

Background: Rgnef (ArhGEF28) is activated downstream of gastrin and the cholecystokinin receptor to promote colon carcinoma tumor progression.
Results: Rgnef activation by Gα13 triggers FAK and paxillin tyrosine phosphorylation in response to gastrin. A C-terminal Rgnef region is necessary for linkage to Gα13.
Conclusion: Rgnef is an effector of Gα13 signaling.
Significance: Gα13 and Rgnef are implicated in colon carcinoma.
Keywords: cell signaling, G protein-coupled receptor (GPCR), guanine nucleotide exchange factor (GEF), heterotrimeric G protein, regulator of G protein signaling, Gastrin
Abstract
The guanine nucleotide exchange factor Rgnef (also known as ArhGEF28 or p190RhoGEF) promotes colon carcinoma cell motility and tumor progression via interaction with focal adhesion kinase (FAK). Mechanisms of Rgnef activation downstream of integrin or G protein-coupled receptors remain undefined. In the absence of a recognized G protein signaling homology domain in Rgnef, no proximal linkage to G proteins was known. Utilizing multiple methods, we have identified Rgnef as a new effector for Gα13 downstream of gastrin and the type 2 cholecystokinin receptor. In DLD-1 colon carcinoma cells depleted of Gα13, gastrin-induced FAK Tyr(P)-397 and paxillin Tyr(P)-31 phosphorylation were reduced. RhoA GTP binding and promoter activity were increased by Rgnef in combination with active Gα13. Rgnef co-immunoprecipitated with activated Gα13Q226L but not Gα12Q229L. The Rgnef C-terminal (CT, 1279–1582) region was sufficient for co-immunoprecipitation, and Rgnef-CT exogenous expression prevented Gα13-stimulated SRE activity. A domain at the C terminus of the protein close to the FAK binding domain is necessary to bind to Gα13. Point mutations of Rgnef-CT residues disrupt association with active Gα13 but not Gαq. These results show that Rgnef functions as an effector of Gα13 signaling and that this linkage may mediate FAK activation in DLD-1 colon carcinoma cells.
Introduction
Intestinal homeostasis is a dynamic process in which stem cells at the base of crypts divide and migrate to the apical region, where they differentiate. During tumor progression, cells can undergo an epithelial to mesenchymal transition associated with increased cell motility. Changes in cytoskeletal dynamics required for cell migration are coordinated in large part by RhoGTPases (1) whose involvement in cancer is well documented (2). Although activation of RhoGTPases can occur by inhibiting GTPase-activating proteins or guanosine nucleotide dissociation inhibitors, a number of studies suggest that the Dbl family of exchange factors known as GEFs5 are the primary mediators of RhoGTPase activation (3).
Rgnef, also termed p190RhoGEF or ArhGEF28, is a member of the Dbl family of RhoGEFs (4). Elevated Rgnef expression promotes colorectal carcinoma cell motility, invasion, and tumor progression (5–7). Rgnef also possesses anti-apoptotic activity (8) and is implicated in the pathogenesis of motor neuron degeneration (9), in the regulation of synapse formation (10), and in signaling associated with dendritic morphogenesis (11, 12). Rgnef knock-out yields homozygous-null mice at less than expected Mendelian ratios, and Rgnef-null fibroblasts exhibit decreased RhoA GTPase activity and cell motility downstream of integrins (6). A link with FAK activity has been described in both normal and tumor cell motility. Rgnef is activated by GPCR signaling cascades (7), but the mechanism of activation downstream of GPCRs remains unclear. In particular, Rgnef responds to gastrin-type 2 cholecystokinin receptor (CCK2R) stimulating signaling events (7).
As a member of the Dbl family (13), p190RhoGEF/Rgnef is characterized by a central tandem DH-PH domain and also contains an N-terminal leucine-rich region and a large C-terminal region that may form an α-helical coiled-coil domain of unknown function. The DH-PH catalytic region is responsible for RhoGEF activity (14, 15). Rgnef has the highest amino acid similarity to p114RhoGEF (ArhGEF18), GEF-H1 (ArhGEF2), and Lbc (ArhGEF13). Other related RhoGEFs with similar subunits are p115RhoGEF (ArhGEF1), PDZ-RhoGEF (ArhGEF11), and LARG (ArhGEF12) (16, 17) that contain a regulator of G protein signaling homology (RH) domain located within the N-terminal region that confers the capability to bind to G proteins. All of these RhoGEFs preferentially activate RhoA. Lbc has also been characterized as a downstream effector of Gα12 (18), and p114RhoGEF can bind to Gβγ subunits (19). Rgnef lacks a recognized RH domain in its N-terminal region and thus has not been considered a downstream effector of Gα12/13.
