C in an atmosphere of 95% air, 5% CO2 in MEM AQ (containing 5.6 mm glucose, Sigma, Poole, UK) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml NGF (Promega, Southampton, UK) and used for experimentation within 48 h. Untransfected CHO cells and CHO cells expressing mouse TRPA1 were grown in MEM AQ supplemented with penicillin (100 units/ml), streptomycin (100 μg/ml), FCS (10%), and hygromycin (200 μg/ml). TRPA1 expression was under the control of a tetracycline-inducible promoter, and expression was induced by addition of tetracycline before experimentation (23). HEK 293t cells were transfected with plasmids of hTRPA1 or mutant hTRPA1 (1 μg each) using Nanofectin (PAA, Pasching, Austria). 24 h after transfection, cells were plated onto poly-l-lysine-coated coverslips and used for calcium imaging experiments the same day. Human TRPA1 cDNA and cDNA of a mutant hTRPA1 lacking cysteine residues with or without lysine 710 in the intracellular domain (C621S/C641S/C665S±K710R) were the kind gift from Dr. Sven-Eric Jordt (Department of Pharmacology, Yale University, New Haven, CT). For some experiments, HEK 293t cells stably expressing hTRPA1 were used.
[Ca2+]i Measurements
CHO, HEK 293t cells, and DRG neurons were loaded with 2.5–3 μm Fura-2 AM in the presence (CHO and DRG) or absence (HEK 293t) of 1 mm probenecid for ∼45–60 min in a physiological saline solution containing (in millimolars) either 140 NaCl, 5 KCl, 10 glucose, 10 HEPES, 2 CaCl2, and 1 MgCl2, buffered to pH 7.4 (NaOH) or NaCl 145, KCl 5, CaCl2 1.25, MgCl2 1, glucose 10, HEPES 10. Images of groups of cells were captured every 1 or 2 s at 340 and 380 nm excitation wavelengths with emission measured at >520 nm with a microscope-based imaging system (either PTI, New Jersey, or Visitron Systems GmbH, Puchheim, Germany). Analyses of emission intensity ratios at 340/380 nm excitation (R, in individual cells) were performed with ImageMaster or VisiView 2.1.1 software. In experiments on HEK cells, the background fluorescence was subtracted before calculation of ratios; cells were stimulated with 250 μm carvacrol to functionally identify transfected cells, and 5 μm ionomycin was applied as a control at the end of each experiment. Averaged results are reported as means (± S.E.) of area under the curve (Δ ratio F340/380 nm).
Electrophysiology
CHO cells and DRG neurons were studied under voltage clamp conditions using an Axopatch 200B amplifier and pClamp 10.0 software (Axon Instruments). Borosilicate glass pipettes (3–6 megohms, 75–80% series resistance compensation) were filled with (in millimolars) 140 KCl (CHO cells) or CsCl (DRG neurons), 1 CaCl2, 2 MgATP, 10 EGTA, and 10 HEPES (pH 7.4 with KOH or CsOH as appropriate). The physiological saline solution detailed for the Ca2+ measurements above was used extracellularly. Cells studied under Ca2+-free conditions were superfused with (in millimolars) 140 NaCl, 5KCl, 1 MgCl2, 1 EGTA, 10 HEPES, and 10 glucose (pH 7.4, NaOH), and the pipettes were filled with 140 KCl, 2 MgATP, 5 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, and 10 HEPES (pH 7.4, KOH). Inside-out patches were recorded with (in millimolars) 140 NaCl, 1 MgCl2, 1 EGTA, 10 HEPES (pH 7.4, NaOH) in the pipette. The intracellular face of the patch was superfused with (in millimolars) 140 KCl, 2 MgATP, 1 EGTA, and 10 HEPES (pH 7.4, KOH). Streptozotocin was diluted as late as possible, usually after a giga-seal had been established. HEK 293t cells were recorded using an EPC10 HEKA amplifier (HEKA Elektronik, Lamprecht, Germany). Pipettes were filled with (in millimolars) KCl 140, MgCl2 2, EGTA 5, HEPES 10 (pH 7.4 with KOH), and the external solution contained (in millimolars) NaCl 140, KCl 5, MgCl2 2, CaCl2 2, HEPES 10, and glucose 10 (pH 7.4 with tetramethylammonium hydroxide).
Mass Spectrometry
A model peptide of the intracellular N-terminal sequence of hTRPA1 comprising amino acids 607–670 (UniProt database, O75762; Thermo Fisher Scientific, Schwerte, Germany) was synthesized. Ultra high resolution ESI-TOF mass spectrometry was performed on a maXis mass spectrometer (Bruker Daltonics, Bremen, Germany) on 50 μm peptide in 20 mm ammonium bicarbonate buffer (pH 7.4) that was treated with 500 μm STZ for 75 min. Additionally, one set of samples was incubated with STZ and 1 mm dimedone for 75 min. After the treatment, samples were incubated with iodoacetamide (45 min at 50 °C), mixed with acetonitrile (1:1, v/v), and 0.1% formic acid and sprayed directly into the ion source. The instrumental parameters were as follows: injection rate 180 μl/h; source temperature 320 °C; capillary voltage 4.5 kV; and collision voltage 10 kV. All experiments were carried out in the positive ion mode, and the obtained spectra were deconvoluted and further processed in data analysis software provided by Bruker Daltonics. For the time-resolved MS, STZ was added to 20 mm ammonium carbonate buffer (pH 7.4), and the spectra were continuously monitored during 45 min.
Cysteine Oxidation, Dihydrorhodamine 123 Oxidation, and Dityrosine Formation
The effect of STZ on cysteine oxidation was assessed by Ellman's reaction, whereas the effects on dihydrorhodamine oxidation and dityrosine formation were studied by following the characteristic emission wavelengths, as described previously (24).
Measurement of TRPA1 Disulfide Formation in Vivo
Dorsal root ganglia were dissected before and 1 and 24 h after STZ injection (180 mg/kg) mice. Ganglia from two animals were pooled together and lysed in N-ethylmaleimide-containing buffer, and TRPA1 was purified by immunoprecipitation (25). The protein was then treated with DTT, desalted on a BioSpin column, and treated with Cy3-N-ethylmaleimide. The samples were run on gradient gels, and the band was visualized by using a ChemiDocTM MP System (Bio-Rad).
Drugs and Chemicals
Fura-2 AM was from Life Technologies, Inc., and dissolved in DMSO with 10% pluronic acid (Life Technologies, Inc.). MnTM-PyP-CL5 was provided by M. R. Filipovic. AP18 was from Maybridge (Tintagel, Cornwall, UK) and HC030031 from TOSLab (Ekaterinburg, Russia). Streptozotocin, salts, and all other reagents were from Sigma. STZ stock solutions were prepared in 0.1 mm citrate (pH 4.5) and adjusted to pH 7.4 just before administration. AP18 was dissolved in 1% DMSO, 0.5% Tween 80, in 0.9% saline for behavioral experiments and in DMSO for cellular assays.
Statistics
Differences in cellular and behavioral response amplitudes, thresholds, and latencies were analyzed by Student's t test, Mann-Whitney U test, or analysis of variance followed by Tukey's HSD test.
Results
Nociceptive Effects of Streptozotocin
We initially performed a dose-response experiment with intraplantar (i.pl.) injections of STZ (1–100 μg in 25 μl) in C57Bl/6J mice to determine whether STZ exerts a local effect on sensory function in vivo. The sensitivities to mechanical (paw pressure test) and cold stimulation (cold plate) were monitored for 24 h after injections to establish the time course of any effects. Intraplantar STZ rapidly exerted a pronociceptive effect and reduced the mechanical withdrawal threshold significantly at each examined dose, whereas vehicle administration was without effect (Fig. 1A). The mechanical hypersensitivity was evident within 30 min of administration and showed no sign of reversal within 6 h, but after 24 h, the paw withdrawal thresholds had returned to the naive, preinjection levels. STZ (1–100 μg, i.pl.) produced a similar but less pronounced reduction in the paw-withdrawal latency in the cold plate test, with a somewhat delayed onset compared with the mechanical hypersensitivity (Fig. 1B).
FIGURE 1.
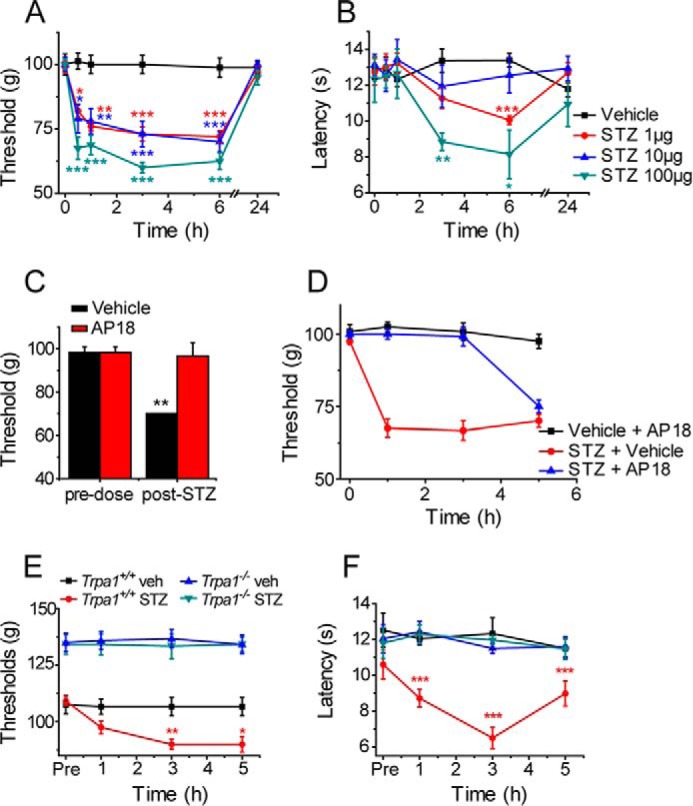
STZ is pronociceptive. Intraplantar injections of STZ reduced the paw pressure test withdrawal threshold (A) and cold-plate withdrawal latency (B) in mice (n = 6). C, effect of STZ (1 μg, i.pl.) on the paw withdrawal threshold in mice, measured 30 min after administration of AP18 (2.5 mg/kg i.p., red columns) or vehicle (black columns). AP18 or vehicle was administered 5 h after STZ (n = 3). D, local intraplantar AP18 (5 μg) inhibited the STZ-evoked mechanical hyperalgesia when co-administered with 1 μg of STZ (n = 6). STZ (1 μg) evoked mechanical (E) and heat (F) hyperalgesia in wild type but not in Trpa1−/− mice (n = 6). Data are mean ± S.E. of the indicated number of mice; *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with vehicle (veh), Tukey's HSD test, or t test (C)).
TRPA1 mediates cellular and behavioral responses to stimulation with a number of oxidants and electrophilic chemicals (20, 26). In vivo, activation of TRPA1 by locally administered agonists evokes a mechanical hyperalgesia and cold hypersensitivity (20, 27) similar to that observed here following local injections of STZ. To determine whether TRPA1 was responsible for the pronociceptive effect of STZ, we examined the effect of the selective TRPA1 antagonist AP18 (12). Intraperitoneal injections of AP18 (2.5 mg/kg, 5 h after 1 μg i.pl. STZ) completely inhibited the STZ-induced reduction in paw withdrawal threshold (Fig. 1C). Furthermore, i.pl. co-injection of AP18 (5 μg) together with STZ (1 μg) prevented the development of mechanical hyperalgesia for at least 3 h, demonstrating that AP18 and STZ act locally in the paw (Fig. 1D). At later time points when AP18 is likely to have been cleared from the tissue (12), mechanical hyperalgesia developed to a similar level to that observed in the vehicle-treated STZ group.
We next examined the effect of i.pl. injections of STZ (1 μg) in Trpa1+/+ and Trpa1−/− mice. STZ produced the expected pronounced mechanical hyperalgesia in wild type mice (Fig. 1E). In contrast, STZ was without any effect on the paw withdrawal threshold in Trpa1−/− mice, suggesting an obligatory role for TRPA1 in the acute STZ-evoked hyperalgesia (Fig. 1E), consistent with the inhibitory effects of AP18 noted above. TRPA1 is primarily expressed in TRPV1-positive, heat-sensitive nociceptive neurons, but it is not involved in the transduction of heat, either in vivo or in vitro. Nevertheless, TRPA1 activation in vivo elicits thermal hyperalgesia (26, 28) in addition to mechanical and cold hypersensitivity (20, 27). Although the baseline sensitivity to noxious mechanical and cold stimulation is reduced in Trpa1−/− mice (Fig. 1E) (20, 26–28), they display normal sensitivity to noxious heat. Sensitivity to noxious heat therefore provides an ideal test to assess involvement of TRPA1 in the absence of phenotypic differences between naive Trpa1+/+ and Trpa1−/− mice. In the hot plate test, local administration of STZ (1 μg i.pl.) reduced the paw withdrawal latency significantly in wild type mice but was without any effect on the latency in TRPA1-null mice (Fig. 1F), demonstrating that TRPA1 is necessary for STZ-mediated induction of acute thermal hyperalgesia.
Systemic STZ Produces Acute Sensory Loss
We next examined the effect of a larger systemic dose of STZ (180 mg/kg, intraperitoneal injection), commonly used to induce diabetes in mice. We first monitored blood glucose levels of wild type and Trpa1−/− mice injected intraperitoneally with STZ (180 mg/kg) to determine whether TRPA1 influenced STZ-mediated induction of diabetes (29). STZ (180 mg/kg) induced hyperglycemia of similar severity, onset, and duration in mice of the two genotypes (Fig. 2A). When the early phase of induction was monitored closely, it was clear that hyperglycemia developed around 3 days after STZ administration in both wild type and Trpa1−/− mice (Fig. 2B). In parallel experiments, we monitored the nociceptive thresholds in mice after STZ injections (180 mg/kg, intraperitoneal). Strikingly, the paw pressure withdrawal threshold was significantly increased in wild type mice within 1 h, and this reduced sensitivity was maintained for at least 10 days (Fig. 2C). In wild type mice, we noticed a similar rapid loss of sensation to stimulation with noxious cold, which lasted for at least 10 days (Fig. 2D). In contrast, the sensitivity to noxious heat was not altered significantly by STZ in wild type mice during the 10-day observation period (Fig. 2E).
FIGURE 2.
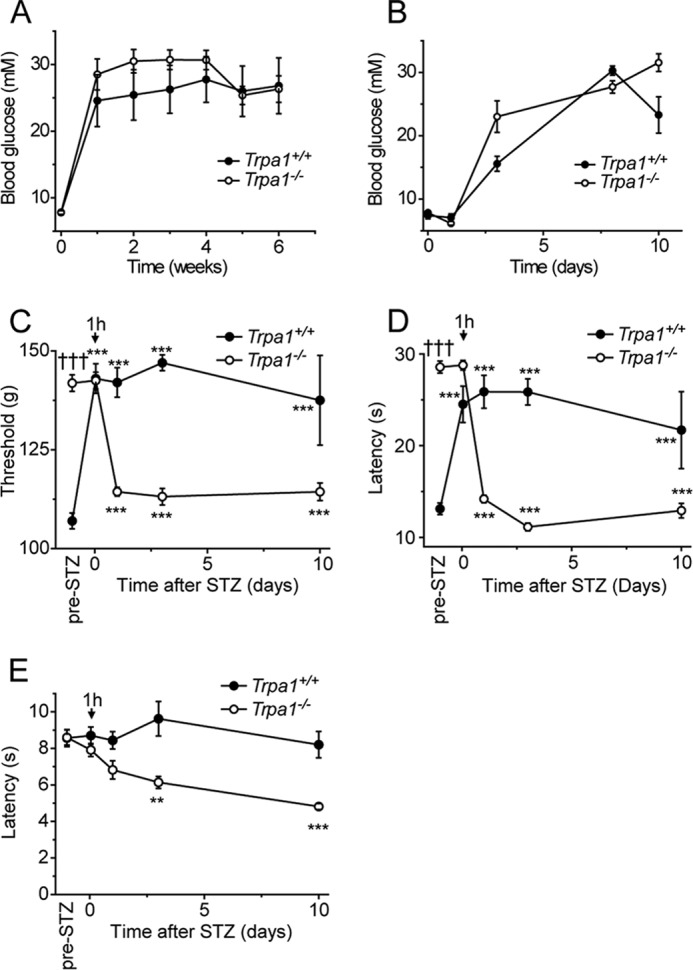
Systemic STZ treatment evokes TRPA1-dependent hypoalgesia. A, intraperitoneal injections of STZ (180 mg/kg) induce hyperglycemia of similar severity and duration in Trpa1+/+ and Trpa1−/− mice. B, early time course of STZ-evoked hyperglycemia is similar in wild type and Trpa1−/− mice. Hyperglycemia is established about 3 days after STZ administration. Within 1 h of STZ (180 mg/kg) injection, wild type mice developed mechanical (C) and cold (D) hyposensitivity, which lasted for at least 10 days. STZ increased the sensitivity to cold and mechanical stimulation in Trpa1−/− mice after 24 h. E, STZ was without effect on the sensitivity to noxious heat in wild type mice but elicited a progressive thermal hyperalgesia in Trpa1−/− mice. Data are mean ± S.E. of n = 4–8; **, p < 0.01; ***, p < 0.001 compared with preinjection value; †††, p < 0.001 compared with wild type, Tukey's HSD test.
Naive Trpa1−/− mice display a marked loss of mechanical and cold sensitivities, as demonstrated previously (Fig. 1E) (20, 21, 27, 28), but normal sensitivity to noxious heat (Fig. 2, C–E). In contrast to our observations in wild type mice, intraperitoneal administration of STZ (180 mg/kg) did not alter the mechanical or cold sensitivity and similarly had no significant effect on heat sensitivity after 1 h in Trpa1−/− mice. However, at later time points, an STZ-mediated significant hypersensitivity developed in Trpa1−/− mice in all three tests (Fig. 2, C–E). These results demonstrated that the systemic dose of STZ used to induce diabetes in mice produces sensory abnormalities through multiple mechanisms during the first 24 h after injection. Importantly, our findings demonstrate that TRPA1 is required for the pronounced early mechanical and cold hypoalgesia seen in wild type mice after systemic administration of STZ.
Streptozotocin Is a TRPA1 Agonist
The observation that TRPA1 is required for the acute nociceptive effects produced by topical STZ prompted us to examine whether STZ stimulates TRPA1 directly. In these experiments we used Fura-2 to monitor changes in intracellular calcium concentration ([Ca2+]i) in cultured DRG neurons and in CHO cells expressing mouse TRPA1. STZ evoked [Ca2+]i increases with an EC50 of 150 μm in CHO cells expressing TRPA1 (Fig. 3A), but it was without effect on the [Ca2+]i in untransfected CHO cells (data not shown). In cultured DRG neurons, STZ elicited concentration-dependent [Ca2+]i responses in a subpopulation of neurons with an EC50 value of 140 μm, almost identical to that produced in TRPA1 CHO cells (Fig. 3B). Pharmacological inhibition of TRPA1 (AP18 20 μm) reduced the proportion of DRG neurons responding to STZ (1 mm) from 22% of DRG neurons (213 of 956 neurons examined) to 5% (22 of 414 neurons). Finally, we examined the effect of STZ (500 μm) on DRG neurons cultured from Trpa1+/+ and Trpa1−/− mice. As TRPA1 is mainly present in TRPV1-expressing, capsaicin-sensitive DRG neurons (23, 30), we restricted the analysis to these neurons. STZ stimulated [Ca2+]i responses in 41% (139 of 341) of capsaicin-sensitive DRG neurons in cultures from Trpa1+/+ mice (Fig. 3C), but it failed to produce any [Ca2+]i responses (0 of 257) in these neurons isolated from Trpa1−/− mice (Fig. 3D). From these experiments, we conclude that TRPA1 mediates STZ responses in DRG neurons as well as the acute pronociceptive effect produced by STZ in vivo.