The heterotrimeric G proteins, Gα12 and Gα13, activate RhoA via RH-containing RhoGEFs resulting in actin stress fiber formation and focal adhesion assembly (20, 21). Soon after their discovery, both Gα12 and Gα13 were demonstrated to induce oncogenic transformation (22–24). Several studies have demonstrated roles for Gα12 or Gα13 in cell proliferation and migration (25). Additionally, Gα12 may alter cell adhesion in a RhoGEF-independent manner (26–28). Invasive breast cancer and prostate adenocarcinoma cells express high levels of Gα12 (25, 29, 30). These findings support the hypothesis that GPCRs may signal through G12 proteins to promote tumor progression (31).
Here, we show that gastrin stimulation of DLD-1 colon carcinoma cells acts through CCK2R to promote Gα13-stimulated FAK Tyr-97 and paxillin Tyr-31 tyrosine phosphorylation, RhoA GTP binding, and the activation of SRE reporter transcription. We find that Gα13 activates Rgnef utilizing a previously uncharacterized domain within the Rgnef C-terminal region in close proximity to the Rgnef-FAK-binding site. These results establish Rgnef as a new effector of Gα13 in the control of cell migration downstream of gastrin.
Experimental Procedures
Antibodies and Reagents
Antibodies to Gα13 (32), Gα12, Gαq/11, and RhoA were from Santa Cruz Biotechnology. Monoclonal anti-Gαq and anti-paxillin were from BD Biosciences. Anti-Rgnef was described previously (7). Anti-paxillin-Tyr(P)-31, and anti-FAK Tyr(P)-397-phosphospecific antibodies were from Invitrogen; anti-FAK, clone 4.47, was from Millipore; anti-FLAG and HA-agarose conjugates were from Sigma; and rat anti-HA was from Roche Applied Science. EE tag antibody was from Cell Signaling. β-Tubulin monoclonal antibody was from Abcam. IRDye 680 and IRDye 800 goat IgG, goat anti-rabbit, and anti-mouse IgGs were from LI-COR. Human amidated gastrin (G17 peptide) was from Calbiochem. YM022 was from Yamanouchi Pharmaceutical. The nonpeptide antagonist of the CCK1R, 1-[2-(4-(2-chlorophenyl)thiazol-2-yl)aminocarbonyl-indoyl]acetic acid (SR-27,897), and the nonpeptide agonist of the CCK1R, 2-[4-(4-chloro-2-,5-dimethoxyphenyl)-5-(2-cyclohexylethyl)thiazol-2-ylcarbamoyl]-5,7-dimethyl-indol-1-yl-1-acetic acid (SR-146,131), were described before (33).
Plasmids
pFGFP-C2-mp190RhoGEF, pCDH-mCherry-p190RhoGEF-FL, pCDH-mCherry-p190RhoGEF-CT, and pCDH-mCherry-p190RhoGEFΔFAK were used as described (5). pcDNA3-HA-190RhoGEF (murine cDNA) was a generous gift from W. H. Moolenaar (The Netherlands Cancer Institute). pcDNA3-HA-RgnefΔNt, pcDNA3.1-HA-RgnefFΔCt, pcDNA3.1-HA-Rgnef-1M (mouse R1487A), and pcDNA3.1-HA-Rgnef-5M were generated using QuikChange Lightning site-directed mutagenesis (Agilent Technologies). Single or multiple site mutagenesis in Rgnef was accomplished using QuikChange mutagenesis. Mouse Rgnef 5M contained alanine changes at His-1393, Glu-1475, Glu-1481, Leu-1483, and Arg-1487 (corresponding to human residues His-1396, Glu-1478, Glu-1484, Leu-1486, and Arg-1490). pLKO.1 scrambled (Scr), pLKO.1-hGα13-sh1, pLKO.1-hGα13-sh2, pLKO.1-hGα13-sh3, pLKO.1-hGα13-sh4, and pLKO.1-hGα13-sh5 were from Sigma. pSRE-Lmut was from D. Toksoz (Tufts Medical Center). pcDNA3.1-HA-γ1, pcDNA3.1-Gα12wt, pcDNA3.1-Gα12(Q229L), pcDNA3.1-Gα13wt, and pcDNA3.1-Gα13(Q226L) were obtained from The Missouri S&T cDNA Resource Center. pcDNA3.1Gαq was described before (34). pGEX-2T-RBD was obtained from Sang-Kyou Han (University of California at San Diego). Plasmid for GST-RhoAG17A was a generous gift from K. Burridge (University of North Carolina).