FIGURE 3.
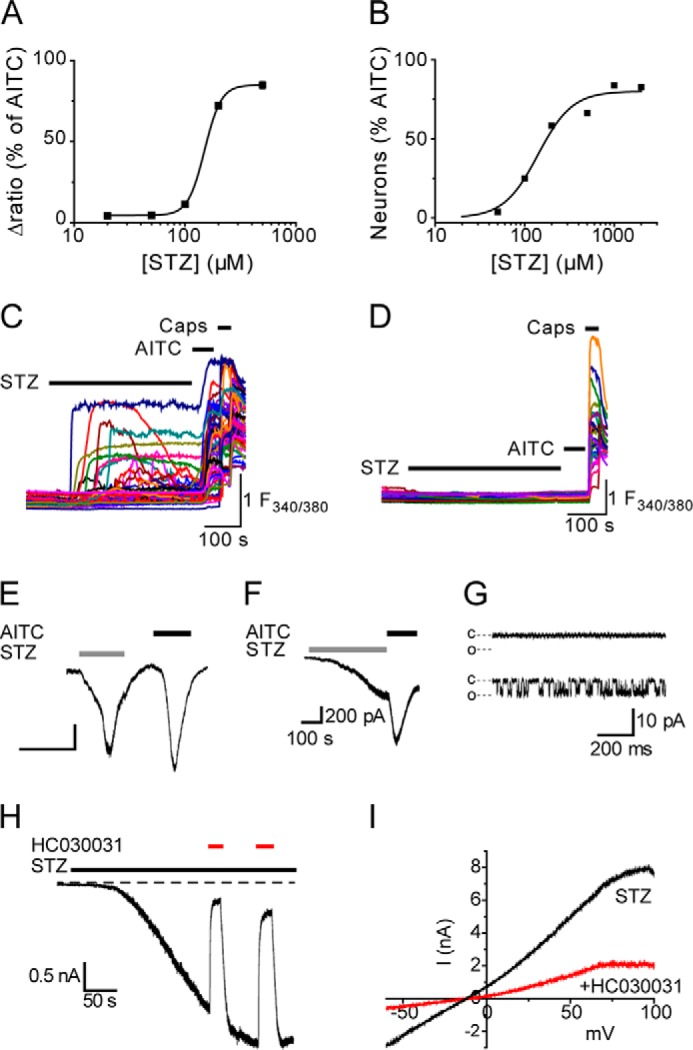
STZ stimulates TRPA1. A, concentration-dependent [Ca2+]i increases evoked by STZ in mTRPA1 CHO cells loaded with Fura-2. Data are the mean percentages ± S.E. (n = 168–648 cells per concentration) of the maximal response amplitude produced by a subsequent challenge with AITC (50 μm). B, concentration-dependent [Ca2+]i responses evoked by STZ in DRG neurons. Data points show the percentage of AITC-sensitive neurons that also responded to an initial challenge with STZ (n = 117–236 AITC-sensitive DRG neurons per concentration). Application of STZ (0.5 mm) stimulated [Ca2+]i responses in TRPA1-positive (AITC-sensitive) neurons cultured from Trpa1+/+ mice (C) but was without effect on [Ca2+]i in neurons from Trpa1−/− mice (D). Fura-2 emission intensity ratio is indicated by F340/380. STZ (1 mm)-induced current responses recorded from mTRPA1 CHO cells (E, n = 5), AITC-sensitive DRG neurons (F, representative of n = 4), and excised inside-out patches from TRPA1 CHO cells (G). In G, the top trace indicates a lack of single channel activity prior to STZ application; the bottom trace shows STZ-evoked channel activity. The closed and open channel current levels are indicated by c and o (the trace is representative of n = 3 patches). Currents were recorded at a holding potential of −60 mV. H, TRPA1 currents evoked by STZ in CHO cells were reversibly inhibited by HC030031 (50 μm, representative of n = 5). I, current-voltage relationship of TRPA1 current evoked by STZ in the presence and absence of HC030031 (50 μm). Experiments with HC030031 were performed under Ca2+-free conditions.
In voltage clamp experiments, application of STZ evoked inward whole-cell currents in mTRPA1 CHO cells (Fig. 3E) and in AITC-sensitive, cultured DRG neurons (Fig. 3F), consistent with activation of TRPA1. Furthermore, recordings from inside-out membrane patches excised from mTRPA1 CHO cells demonstrated that STZ stimulated single channel activity by a membrane-delimited mechanism (Fig. 3G). STZ-evoked TRPA1 currents were completely and reversibly inhibited by co-application of the selective TRPA1 antagonist HC030031 (50 μm; Fig. 3, H and I).
Covalent Modification of N-terminal Cysteine Residues in TRPA1
Oxidants and electrophilic agonists are thought to stimulate TRPA1 through covalent modification of cysteine residues located in the intracellular N terminus (7, 8, 31). To investigate the importance of these N-terminal cysteine residues in streptozotocin-induced activation of TRPA1, a custom-made synthetic peptide consisting of 64 amino acids of the human TRPA1 channel orthologue (residues 607–670) was used. This peptide contained three essential cysteine residues (Cys-621, Cys-641, and Cys-665) as well as three additional neighboring cysteines, all of which are conserved in the mouse TRPA1 channel. The peptide was treated with 10-fold molar excess of STZ for 75 min, then incubated with 50-fold excess of iodoacetamide (IA), and analyzed by HPLC-ESI-TOF-MS. The control untreated peptide showed only one species with a monoisotopic peak at m/z 8003.5, which corresponds to molecular weight of the peptide with all six cysteines alkylated by IA (Δm/z = 6 × 57; m/z 7661.53 for unmodified peptide, Figs. 4A and 5). STZ treatment led to the appearance of several new species with a lower m/z suggesting that some cysteines were modified by the STZ treatment and were not available for the reaction with IA. Two key species were identified, one species with four alkylated cysteines and the remaining two cysteines in the form of a disulfide and one species with two alkylated cysteines and two disulfides (Fig. 4B). In addition, we identified another species with three alkylated cysteines, one disulfide, and one cysteine in the form of a cysteinyl radical. As cysteinyl radicals could be further oxidized to sulfenic acids and/or sulfonic acids, which could be an irreversible form of TRPA1 activation (32), an experiment was performed where the sulfenic acid trap dimedone was added to the reaction mixture containing the peptide and STZ. After 75 min, the reaction mixture was analyzed to reveal the presence of dimedone-labeled peptide confirming that sulfenic acids are formed during STZ-induced cysteine oxidation (Figs. 4C and 6). To identify the exact position of these modifications, MS/MS analysis of the peaks was performed. The data are suggestive of disulfides being formed between Cys-621 and Cys-633 and between Cys-641 and Cys-651 (Fig. 4D and Table 1). The presence of disulfides, cysteinyl radicals, and sulfenic acids implies that STZ decomposition products act as thiol oxidizing agents, which could be the mechanism by which STZ activates TRPA1 channels.
FIGURE 4.

STZ oxidizes critical cysteine residues of TRPA1 N terminus. A, MS spectrum of the TRPA1 peptide used in the study. B, MS spectrum of peptide (50 μm) treated with STZ (500 μm) for 75 min. After the reaction, the remaining free cysteines were blocked with IA. C, dimedone trapping of sulfenic acid formed in the reaction of STZ and the peptide. AU, arbitrary units. D, actual positions of the two disulfide bonds formed when TRPA1 peptide was exposed to STZ. The positions of critical cysteines are indicated by the numbers given in parentheses. Cysteines marked yellow are found to form intramolecular disulfides. The cysteine residue, which also forms sulfenic acid and is observed in MS analysis as labeled with dimedone, is marked green. For more details see Figs. 5 and 6 and Table 1. E, TRPA1 protein purified from DRGs collected from control mice (0 h) contained a basal level of disulfides. STZ increased the content of oxidized thiols in TRPA1 protein at 1 and 24 h after injection. The lower panel indicates the total protein load.
FIGURE 5.