Cells
HEK293, HEK293T, and DLD-1 cells were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen), 2 mm l-glutamine (Invitrogen) and 100 units of penicillin/streptomycin (Lonza) in a humidified atmosphere of 5% CO2 in air at 37 °C. Prior to cell treatments, cells were serum-starved for 16 h. Cells were stimulated with gastrin at 200 nm in serum-free medium for the indicated time periods. For transient transfections, cells were plated at 60–70% confluence, and after 24 h, cells were transfected utilizing FuGENE 6 (Roche Applied Science) or Metafectene Pro (Biontex) according to the manufacturer's instructions. Protein expression was evaluated 24–48 h after transfection.
shRNA Knockdown
Gα13 short hairpin-interfering RNA (shRNA) targeted against human GNA13 (TRCN0000036885, Mission shRNA, Sigma) was used for stable knockdown in DLD-1 cells. For lentivirus production, HEK293T were co-transfected with pLKO.1 vectors containing the shRNA vector together with pCMVΔR8.91 (HIV Gag and Pol) and pVSVG at a ratio of 4:3:1, respectively. Medium was collected every 24 h for 2 days and sterile-filtered (0.45-μm MCE filter, Millipore). Viral particles were stored at −80 °C. To infect cells by spinoculation, 1 ml of the viral particle supernatant was added to cells, and plates were centrifuged at 1200 × g for 2 h at room temperature. After 24 h, infected cells were selected by adding 2 μg/ml of puromycin (Sigma). The efficiency of shRNA knockdown was confirmed by SDS-PAGE and Western blot analysis of protein extracts.
Immunoprecipitation and Western Blots Analysis
Cells were lysed in RIPA buffer (50 mm Tris, pH 7.4, 0.3 m NaCl, 0.1% SDS, 0.5% DOC, 10 mm MgCl2, 1 mm Na3VO4, 10 mm NaF, 1% n-dodecyl β-d-maltoside, and protease inhibitors). To undergo AlF4− treatment, 30 μm AlCl3 and 5 mm NaF were added to lysis and wash buffers. Lysates were pre-cleared with 50 μl of IgG-free BSA (10 mg/ml) (Sigma) and 50 μl of protein A-Sepharose (Roche Applied Science) for 1.5 h at 4 °C on a rotating wheel followed by centrifugation. Specific antibodies were added to lysate supernatants and incubated overnight at 4 °C with rotation. Protein A-Sepharose (Sigma) or protein G-agarose (Roche Applied Science) beads were added and incubated for 1.5 h at 4 °C with rotation. Samples were centrifuged for 5 min at 425 × g, and the beads were washed three times with gentle shaking in RIPA wash buffer (50 mm Tris, pH 7.4, 0.3 m NaCl, 0.1% SDS, 0.5% DOC, 10 mm MgCl2, 1 mm Na3VO4), followed by aspiration, resuspension in SDS loading buffer, and heating at 100 °C. Cell lysates and immunoprecipitates were loaded in SDS-PAGE, immunoblotted to Immobilon-FL membranes (Millipore), and analyzed with the indicated antibodies. Western blots were visualized by infrared detection (Odyssey System) and quantified by Image Studio software (LI-COR) (35).
Expression and Purification of GST-RBD and GST-RhoAG17A Constructs
pGEX plasmids were transformed in BL21 Escherichia coli (36). Protein expression was induced by the addition of 0.5 mm isopropyl β-d-thiogalactoside for GST-RBD or 0.1 mm for GST-RhoAG17A and incubated for 16 h at room temperature. Bacteria pellets were resuspended in ice-cold RBD1 (50 mm Tris-HCl, pH 7.2, 150 mm NaCl, 10 mm MgCl2, 1% Triton X-100, 0.5% NaDOC, 0.1% SDS, and protein inhibitors) or RhoAG17A (20 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1% Triton X-100, 1 mm DTT, and protease inhibitors) lysis buffers. Bacteria were lysed by sonication on ice for 1 min and centrifuged at 20,000 × g for 15 min at 4 °C. Supernatants were incubated with 200 μl of 50% glutathione-Sepharose 4B slurry (GE Healthcare) for 45–60 min at 4 °C with rotation. GST-RBD beads were washed six times in RBD wash buffer (50 mm Tris-HCl, pH 7.2, 150 mm NaCl, 10 mm MgCl2, 1% Triton X-100, and protease inhibitors), and GST-RhoAG17A beads were washed twice in RhoAG17A lysis buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1% Triton X-100, 1 mm DTT, and protein inhibitors) and two more times with HBS wash buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT). Bead-associated GST-RBD and GST-RhoAG17A protein concentration was estimated by SDS-PAGE and Coomassie Blue staining alongside BSA protein standards. Bead aliquots were stored at −80 °C in 10% glycerol.
RhoA Activation Assays
Cells were transfected with Rgnef and/or Gα13 plasmids utilizing MetafecteneTM Pro (Biontex) and serum-starved overnight 24 h post-transfection. After cell stimulation with gastrin, cells were lysed in RBD2 lysis buffer (50 mm Tris-HCl, pH 7.2, 1% Triton X-100, 0.5% NaDOC, 0.1% SDS, 500 mm NaCl, 10 mm MgCl2, and protease inhibitors) and clarified by centrifugation (13,000 × g for 5 min at 4 °C). Modified Bradford assays were used to determine protein concentration (Bio-Rad). Aliquots were mixed with SDS sample buffer and stored at −20 °C as total protein lysates. Lysates of equal protein content were incubated with 30–40 μg of GST-Rhotekin RBD immobilized to glutathione-Sepharose 4B beads for 45–60 min at 4 °C by rotation. Beads were washed four times with 4 °C RBD wash buffer and activated RhoA eluted with SDS sample buffer addition and detected by monoclonal anti-RhoA immunoblotting.