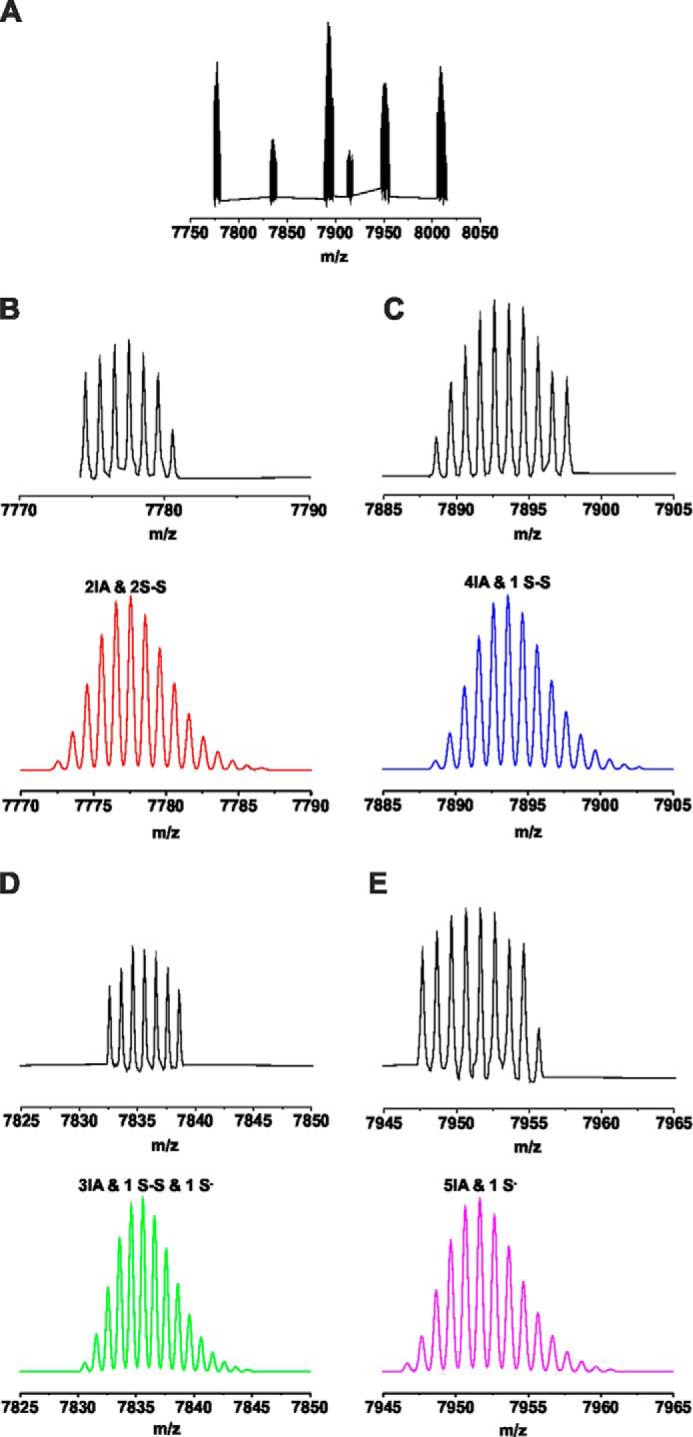
Speciation of the peaks observed by MS analysis of STZ-treated TRPA1 peptide. A, observed spectrum. B–E, peak assignment based on the isotopic distribution prediction. Upper panels (black) correspond to the measured peaks, and the lower panels (red, blue, green, and pink) represent the isotopic distribution prediction for the modification marked on each panel, obtained by the Data Analysis software (Bruker Daltonics).
FIGURE 6.
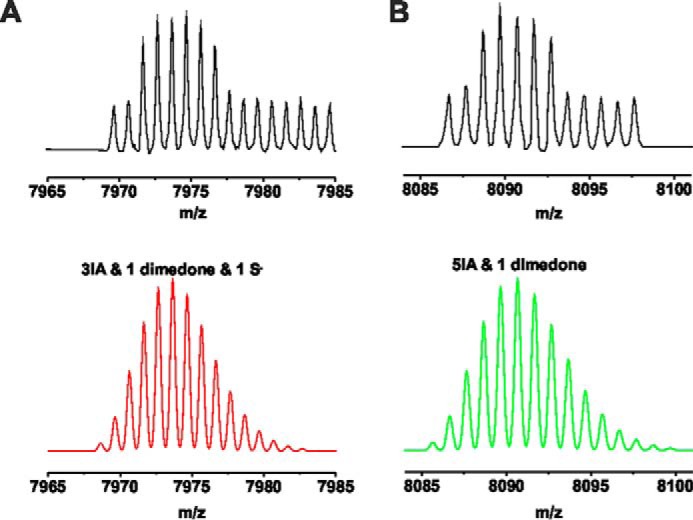
Peak assignments for the STZ + dimedone-treated TRPA1 peptide. A and B, upper figure (black) corresponds to the measured peaks, and the lower figure (red and green) represents the isotopic distribution prediction for the modification marked on each panel, obtained by the Data Analysis software (Bruker Daltonics).
TABLE 1.
Rationale for the disulfide bond assignments
Peptide fragments were detected in the MS/MS analysis of the STZ-treated peptide and/or STZ + dimedone-treated peptide. Cysteine residues that form disulfides are marked blue; the cysteine residue that is oxidized to sulfenic acid is marked green, and the cysteine residue modified by IA is marked red.
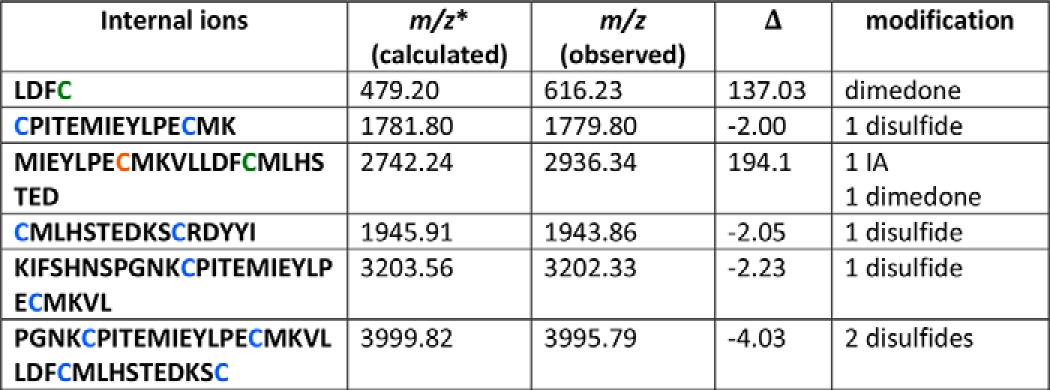
* Data were calculated for unmodified peptide.
In a separate experiment, we collected DRG from untreated control mice and from STZ-treated mice 1 and 24 h after injection to assess whether STZ (180 mg/kg) treatment produced TRPA1 oxidation in vivo (Fig. 4E). TRPA1 protein was purified from DRGs and analyzed by a modified biotin-switch assay (25). The observed basal level of disulfides present in TRPA1 is in good agreement with our recent findings (25). We noticed an obvious increase in the fluorescence labeling of TRPA1 purified from mice 1 and 24 h after STZ treatment, compared with TRPA1 purified from ganglia collected from untreated mice (Fig. 4E). This observation demonstrates that treatment with STZ acutely increases the content of oxidized thiols in TRPA1 in vivo, an effect that lasts for at least 24 h (Fig. 4E).
Previous studies showed that decomposition of STZ leads to formation of both superoxide and nitric oxide (NO) (33–35). As these two react in a diffusion-controlled manner to form peroxynitrite (36), which is known to oxidize cysteines forming sulfenic acid and/or disulfides (37), we examined a direct reaction of STZ with cysteines. Decomposition of STZ in buffered solution (pH 7.4) was monitored by time-resolved ESI-TOF-MS. Our data clearly show that STZ spontaneously decomposes with time as observed by the drop of the STZ parent ion ([STZ + Na]+, m/z 288.08, Fig. 7A). When STZ was incubated with cysteine for 15, 30, and 60 min and the total amount of free thiols assessed by Ellman's reaction, an obvious time-dependent decrease in free thiols was observed (Fig. 7, B and C). The possibility that peroxynitrite might be formed was tested using the standard fluorescence sensor for peroxynitrite, dihydrorhodamine 123. As shown in Fig. 7D, incubation of dihydrorhodamine 123 with STZ led to formation of fluorescent rhodamine 123. In addition, formation of dityrosine, an oxidation product of the reaction of peroxynitrite with tyrosine, was monitored by spectrofluorometry. Fig. 7E confirms that dityrosine was formed when STZ was incubated with tyrosine, suggesting that peroxynitrite is indeed the active compound. Furthermore, it was not possible to detect free superoxide using the superoxide-specific fluorescence sensor hydroethidine, an observation that provides additional support for peroxynitrite formation from spontaneous STZ decomposition (data not shown). We also examined the possible involvement of superoxide and H2O2 for the TRPA1 agonist activity of STZ using the superoxide dismutase mimetic MnTM-PyP-Cl5 (38). As shown in Fig. 7F, MnTM-PyP-Cl5 (10 μm) did not reduce [Ca2+]i responses evoked by 0.5 mm STZ in mTRPA1 CHO cells, suggesting that superoxide is not the activator of TRPA1, which is in agreement with the lack of hydroethidine oxidation. MnTM-PyP-Cl5 may also possess moderate catalase activity (39). If H2O2 was responsible for the STZ-induced TRPA1 activation, the treatment with MnTM-PyP-Cl5 should inhibit this effect. The mimetic indeed inhibited [Ca2+]i increases stimulated by H2O2 alone (Fig. 7G), but it had no effect on STZ-induced responses, further strengthening the evidence for peroxynitrite rather than a superoxide/H2O2 pathway.
FIGURE 7.