Affinity Precipitation of Activated GEFs
HEK293 cells expressing Rgnef and Gα proteins were lysed in ice-cold RhoAG17A lysis buffer. Lysates were clarified by centrifugation (16,000 × g for 1 min at 4 °C) and pre-cleared by incubation with 50 μl of GST bound to glutathione-Sepharose (1 mg/ml) for 10 min at 4 °C. Modified Bradford assays were used to determine protein concentration (Bio-Rad). GST-RhoAG17A beads (10 μg) were added to each lysate, rotated for 45–60 min at 4 °C, and washed three times with GST-RhoAG17A lysis buffer. Bead-associated protein complexes were separated by SDS-PAGE followed by anti-HA tag or Rgnef immunoblotting.
SRE Luciferase Assays
The SRE reporter was designed to monitor the activity of serum-response factor (SRF)-mediated signal transduction. pSRE.L luciferase reporter plasmid encodes for firefly luciferase positioned downstream of a mutant SRE that contains SRF-binding sites but eliminates the ternary complex factor-binding site (37). RhoA activation stimulates SRE transcriptional activation (38). HEK293 cells were transfected with the indicated plasmids with pSRE.L (0.1 μg) and the Renilla luciferase thymidine kinase (pRL-TK) (0.01 μg). Cells were serum-starved for 24 h and then stimulated with ligands for the indicated time. Cells were washed with PBS and lysed in 200 μl of 1× Passive® lysis buffer (Promega) for 20 min at 4 °C by agitation. Lysates were clarified by centrifugation at 18,000 × g for 15 min. Cell extracts (20 μl) were mixed with 25 μl of Luciferase Assay Reagent II (Promega) and luminescence-quantified (TD-20/20 luminometer, Sirius). Reactions were quenched by 25 μl of Stop & Glo reagent addition (Promega), and Renilla luciferase activity was quantified. Values were normalized and expressed as arbitrary units.
Modeling the Rgnef-Gα13 Complex
RH domain sequences for human p115RhoGEF (ArhGEF1), murine Lsc (ArhGEF1), PDZ-RhoGEF (ArhGEF11), and LARG (ArhGEF12) were retrieved from the Uniprot (UniProt-Consortium, 2010). Entries tagged as reviewed were saved (Q9NZN5, Q8R4H2, Q92888, O15085, Q9ES67, Q61210, and Q9Z1I6) and aligned using ClustalW (39). Next, we aligned the C-terminal portion of Rgnef (entries P97433, Q8N1W1, and P0C6P5) to the RH domains of the p115RhoGEF family. Relying on this alignment, we constructed a computational model of the Rgnef RH-like domain bound to Gα13 using Modeler (40) based on the homology to the RH-like domain of ArhGEF11, for which the structure in complex with Gα13 (Protein Data Bank code 3CX8) is available (41, 42). The molecular complex was refined using energy minimization with AMBER 12.0 employing the Amberff99SB-ILDN force field (43).
Statistical Analysis
Data are presented as mean values or as fold induction ± S.E. or ± S.D. as indicated in each figure legend. The sums of all the individual stimulatory effects were compared with the combined effects by using the unpaired t test with two-tailed p values. Differences were considered statistically significant when p < 0.05 (noted with *), p < 0.005 (noted with **), and p < 0.001 (noted with ***).
Results
Gastrin Enhances Paxillin Tyrosine Phosphorylation through CCK2R
DLD-1 colon carcinoma cells will form focal adhesions with enhanced paxillin tyrosine phosphorylation associated with increased cell motility in response to gastrin stimulation (7). To evaluate the involvement of the G protein-coupled CCK2R in gastrin-induced motility-associated signaling, paxillin Tyr-31 phosphorylation was determined by phosphospecific immunoblotting (Fig. 1). Gastrin increased paxillin Tyr(P)-31 phosphorylation within 15–30 min that was sustained to 60 min (Fig. 1A). The CCK2R antagonist YM022 inhibited gastrin-induced paxillin Tyr-31 phosphorylation (Fig. 1, B and C), suggesting that this linkage was mediated by CCK2R. Conversely, addition of the CCK1R antagonist SR27897 increased paxillin Tyr(P)-31 phosphorylation by 54% with gastrin co-stimulation. Accordingly, addition of the CCK1R agonist SR146131 did not enhance paxillin Tyr(P)-31 phosphorylation (Fig. 1, D and E), suggesting that gastrin induction of paxillin phosphorylation is dependent on CCK2R (but not CCK1R) in DLD-1 cells.
FIGURE 1.
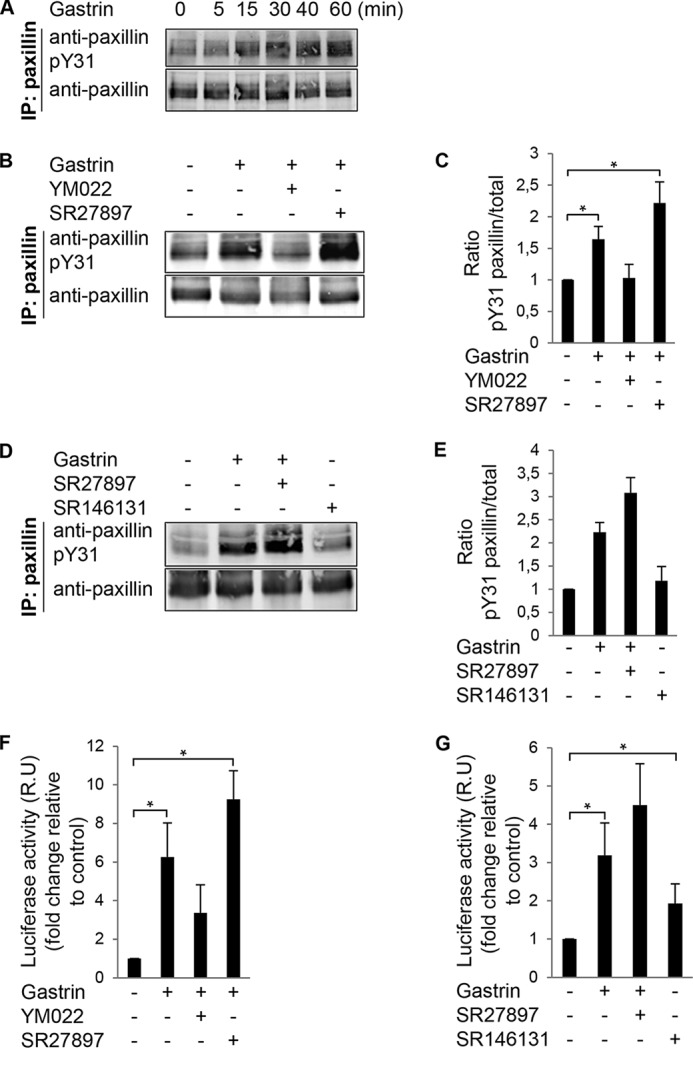
Gastrin stimulation of paxillin tyrosine phosphorylation and SRE activation in DLD-1 cells. A, gastrin (200 nm) was added to DLD-1 cells for the indicated times. Protein lysates were analyzed by paxillin IP and phosphospecific paxillin (pY31) or total paxillin immunoblotting. B, DLD-1 cells were pretreated with 2 μm SR27987 (CCK1R antagonist) or 2 μm YM022 (CCK2R antagonist) for 20 min. Gastrin (200 nm) was added, and lysates were prepared for paxillin IP after 40 min as in A. C, LI-COR image quantification of Tyr(P)-31 paxillin to total paxillin ratio from experiments in B. Values were set to 1 and are means ± S.E. fold increase of three experiments conducted in triplicate (*, p < 0.05, two-tailed t test). D, DLD-1 cells were pretreated with 2 μm SR27987 (CCK1R antagonist). Cells were pretreated with gastrin (200 nm) or SR146131 (CCK1R agonist) for 40 min before anti-paxillin IP as in A. E, image quantification of Tyr(P)-31 paxillin to total paxillin ratio from experiments in D. Values were set to 1. Data are the mean ± S.E. fold increase for three independent experiments. F, DLD-1 cells were transfected with pSRE.L and Renilla luciferase (RLuc) vectors. Cells were serum-starved and then pretreated with 2 μm SR27987 (CCK1R antagonist) or 2 μm YM022 (CCK2R antagonist) for 20 min. Gastrin (200 nm) or vehicle was added for 5 h, and lysates were prepared for luciferase detection. G, DLD-1 cells were transfected as above and were pretreated with 2 μm SR27987 (CCK1R antagonist). Gastrin (200 nm) or SR146131 (CCK1R agonist) was added as indicated for 5 h, and lysates were prepared for luciferase detection. F and G, values (mean ± S.E. of three independent experiments in triplicate) were normalized based on expression of RLuc and expressed (relative units, R.U.) as fold induction over serum-starved conditions (*, p < 0.05, two-tailed t test).