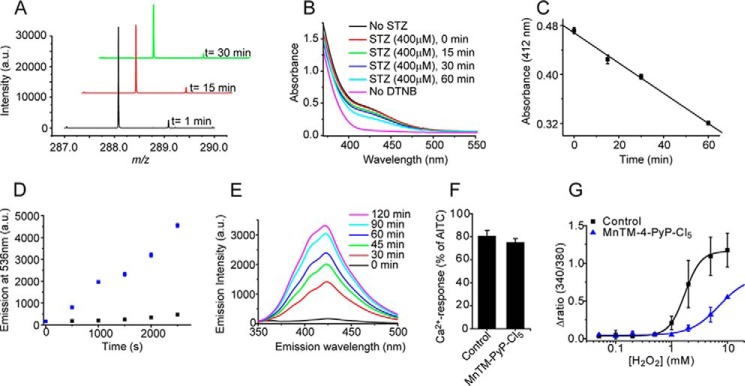
STZ decomposes spontaneously to give a peroxynitrite-like product. A, time-resolved MS spectrum of the parent STZ ion, [STZ + Na]+ at m/z 288.08. B and C, STZ-induced oxidation of cysteine thiols. Cysteine (40 μm) was incubated with STZ (400 μm), and the amount of free thiols was determined by Ellman's reagent (5,5′-dithiobis(nitrobenzoic acid) (DTNB)) at different time points. The time-dependent decay of the 412-nm product indicates STZ-induced oxidation of thiols (n = 3 experiments, data points are mean ± S.E.). D, STZ-induced oxidation of dihydrorhodamine to the fluorescent product, rhodamine 123 (n = 3 experiments, data points are mean ± S.E.). E, fluorescence spectra of tyrosine incubated with STZ, indicating the formation of dityrosine. Black is the control spectrum of 1 mm tyrosine before the addition of STZ. Red to pink, spectra collected at different time points after the addition of 10 mm STZ. F, superoxide dismutase mimetic MnTM-PyP-Cl5 (10 μm) did not reduce STZ (0.5 mm)-evoked [Ca2+]i responses in TRPA1 CHO cells (p > 0.05, n = 4 coverslips with at least 20 cells). G, MnTM-PyP-Cl5 inhibits H2O2 evoked [Ca2+]i responses in CHO cells expressing mTRPA1 (n = 3 experiments, each run in triplicate wells). Data points in G are the mean ± S.E. from one representative experiment. a.u., arbitrary units.
The rate of STZ decomposition and NO release can be accelerated by UV irradiation (33). Brief UV irradiation (10 s, 380 nm) during applications of STZ at concentrations (100–200 μm), which on their own failed to elicit macroscopic currents in hTRPA1-expressing HEK293 cells, rapidly stimulated large TRPA1 currents (Fig. 8, A–D). More extended periods of irradiation with 380 nm light (60 s) evoked TRPA1 currents in the absence of other stimuli but with slower onset and with significantly smaller amplitudes than in combination with STZ (Fig. 8C), in agreement with an earlier study (40).
FIGURE 8.
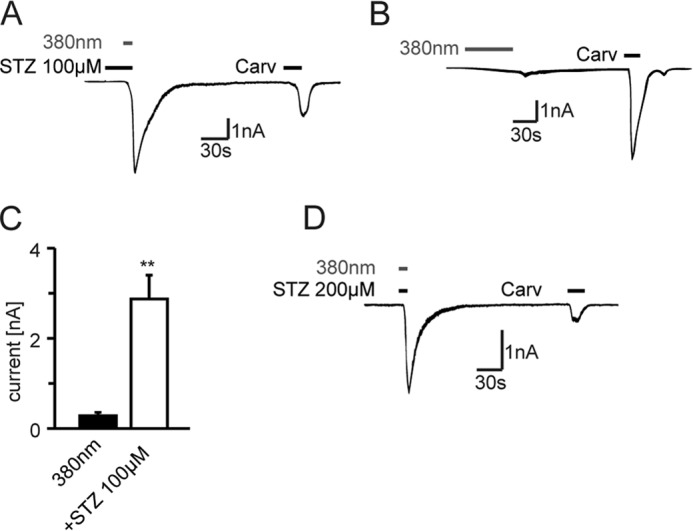
Irradiation with UV light (380 nm) enhances STZ-induced inward currents. A, STZ (100 μm) induced immediate inward currents in hTRPA1-transfected HEK cells, when cells were irradiated with 380 nm for 10 s. No inward currents were recorded by a brief application of STZ 100 μm (20 s) alone. B, continuous irradiation with UV light (380 nm, 60 s) activates TRPA1, but with a slower onset and to a lesser extent (C) than in combination with STZ 100 μm (**, p < 0.004, Mann-Whitney U test, n = 6). D, combination of STZ (200 μm) and fluorescent light (380 nm, 10 s) leads to immediate inward currents in TRPA1-expressing HEK cells. Carv, carvacrol.
Functional Importance of N-terminal Cysteine Residues
We next examined HEK 293t cells expressing hTRPA1 using [Ca2+]i measurements to confirm that STZ also activates the human TRPA1 channel. Application of STZ (50 μm, 30 s) evoked robust increases in [Ca2+]i in 52.9% of hTRPA1-positive cells (expression confirmed using the nonelectrophilic hTRPA1 agonist carvacrol (250 μm, 30 s, Fig. 9, A and G). Neither STZ (1 mm) nor carvacrol (250 μm) evoked [Ca2+]i responses in untransfected HEK 293t cells. The TRPA1 antagonist HC030031 (50 μm) completely blocked STZ-induced responses in hTRPA1 cells (Fig. 9, B and G). However, we noticed an immediate [Ca2+]i response after washout of HC030031, suggesting that the STZ-mediated modifications of hTRPA1 were sustained and not immediately reversible (Fig. 9B). The presence of a stoichiometric excess of dithiothreitol (DTT) provides thiol groups that may scavenge reactive STZ decomposition products such as peroxynitrite. The presence of 5 mm DTT abolished hTRPA1 activation by 50 μm STZ (Fig. 9, C and G), without affecting the basal [Ca2+]i in cells before STZ stimulation (p = 0.6).
FIGURE 9.
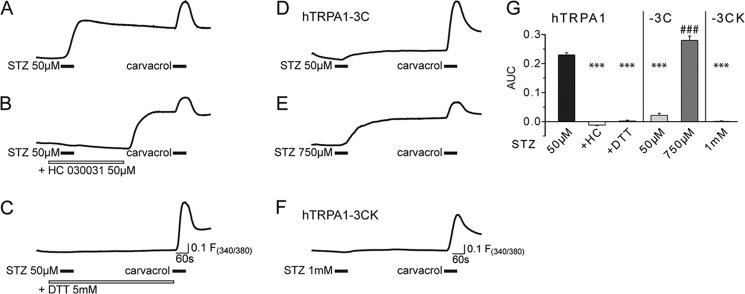
STZ activation of TRPA1 involves modification of intracellular cysteine residues. STZ (50 μm) stimulated [Ca2+]i responses in hTRPA1-expressing HEK293 cells loaded with Fura-2 (A). Application of the TRPA1 antagonist HC030031 inhibited [Ca2+]i responses, but a marked rebound increase in [Ca2+]i was seen after the antagonist was removed (B). The reducing agent dithiothreitol (5 mm) abolished STZ-evoked [Ca2+]i responses (C). In HEK 293t cells transfected with the human TRPA1, the C621S/C641S/C665S mutant channel (−3C) [Ca2+]i responses to stimulation with STZ (50 μm) were strongly reduced (D). Higher concentrations of STZ (750 μm) still produced [Ca2+]i responses in cells expressing the triple cysteine mutant channel (E). STZ (1 mm) failed to induce [Ca2+]i responses in cells expressing the hTRPA1-3C/K710R (−3CK) mutant channel (F). Application of carvacrol (250 μm, for 30 s) was used as a positive control to confirm channel function in A–F. G, quantification of the results presented in A–F. The bar chart demonstrates the area under the curve (AUC) of the fluorescence ratio signals 60 s following STZ stimulation in wild type hTRPA1, the mutant hTRPA1-3C, and hTRPA1-3CK cells. The data in A–G show the mean responses of n = 120–448 cells, S.E. have been omitted in A–F for clarity. ***, p < 0.001 compared with control; ###, p ≤ 0.001 compared with hTRPA1-3C 50 μm STZ; or analysis of variance followed by HSD post hoc test.
We examined the agonist activity of STZ on HEK 293t cells transfected with the hTRPA1 C621S/C641S/C665S mutant (hTRPA1-3C) to determine the functional importance of the cysteine residues identified in the mass spectrometry studies above (7). These three cysteine residues have previously been identified as critical for gating of the human TRPA1 channel by electrophilic compounds, with channels carrying the triple mutation exhibiting a very substantially reduced responsiveness to electrophilic agonists (7). STZ (50 μm) only induced [Ca2+]i increases in a relatively small proportion of hTRPA1-3C cells (12.1%), whereas higher concentrations of STZ (750 μm) evoked intense responses in 49.2% cells (Fig. 9, D and E). The observation that the effect of the triple cysteine mutant was surmountable indicates that STZ must target other amino acid residues in addition to Cys-621, Cys-641, and Cys-665. Peroxynitrite liberated from STZ could oxidize intracellular glucose leading to the formation of products that are shown to modify lysine residues to form Nϵ-(carboxymethyl)lysine (41). Lysine 710, together with the three cysteines, has been shown to be important for the agonist activity of AITC (7). In keeping with such a mechanism, cells expressing the hTRPA1 C621S/C641S/C665S/K710R (hTRPA1-3CK) quadruple mutant were completely insensitive to a high concentration (1 mm) of STZ (Fig. 9F). This observation confirms earlier reports that Lys-710 is specifically targeted by TRPA1 agonists that act through electrophilic modification (7). Importantly, all hTRPA1-3CK cells did respond to the noncovalent hTRPA1 agonist carvacrol, so although it is possible that some of the loss of function seen with the quadruple mutation is due to structural changes of the channel protein, the channel remains functional and able to respond to some agonists.
Discussion
Here, we have demonstrated that STZ stimulates TRPA1 in vivo and in vitro. STZ evokes TRPA1-dependent polymodal hyperalgesia after topical administration but acute sensory loss after systemic administration. In vitro, we show that STZ stimulates TRPA1 in cultured DRG neurons, cell lines, and excised membrane patches. Spontaneous decomposition of STZ generates the oxidant peroxynitrite, and we show that STZ treatment leads to oxidation of TRPA1 cysteine residues in vivo and in vitro.
STZ Targets TRPA1 in Vivo
TRPA1 has been identified as a potential pharmacological target both for disease modification and for treatment of the pain and sensory abnormalities associated with diabetic neuropathy (14, 15). These conclusions were based on the observations that TRPA1 inhibition reduces the loss of epidermal nerve fibers and inhibits the mechanical hyperalgesia seen in STZ-treated diabetic rats. A second line of work further implicated TRPA1 as an effector molecule in diabetic sensory neuropathy. The electrophilic glucose metabolite methylglyoxal, which is produced endogenously during hyperglycemia, and reactive oxygen species and lipid peroxidation products, which also occur at increased levels in diabetes, evoke pain and hypersensitivity by activation of TRPA1 (14, 16–18, 42). TRPA1 thus appears to play a central role both for the development of sensory diabetic neuropathy in STZ models and as a molecular pronociceptive target for metabolites formed in diabetes during hyperglycemic episodes.
Here, we show that activation of TRPA1 by topical administration of very low doses of STZ elicits mechanical and thermal hypersensitivities in vivo. These STZ-induced hypersensitivities are lost in TRPA1-null mice and completely inhibited by the selective TRPA1 antagonist AP18 in wild type mice, demonstrating that peripheral activation of TRPA1 is responsible for the acute pronociceptive response. Systemic administration of STZ produced an acute sensory loss to cold and mechanical stimulation in wild type animals. Mechanical and cold hypoalgesia was established within an hour of STZ administration, while the mice developed hyperglycemia around day 3. We also show that the onset of hypoalgesia is associated with oxidation of TRPA1 cysteine residues in vivo (at 1 and 24 h after STZ). Because the STZ-induced hypoalgesia appeared before the onset of hyperglycemia, our results suggest that an effect of STZ on TRPA1 explains the rapid appearance of sensory behavioral changes. STZ treatment produces a qualitatively different behavioral profile in rats and mice. Rats generally develop hyperalgesia after STZ, whereas mice, in agreement with this report, develop hypoalgesia (5). However, it is not likely that species differences in the TRPA1 sequence are responsible for this discrepancy. TRPA1 is highly conserved between rats and mice (97% identity), and the residues previously proposed to be important for channel activation are identical (6). Previous reports of thermal and mechanical hyperalgesia, which develop both in hyperglycemic and normoglycemic STZ-treated rats, therefore support our behavioral findings (43, 44). In this study, the magnitudes of the sensory deficits were similar for at least 10 days following STZ administration. This observation suggests that it would be difficult to distinguish sensory abnormalities produced directly by STZ from those that develop as a consequence of the ensuing diabetic neuropathy. In contrast to the sensory loss observed in wild type mice, STZ treatment produced hypersensitivity to mechanical, heat, and cold stimulation within 24 h in Trpa1−/− mice. The STZ-induced hypersensitivities observed in TRPA1-null mice suggest that STZ can evoke complex sensory changes but that TRPA1-mediated sensory loss is the dominant mechanism in wild type animals.
Oxidative stress and an increased production of reactive carbonyl compounds such as methylglyoxal are prominent factors leading to the development of diabetic neuropathy and other diabetic complications (45–47). Methylglyoxal, oxidants, and electrophilic agents formed during oxidative stress all stimulate TRPA1 (14, 16–18), and earlier studies have demonstrated that inhibition of TRPA1 reduces or prevents both the loss of intra-epidermal nerve fibers and the behavioral manifestations of STZ-induced diabetic neuropathy in the rat (14, 15). In models of STZ-induced diabetes, TRPA1 is thus targeted both acutely by STZ, as demonstrated here, and by glucose metabolites during the later phase of diabetic neuropathy.
Topical administration of TRPA1 agonists, e.g. to the paw, evokes pain or hyperalgesia (12, 26, 48, 49). In contrast, intrathecal administration of TRPA1 or TRPV1 agonists or systemic administration of TRPV1 agonists (TRPV1 is expressed in the majority of TRPA1 containing sensory neurons) results in analgesia or hypoalgesia (28, 50–52). Persistent stimulation of TRPV1 leads to desensitization or even damage to central or peripheral terminal (51, 53), which has also been proposed to be the consequence of persistent stimulation of TRPA1 (14). In addition, depolarization of the central terminals of nociceptive neurons by intrathecal administration of TRPA1 or TRPV1 agonists produce analgesia through mechanisms akin to depolarization block or presynaptic inhibition (28, 50, 54). Our observations following topical intraplantar (hyperalgesia) and systemic intraperitoneal injections (hypoalgesia) of STZ are therefore in excellent agreement with earlier observations implicating TRPA1 activation with the observed behavioral changes.
Mechanism of Action
In this report we demonstrate that STZ stimulates TRPA1 at concentrations below those thought to be required to kill cultured pancreatic β-cells in vitro (∼2 mm (55, 56)). STZ stimulates TRPA1 in excised membrane patches, which indicates that channel activation occurs through a direct membrane delimited mechanism. Our studies of the spontaneous decomposition of STZ provide strong evidence for formation of peroxynitrite and direct modification of the TRPA1 protein. In agreement with peroxynitrite liberation mediating TRPA1 stimulation by STZ, we found that STZ covalently modified an N-terminal peptide by oxidizing several cysteine residues both to disulfides and sulfenic acid (see Scheme 1). In vivo, STZ treatment increased the content of oxidized thiols (cysteine residues) in TRPA1 protein purified from DRGs. It should be noted that the STZ concentrations achieved in vivo following intraperitoneal injections of STZ (180 mg/kg, 0.68 mmol/kg) are likely to be comparable with the 500 μm, which was used to modify cysteine residues in our MS experiments.
SCHEME 1.
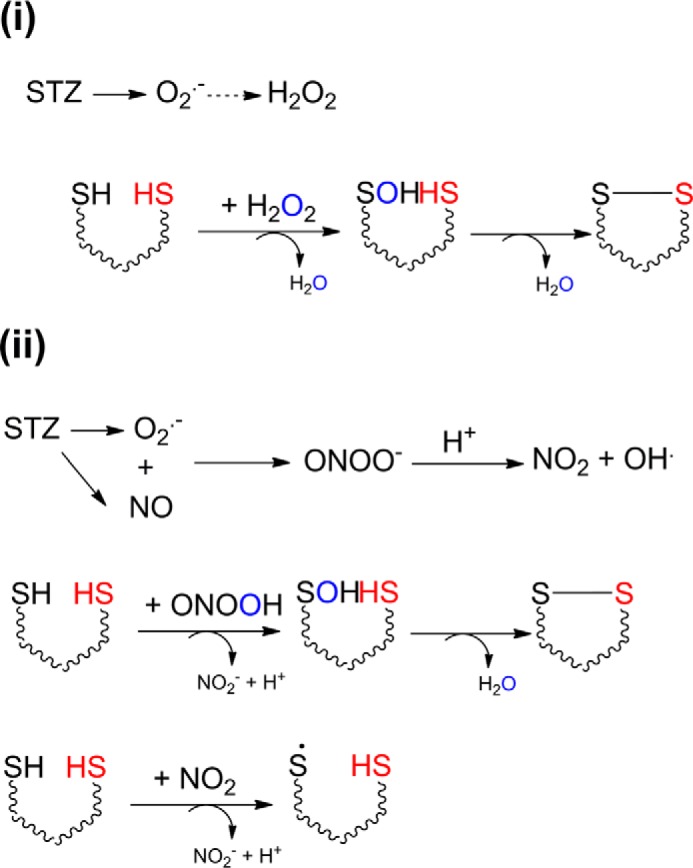
Two possible pathways for STZ decomposition, which could lead to TRPA1 activation, are as follows: (i) STZ decomposes to give superoxide, which forms H2O2, or (ii) STZ decomposes to give both NO and superoxide, which react immediately to give peroxynitrite.
Functional analysis of TRPA1 mutants demonstrated that the cysteine residues modified by incubation with STZ are also important for the agonist activity of STZ. In addition, our studies suggest a role for Lys-710 in STZ-induced channel activation. Although peroxynitrite does not react with lysine directly, it has been shown that it does oxidize glucose and that oxidation products formed in this reaction modify lysine leading to the formation of Nϵ-(carboxymethyl)lysine, one of the prototypical advanced glycation end products (41). This reaction is also thought to be of importance in diabetic neuropathy (57). Although high concentrations of STZ could still activate the triple cysteine mutant channel (C621S, C641S, and C665S), this agonism was lost in channels containing the additional K710R mutation. This could either suggest a reaction involving Lys-710 or, perhaps more likely, an indirect influence of this residue on the sensitivity to oxidants and electrophilic agents (58).