The signaling pathway downstream gastrin, CCK2R, and RhoA have not been established. Increased transcriptional activity of an SRE-luciferase reporter containing SRF-binding sites is a defined signaling linkage downstream of RhoA GTPase activation (44). In DLD-1 cells, gastrin-stimulated SRE activity was reduced by the YM022 CCK2R antagonist but not by the SR27897 CCK1R antagonist (Fig. 1F). Compared with gastrin, addition of the SR146131 CCK1R agonist only weakly enhanced SRE promoter activity (Fig. 1G). Together, these results support the notion that gastrin-treated DLD-1 cells signal through CCK2R to stimulate paxillin tyrosine phosphorylation and likely RhoA activation leading to an enhanced SRE transcriptional response.
Gastrin-stimulated Rho Activation, FAK, and Paxillin Phosphorylation Involved Gα13
To test whether Gα13 could have any role in paxillin-Tyr(P)-31 phosphorylation and in Rho activation of SRE transcription activity following CCK2R stimulation, we generated Gα13 shRNA targeted against the coding region of Gα13 for stable knockdown in DLD-1 cells. Expression of Gα13-shRNA resulted in >75% stable reduction in Gα13 protein expression compared with Scr shRNA-expressing and Mock-transduced cells (Fig. 2A). Gα13-shRNA did not affect other Gα subunit expressions (Fig. 2A). DLD-1-depleted Gα13 cells were analyzed for effects on gastrin-stimulated paxillin Tyr-31 phosphorylation. Compared with Mock-transduced and Scr shRNA-expressing DLD-1 cells, Gα13-shRNA paxillin Tyr-31 phosphorylation was selectively reduced upon Gα13 knockdown (Fig. 2, B and C). Together, these results support a gastrin, CCK2R, and Gα13 signaling linkage upstream of paxillin tyrosine phosphorylation in DLD-1 cells.
FIGURE 2.

Gα13 is necessary for gastrin-stimulated paxillin tyrosine phosphorylation and SRE activation. A, lysates of Mock, Gα13, or Scr shRNA-expressing DLD-1 cells were immunoblotted with antibodies to Rgnef, Gα12, Gα13, Gαq, and β-tubulin. B, representative paxillin IPs from lysates of Mock, Scr-, and Gα13 shRNA-expressing DLD-1 cells pretreated with gastrin (200 nm, 40 min). Phosphospecific paxillin Tyr(P)-31 (pY31) was followed by detection of total paxillin levels. C, image quantification from paxillin IPs shown in B. Control was set to 1, and values are means ± S.E. for fold induction of three independent experiments. D, representative FAK IPs from lysates of Scr- and Gα13 shRNA-expressing DLD-1 cells that stimulated with gastrin (200 nm, 30 min). Phosphospecific FAK-Tyr(P)-397 (pY397) was followed by detection of total FAK levels by immunoblotting. Phosphospecific paxillin Tyr(P)-31 was followed by detection of total paxillin levels by immunoblotting on the corresponding lysates. E, DLD-1 cells transduced with anti-Gα13 or Scr shRNA were transfected with pSRE.L and RLuc vectors. After 24 h, cells were serum-starved and then treated with vehicle or gastrin (200 nm) for 6 h, and lysates were prepared for luciferase detection. Values were set to 1, and data are means ± S.E. fold induction (relative units, R.U.) from three independent experiments (*, p < 0.05, two-tailed t test).
Phosphorylation of FAK at Tyr-397 also occurs in response to gastrin stimulation of DLD-1 cells, and pharmacological FAK inhibition can prevent gastrin-stimulated paxillin tyrosine phosphorylation and cell motility (7). Gα13-shRNA expression reduced gastrin-stimulated FAK Tyr-397 phosphorylation without effects on total FAK expression (Fig. 2D). Gastrin-stimulated SRE-luciferase activity was also reduced in the presence of Gα13 shRNA compared with mock DLD-1-infected cells (Fig. 2E). Together, these results support a gastrin, CCK2R, Gα13, FAK, and paxillin signaling linkage that may impact gastrin-stimulated SRE transcriptional activity.
Gα13 Activates Rgnef and RhoA in a Linkage to Enhanced SRE Transcription
Rgnef functions to promote paxillin tyrosine phosphorylation downstream of integrins in fibroblasts and gastrin-CCK2R stimulation in DLD-1 colon carcinoma cells (5–7). Because we found that Gα13 is important for gastrin-stimulated paxillin tyrosine phosphorylation and enhanced SRE activity, we investigated whether Gα13 could activate Rgnef via affinity binding to a purified nucleotide-free point mutant (G17A) of RhoA (Fig. 3). In cells expressing CCK2R, gastrin stimulation increased Rgnef binding to RhoAG17A (Fig. 3A). In cells expressing Gα13 or Gαq, Rgnef possessed a higher basal level of activity as determined by RhoAG17A binding. AlF4− addition to activate the Gα proteins resulted in strong Rgnef binding to RhoAG17A (Fig. 3A). Importantly, expression of constitutively active Gα13Q226L and GαqR183C point mutants promoted Rgnef binding to RhoAG17A (Fig. 3B).
FIGURE 3.

Gastrin, Gα13, and Gαq promote Rgnef and RhoA activation. A and B, affinity binding to a nucleotide-free mutant of RhoA (G17A) was used to evaluate Rgnef activation. HEK293 cells were transfected with expression vectors for HA-Rgnef, CCK2R, Gα13wt, or Gαqwt (A) or HA-Rgnef and Gα13Q226L or GαqR183C (B) as indicated. Cells were either left unstimulated or stimulated with 100 nm gastrin or 10 μm AlF4− for 40 min. Active Rgnef bound to GST-RhoAG17A beads was visualized by anti-HA immunoblotting (top blots), and protein expression in total cell lysates was evaluated by anti-HA, -Gα13, and -Gαq immunoblotting. Experiments were repeated three times with similar results. C and D, active RhoA was measured by the GST-Rhotekin RBD pulldown assay. HEK293 cells were transfected with expression vectors for HA-Rgnef, Gα13wt, or Gαqwt (C) or mCherry-Rgnef, FLAG-Gβ1, Gγ1, or Gα13wt (D) as indicated. Active RhoA in the pulldown was visualized by anti-RhoA immunoblotting (top blots), and protein expression in total cell lysates was evaluated by anti-RhoA, -mCherry, -FLAG, and -Gα13 immunoblotting. Experiments were repeated three times with similar results. E and F, SRE reporter transcriptional activity was measured in HEK293 cells transfected with expression vectors for HA-Rgnef, Gα13Q226L, and GαqR183C (E) or expression vectors for Gα13QL, PDZ-RhoGEF, LARG, or Rgnef (F) as indicated. Cells were serum-starved overnight, and luciferase activity was measured. Control empty vector-transfected cell values were set to 1. Data are means ± S.E. fold induction (relative units, R.U.) from three independent experiments (*, p < 0.05; two-tailed t test). Anti-HA (Rgnef), -Gα13, and -Gαq immunoblotting was used to verify transfected construct expression.
To analyze whether Gα13 could activate Rho downstream of Rgnef, we measured the activity of RhoA in an affinity binding pulldown assay (Fig. 3C). A combined expression of Gα13 plus Rgnef promoted RhoA activation (Fig. 3C). Importantly, Rgnef expression in combination with Gα12 expression did not result in significant RhoA activation compared with the combination of Gα13 and Rgnef (Fig. 3D). We evaluated the effect of Gβ1γ1 expression (Fig. 3D) because p114RhoGEF, another member of the Rgnef subfamily, was shown to activate RhoA via Gβγ (19). However, for Rgnef we could not detect additional Rho activity in the presence of Gβ1γ1. The decreased activity observed, which was not always consistent, could be an indirect effect of Gβ1γ1 expression. These results support the notion that there may be a selective signaling linkage between Gα13, Gαq, and Rgnef.
We next performed SRE-luciferase assay in the presence of Rgnef and active forms of Gα13 or Gαq proteins as alternative measure of RhoA signaling (Fig. 3E). Combined Rgnef expression with active Gα13 or Gαq induced a synergistic increase in SRE-luciferase activity. As internal control, we analyzed the expression of active Gα13 with its well known downstream RhoGEF effectors. Expression of LARG or Rgnef resulted in increased SRE-luciferase activity compared with expression of active Gα13 alone (Fig. 3F). Taken together, these results suggest that Gα13 and Gαq, but not Gα12 or Gβ1γ1, stimulate Rgnef activity leading to RhoA activation.
Gα13 Immunoprecipitated with the N- and C-terminal Region of Rgnef
To determine whether Rgnef might act as a downstream effector for Gα13, co-immunoprecipitation (IP) analyses were performed with endogenous and exogenous proteins. In DLD-1 cells endogenous Gα13 protein was associated with a 190-kDa anti-Rgnef immunoreactive protein (Fig. 4A). A similar band was observed with exogenous HA-Rgnef that immunoprecipitated with endogenous Gα13 in DLD-1 cells. Additionally, AlF4− stimulation of cells enhanced the amount of Gα13 co-immunoprecipitating with antibodies to Rgnef (Fig. 4B). This result suggests that Gα13 needs to be activated downstream of GPCRs to immunoprecipitated with Rgnef. Specificity of Gα and Rgnef immunoprecipitations was tested by co-transfection and co-IP studies. HA-Rgnef associated with active forms of Gα13 and Gαq but not with GαZ (Fig. 4, C and D). No consistent immunoprecipitation was observed with the active forms of Gα12 (Fig. 4C), in agreement with the activity data (Fig. 4C). Taken together, the results suggest that active Gα13 and Gαq form a complex with Rgnef and that Gα13 and Gαq are upstream activators of Rgnef.
FIGURE 4.