There are two major pathways that could account for the observed cysteine oxidation products (Scheme 1). Decomposition of STZ could lead to superoxide and finally H2O2 (Scheme 1-i) (35, 59), which is a two-electron oxidant known to activate TRPA1 channels (17). However, STZ decomposes to give both NO and superoxide (33–35, 59), which are known to react in a diffusion-controlled manner to give peroxynitrite, a powerful oxidant that could produce all the cysteine modifications that we observed, i.e. cysteinyl radical, sulfenic acid, and disulfides (Scheme 1-ii) (37). Under physiological conditions, i.e. in the absence of oxidative stress, disulfides could be reduced by the reductive cellular environment or by the action of disulfide reductases. In contrast, cysteine residues oxidized further to sulfenic acids would be irreversibly modified. Our experiments with the TRPA1 antagonist HC030031 in hTRPA1 HEK cells showed that although this compound inhibited STZ-evoked responses completely, a sudden Ca2+-influx response was evident when HC030031 was removed from the extracellular solution several minutes after the removal of STZ, demonstrating that the agonist effect of STZ is not rapidly reversible. In vivo, local co-administration of the TRPA1 antagonist AP18 together with STZ completely inhibited the STZ-evoked hypersensitivity for 3 h, but thereafter the mechanical hyperalgesia developed to the same level as in mice that received vehicle instead of AP18, presumably reflecting tissue clearance of the antagonist. Finally, administration of STZ oxidized TRPA1 cysteine residues in vivo, an effect that was evident for at least 24 h. These observations are compatible with slowly reversible (disulfides) or irreversible (sulfenic acids) modification of TRPA1, resulting in sustained channel activation. It is possible that STZ, methylglyoxal, H2O2, nitroxyl (HNO) (16–18, 25), and other agents that stimulate TRPA1 through disulfide formation produce a sustained mode of TRPA1 activation. Persistent TRPA1 channel activity may contribute to the development of long lasting sensory abnormalities seen in STZ-treated diabetic (14) and nondiabetic (43) animals, but also in otherwise healthy mice with elevated methylglyoxal levels (16, 19). Formation of dityrosine and oxidation of dihydrorhodamine, as well as undetectable superoxide levels, suggest that peroxynitrite is the most probable active principle for STZ-induced TRPA1 stimulation. Treatment with a peroxynitrite decomposition catalyst for 3 weeks, starting 3 weeks after diabetes induction with STZ, has previously been shown to counteract the development of sensory neuropathy in STZ-treated mice (60). Peroxynitrite may therefore contribute both to the acute effects of STZ and the later development of diabetic sensory neuropathy. The results from our detailed characterization of STZ decomposition support an earlier demonstration that STZ can produce nitric oxide under certain conditions (33). Nitric oxide reacts very slowly with thiols (61, 62), and accordingly, TRPA1 is resistant even to high concentrations of the NO donor diethylamine NONOate (25). It is therefore unlikely that nitric oxide itself contributes to the agonist activity of STZ. The rate of STZ decomposition is increased by UV light, and in our measurements, irradiation with 380 nm produced a marked potentiation of the effects of STZ. In the presence of UV irradiation, brief applications of modest concentrations of STZ elicited large, almost instantaneous, TRPA1 currents, and it is tempting to speculate that both NO and superoxide are released from irradiated STZ, with diffusion-controlled, rapid formation of peroxynitrite as a consequence. TRPA1 can itself be activated by UV light and photosensitizing agents (40), and we did observe small, slowly developing currents in response to continuous illumination with 380 nm, but these responses were much smaller than those observed in the presence of STZ.
Conclusions
Here, we report the identification of TRPA1 as a molecular target for STZ, which may explain the rapid onset of sensory abnormalities seen in studies of STZ-induced diabetic sensory neuropathy before glucose levels are elevated. We demonstrate a TRPA1-dependent, rapid, and sustained loss of cold and mechanical sensitivity following systemic STZ treatment. The acute sensory abnormality produced by systemic STZ may not be readily distinguished from hypoalgesia developing later as a consequence of diabetic neuropathy. This report further provides new details of the chemical decomposition of STZ and identifies peroxynitrite-mediated oxidation of TRPA1 as an effector mechanism for STZ in vivo and in vitro. In studies of STZ-evoked diabetic neuropathy, TRPA1 may be targeted both directly by STZ and later by the oxidative stress and formation of electrophilic glucose metabolites such as methylglyoxal associated with hyperglycemia. Direct activation of TRPA1 seriously complicates the interpretation of STZ-induced models of diabetic sensory neuropathy, and our results argue that more refined models of diabetic neuropathy should replace the use of STZ.
Acknowledgments
We thank Jan Miljkovic and Rudolf Wedmann for their technical assistance with the fluorescence measurements.
This work was supported in part by the Diabetes UK Alec and Beryl Warren Award BDA 13/0004649 (to D. A. A. and S. B.).
- STZ
- streptozotocin
- DRG
- dorsal root ganglion
- [Ca2+]i
- intracellular calcium concentration
- hTRPA1
- human TRPA1
- mTRPA1
- mouse TRPA1
- ESI
- electrospray ionization
- MnTM-PyP-CL5
- Mn(III)mesotetrakis(N-methylpyridinium-4-yl)porphyrin (pentachloride)
- AITC
- allyl isothiocyanate.
References
- 1. Edwards J. L., Vincent A. M., Cheng H. T., Feldman E. L. (2008) Diabetic neuropathy: mechanisms to management. Pharmacol. Ther. 120, 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Forbes J. M., Cooper M. E. (2013) Mechanisms of diabetic complications. Physiol. Rev. 93, 137–188 [DOI] [PubMed] [Google Scholar]
- 3. Vincent A. M., Callaghan B. C., Smith A. L., Feldman E. L. (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 7, 573–583 [DOI] [PubMed] [Google Scholar]
- 4. Dworkin R. H., O'Connor A. B., Backonja M., Farrar J. T., Finnerup N. B., Jensen T. S., Kalso E. A., Loeser J. D., Miaskowski C., Nurmikko T. J., Portenoy R. K., Rice A. S., Stacey B. R., Treede R. D., Turk D. C., Wallace M. S. (2007) Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 132, 237–251 [DOI] [PubMed] [Google Scholar]
- 5. Obrosova I. G. (2009) Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics 6, 638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zygmunt P. M., Högestätt E. D. (2014) Trpa1. Handb. Exp. Pharmacol. 222, 583–630 [DOI] [PubMed] [Google Scholar]
- 7. Hinman A., Chuang H. H., Bautista D. M., Julius D. (2006) TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. U.S.A. 103, 19564–19568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Macpherson L. J., Dubin A. E., Evans M. J., Marr F., Schultz P. G., Cravatt B. F., Patapoutian A. (2007) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545 [DOI] [PubMed] [Google Scholar]
- 9. Dai Y., Wang S., Tominaga M., Yamamoto S., Fukuoka T., Higashi T., Kobayashi K., Obata K., Yamanaka H., Noguchi K. (2007) Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J. Clin. Invest. 117, 1979–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang S., Dai Y., Fukuoka T., Yamanaka H., Kobayashi K., Obata K., Cui X., Tominaga M., Noguchi K. (2008) Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain 131, 1241–1251 [DOI] [PubMed] [Google Scholar]
- 11. Obata K., Katsura H., Mizushima T., Yamanaka H., Kobayashi K., Dai Y., Fukuoka T., Tokunaga A., Tominaga M., Noguchi K. (2005) TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J. Clin. Invest. 115, 2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petrus M., Peier A. M., Bandell M., Hwang S. W., Huynh T., Olney N., Jegla T., Patapoutian A. (2007) A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol. Pain 3, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eid S. R., Crown E. D., Moore E. L., Liang H. A., Choong K. C., Dima S., Henze D. A., Kane S. A., Urban M. O. (2008) HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol. Pain 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koivisto A., Hukkanen M., Saarnilehto M., Chapman H., Kuokkanen K., Wei H., Viisanen H., Akerman K. E., Lindstedt K., Pertovaara A. (2012) Inhibiting TRPA1 ion channel reduces loss of cutaneous nerve fiber function in diabetic animals: sustained activation of the TRPA1 channel contributes to the pathogenesis of peripheral diabetic neuropathy. Pharmacol. Res. 65, 149–158 [DOI] [PubMed] [Google Scholar]
- 15. Wei H., Hämäläinen M. M., Saarnilehto M., Koivisto A., Pertovaara A. (2009) Attenuation of mechanical hypersensitivity by an antagonist of the TRPA1 ion channel in diabetic animals. Anesthesiology 111, 147–154 [DOI] [PubMed] [Google Scholar]
- 16. Andersson D. A., Gentry C., Light E., Vastani N., Vallortigara J., Bierhaus A., Fleming T., Bevan S. (2013) Methylglyoxal evokes pain by stimulating TRPA1. PLoS One 8, e77986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andersson D. A., Gentry C., Moss S., Bevan S. (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J. Neurosci. 28, 2485–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eberhardt M. J., Filipović M. R., Leffler A., de la Roche J., Kistner K., Fischer M. J., Fleming T., Zimmermann K., Ivanović-Burmazovic I., Nawroth P. P., Bierhaus A., Reeh P. W., Sauer S. K. (2012) Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J. Biol. Chem. 287, 28291–28306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bierhaus A., Fleming T., Stoyanov S., Leffler A., Babes A., Neacsu C., Sauer S. K., Eberhardt M., Schnölzer M., Lasitschka F., Lasischka F., Neuhuber W. L., Kichko T. I., Konrade I., Elvert R., Mier W., Pirags V., Lukic I. K., Morcos M., Dehmer T., Rabbani N., Thornalley P. J., Edelstein D., Nau C., Forbes J., Humpert P. M., Schwaninger M., Ziegler D., Stern D. M., Cooper M. E., Haberkorn U., Brownlee M., Reeh P. W., Nawroth P. P. (2012) Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 18, 926–933 [DOI] [PubMed] [Google Scholar]
- 20. Kwan K. Y., Allchorne A. J., Vollrath M. A., Christensen A. P., Zhang D. S., Woolf C. J., Corey D. P. (2006) TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50, 277–289 [DOI] [PubMed] [Google Scholar]
- 21. Gentry C., Stoakley N., Andersson D. A., Bevan S. (2010) The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol. Pain 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bevan S., Winter J. (1995) Nerve growth factor (NGF) differentially regulates the chemosensitivity of adult rat cultured sensory neurons. J. Neurosci. 15, 4918–4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Story G. M., Peier A. M., Reeve A. J., Eid S. R., Mosbacher J., Hricik T. R., Earley T. J., Hergarden A. C., Andersson D. A., Hwang S. W., McIntyre P., Jegla T., Bevan S., Patapoutian A. (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829 [DOI] [PubMed] [Google Scholar]
- 24. Filipović M. R., Stanić D., Raicević S., Spasić M., Niketić V. (2007) Consequences of MnSOD interactions with nitric oxide: nitric oxide dismutation and the generation of peroxynitrite and hydrogen peroxide. Free Rad. Res. 41, 62–72 [DOI] [PubMed] [Google Scholar]
- 25. Eberhardt M., Dux M., Namer B., Miljkovic J., Cordasic N., Will C., Kichko T. I., de la Roche J., Fischer M., Suárez S. A., Bikiel D., Dorsch K., Leffler A., Babes A., Lampert A., et al. (2014) H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 5, 4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bautista D. M., Jordt S. E., Nikai T., Tsuruda P. R., Read A. J., Poblete J., Yamoah E. N., Basbaum A. I., Julius D. (2006) TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282 [DOI] [PubMed] [Google Scholar]
- 27. Andersson D. A., Gentry C., Moss S., Bevan S. (2009) Clioquinol and pyrithione activate TRPA1 by increasing intracellular Zn2+. Proc. Natl. Acad. Sci. U.S.A. 106, 8374–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersson D. A., Gentry C., Alenmyr L., Killander D., Lewis S. E., Andersson A., Bucher B., Galzi J. L., Sterner O., Bevan S., Högestätt E. D., Zygmunt P. M. (2011) TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ(9)-tetrahydrocannabiorcol. Nat. Commun. 2, 551. [DOI] [PubMed] [Google Scholar]
- 29. Cao D. S., Zhong L., Hsieh T. H., Abooj M., Bishnoi M., Hughes L., Premkumar L. S. (2012) Expression of transient receptor potential ankyrin 1 (TRPA1) and its role in insulin release from rat pancreatic beta cells. PLoS One 7, e38005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jordt S. E., Bautista D. M., Chuang H. H., McKemy D. D., Zygmunt P. M., Högestätt E. D., Meng I. D., Julius D. (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265 [DOI] [PubMed] [Google Scholar]
- 31. Takahashi N., Mizuno Y., Kozai D., Yamamoto S., Kiyonaka S., Shibata T., Uchida K., Mori Y. (2008) Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels 2, 287–298 [DOI] [PubMed] [Google Scholar]
- 32. Engel M. A., Leffler A., Niedermirtl F., Babes A., Zimmermann K., Filipović M. R., Izydorczyk I., Eberhardt M., Kichko T. I., Mueller-Tribbensee S. M., Khalil M., Siklosi N., Nau C., Ivanović-Burmazovic I., Neuhuber W. L., et al. (2011) TRPA1 and substance P mediate colitis in mice. Gastroenterology 141, 1346–1358 [DOI] [PubMed] [Google Scholar]
- 33. Kwon N. S., Lee S. H., Choi C. S., Kho T., Lee H. S. (1994) Nitric oxide generation from streptozotocin. FASEB J. 8, 529–533 [DOI] [PubMed] [Google Scholar]
- 34. Raza H., John A. (2012) Streptozotocin-induced cytotoxicity, oxidative stress and mitochondrial dysfunction in human hepatoma HepG2 cells. Int. J. Mol. Sci. 13, 5751–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nukatsuka M., Sakurai H., Yoshimura Y., Nishida M., Kawada J. (1988) Enhancement by streptozotocin of O2− radical generation by the xanthine oxidase system of pancreatic beta-cells. FEBS Lett. 239, 295–298 [DOI] [PubMed] [Google Scholar]
- 36. Kissner R., Nauser T., Bugnon P., Lye P. G., Koppenol W. H. (1997) Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 10, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 37. Alvarez B., Ferrer-Sueta G., Freeman B. A., Radi R. (1999) Kinetics of peroxynitrite reaction with amino acids and human serum albumin. J. Biol. Chem. 274, 842–848 [DOI] [PubMed] [Google Scholar]
- 38. Friedel F. C., Lieb D., Ivanović-Burmazovic I. (2012) Comparative studies on manganese-based SOD mimetics, including the phosphate effect, by using global spectral analysis. J. Inorg. Biochem. 109, 26–32 [DOI] [PubMed] [Google Scholar]
- 39. Day B. J., Fridovich I., Crapo J. D. (1997) Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch. Biochem. Biophys. 347, 256–262 [DOI] [PubMed] [Google Scholar]
- 40. Hill K., Schaefer M. (2009) Ultraviolet light and photosensitising agents activate TRPA1 via generation of oxidative stress. Cell Calcium 45, 155–164 [DOI] [PubMed] [Google Scholar]
- 41. Nagai R., Unno Y., Hayashi M. C., Masuda S., Hayase F., Kinae N., Horiuchi S. (2002) Peroxynitrite induces formation of N(ϵ)-(carboxymethyl) lysine by the cleavage of Amadori product and generation of glucosone and glyoxal from glucose: novel pathways for protein modification by peroxynitrite. Diabetes 51, 2833–2839 [DOI] [PubMed] [Google Scholar]
- 42. Bessac B. F., Sivula M., von Hehn C. A., Escalera J., Cohn L., Jordt S. E. (2008) TRPA1 is a major oxidant sensor in murine airway sensory neurons. J. Clin. Invest. 118, 1899–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bishnoi M., Bosgraaf C. A., Abooj M., Zhong L., Premkumar L. S. (2011) Streptozotocin-induced early thermal hyperalgesia is independent of glycemic state of rats: role of transient receptor potential vanilloid 1(TRPV1) and inflammatory mediators. Mol. Pain 7, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cunha J. M., Funez M. I., Cunha F. Q., Parada C. A., Ferreira S. H. (2009) Streptozotocin-induced mechanical hypernociception is not dependent on hyperglycemia. Braz. J. Med. Biol. Res. 42, 197–206 [DOI] [PubMed] [Google Scholar]
- 45. Jack M. M., Ryals J. M., Wright D. E. (2012) Protection from diabetes-induced peripheral sensory neuropathy–a role for elevated glyoxalase I? Exp. Neurol. 234, 62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fleming T., Cuny J., Nawroth G., Djuric Z., Humpert P. M., Zeier M., Bierhaus A., Nawroth P. P. (2012) Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia 55, 1151–1155 [DOI] [PubMed] [Google Scholar]
- 47. Tomlinson D. R., Gardiner N. J. (2008) Glucose neurotoxicity. Nat. Rev. Neurosci. 9, 36–45 [DOI] [PubMed] [Google Scholar]
- 48. McNamara C. R., Mandel-Brehm J., Bautista D. M., Siemens J., Deranian K. L., Zhao M., Hayward N. J., Chong J. A., Julius D., Moran M. M., Fanger C. M. (2007) TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. U.S.A. 104, 13525–13530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bandell M., Story G. M., Hwang S. W., Viswanath V., Eid S. R., Petrus M. J., Earley T. J., Patapoutian A. (2004) Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857 [DOI] [PubMed] [Google Scholar]
- 50. Jeffry J. A., Yu S. Q., Sikand P., Parihar A., Evans M. S., Premkumar L. S. (2009) Selective targeting of TRPV1 expressing sensory nerve terminals in the spinal cord for long lasting analgesia. PLoS One 4, e7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wrigglesworth R., Walpole C. S., Bevan S., Campbell E. A., Dray A., Hughes G. A., James I., Masdin K. J., Winter J. (1996) Analogues of capsaicin with agonist activity as novel analgesic agents: structure-activity studies. 4. Potent, orally active analgesics. J. Med. Chem. 39, 4942–4951 [DOI] [PubMed] [Google Scholar]
- 52. Szolcsányi J. (2004) Forty years in capsaicin research for sensory pharmacology and physiology. Neuropeptides 38, 377–384 [DOI] [PubMed] [Google Scholar]
- 53. Jancsó N., Jancsó-Gábor A., Szolcsányi J. (1967) Direct evidence for neurogenic inflammation and its prevention by denervation and by pretreatment with capsaicin. Br. J. Pharmacol. Chemother. 31, 138–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Willis W. D. (2006) John Eccles' studies of spinal cord presynaptic inhibition. Prog. Neurobiol. 78, 189–214 [DOI] [PubMed] [Google Scholar]
- 55. Eizirik D. L., Strandell E., Sandler S. (1988) Culture of mouse pancreatic islets in different glucose concentrations modifies B cell sensitivity to streptozotocin. Diabetologia 31, 168–174 [DOI] [PubMed] [Google Scholar]
- 56. Pipeleers D., Van de Winkel M. (1986) Pancreatic B cells possess defense mechanisms against cell-specific toxicity. Proc. Natl. Acad. Sci. U.S.A. 83, 5267–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sugimoto K., Yasujima M., Yagihashi S. (2008) Role of advanced glycation end products in diabetic neuropathy. Curr. Pharm. Des. 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 58. Moparthi L., Survery S., Kreir M., Simonsen C., Kjellbom P., Högestätt E. D., Johanson U., Zygmunt P. M. (2014) Human TRPA1 is intrinsically cold- and chemosensitive with and without its N-terminal ankyrin repeat domain. Proc. Natl. Acad. Sci. U.S.A. 111, 16901–16906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takasu N., Komiya I., Asawa T., Nagasawa Y., Yamada T. (1991) Streptozocin- and alloxan-induced H2O2 generation and DNA fragmentation in pancreatic islets. H2O2 as mediator for DNA fragmentation. Diabetes 40, 1141–1145 [DOI] [PubMed] [Google Scholar]
- 60. Drel V. R., Pacher P., Vareniuk I., Pavlov I., Ilnytska O., Lyzogubov V. V., Tibrewala J., Groves J. T., Obrosova I. G. (2007) A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin-diabetic mice. Eur. J. Pharmacol. 569, 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wink D. A., Nims R. W., Darbyshire J. F., Christodoulou D., Hanbauer I., Cox G. W., Laval F., Laval J., Cook J. A., Krishna M. C. (1994) Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 7, 519–525 [DOI] [PubMed] [Google Scholar]
- 62. Lancaster J. R., Jr. (2006) Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chem. Res. Toxicol. 19, 1160–1174 [DOI] [PubMed] [Google Scholar]