Rgnef association with Gα13 and Gαq proteins. A, DLD-1 cells were transfected with expression vectors for Gα13Q226L and HA-Rgnef as indicated. IP analyses with anti-Gα13 antibodies were used to detect complexes of endogenous and exogenous Gα13 with Rgnef. Protein expression in total cell lysates was evaluated by anti-HA (Rgnef), -Gα13, and β-tubulin immunoblotting. B, HEK293 cells transfected with Gα13 and HA-Rgnef in the presence or absence of 10 μm AlF4−. IP analyses with anti-HA antibodies were used to detect complexes with Gα13 with anti-HA and -Gα13 immunoblotting. Protein expression levels were evaluated immunoblotting of total lysates. C and D, HEK293 cells were transfected with expression vectors encoding HA-Rgnef, Gα12Q229L, Gα13Q226L, or GαqR183C (C) or HA-Rgnef, Gα13Q226L, or Gαz-EE (D) as indicated. C, IP analyses with anti-HA antibodies were used to detect complexes with Gα12, Gα13, and Gαq by anti-HA and -Gα12, -Gα13, and -Gαq immunoblotting. D, IP analyses with anti-HA antibodies were used to detect complexes with Gα13 and Gαz by anti-HA, -Gα13, and -EE tag immunoblotting. Protein expression levels were evaluated by immunoblotting of total cell lysates. A–D, experiments were repeated at least three times with similar results.
Rgnef C-terminal Domain Immunoprecipitated with Activated Gα13
To determine the Rgnef region that mediates Gα13 immunoprecipitation, a series of HA- or mCherry-tagged N- or C-terminal truncated Rgnef constructs was created and verified to express proteins of different but expected sizes (Fig. 5A). Anti-HA co-IP analyses show that Gα13 immunoprecipitated with the full-length and the C-terminal (1185–1693) region of Rgnef (Fig. 5B). A weaker co-immunoprecipitation was also observed with the N-terminal region (1–1184) of Rgnef. Because the Rgnef C-terminal region interacts with Gα13, we tested whether Rgnef-CT overexpression could block Gα13 activity. Co-expression of HA-Rgnef-Ct(1185–1693), HA-Rgnef-Ct*(1279–1582), or HA-Rgnef-CtΔFAK(1302–1582) in HEK293 cells produced a significant reduction of Gα13Q226L-induced SRE-luciferase activity (Fig. 5C). These results support the hypothesis that Rgnef may be proximally activated by Gα13Q226L and that overexpression of the putative Rgnef-Ct interacting region (1279–1582) blocks Gα13Q226L-induced SRE activity independently of FAK binding to Rgnef (Fig. 5C).
FIGURE 5.

Rgnef C-terminal domain association with activated Gα13Q226L. A, schematic of full-length and truncated Rgnef constructs as HA- or mCherry- fusion proteins. Lower panel, expression of HA-tagged full-length, Nt(1–1184), and Ct(1185–1693) Rgnef constructs in HEK293 cells by anti-HA immunoblotting. B, HEK293 cells were transfected with HA-tagged vectors encoding for Rgnef full-length, Rgnef-Nt(1–1184), Rgnef-Ct(1185–1693), and/or Gα13Q226L. Cell lysates were immunoprecipitated as described before. Data are representative of at least three independent experiments. Arrow points to Rgned-Ct. C, Rgnef-Ct domain constructs inhibit Gα13Q226L-mediated SRF activation. HEK293 cells were transfected with pSRE.L and pRL-TK, together with either empty vector, vector encoding Gα13QL, and/or Rgnef -Ct(1185–1693), Rgnef-Ct*(1279–1582), and Rgnef ΔFAK(1302–1582). After 24 h of transfection, cells were serum-starved overnight, and then SRF activities of cell lysates were measured using the Dual-Luciferase assay kit (Promega). Anti-Gα13 immunoblotting shows equal expression in cell lysates. Data are means ± S.E. of four independent experiments, each conducted in triplicate (*, p < 0.05; **, p < 0.005; two-tailed t test). D, HEK293 cells expressing Gα13 in presence or absence of mCherry-Rgnef-FL, mCherry-Rgnef-Ct*, and mCherry-RgnefΔFAK were immunoprecipitated using anti-Gα13 antibody and subjected to Western blot analysis. Cells lysates were analyzed in parallel. Full-length Rgnef, Rgnef-Ct, and ΔFAK were detected by anti-Rgnef, and Gα13 was detected using anti-Gα13 immunoblotting. Data are representative of four independent experiments.
To determine whether the Rgnef region that blocks Gα13Q226L-induced SRE activity can also form a complex with Gα13Q226L, HEK293 co-transfection and co-IP experiments were performed (Fig. 5D). mCherry-Rgnef-Ct* and mCherry-RgnefΔFAK associated with antibodies to Gα13. Additionally, mCherry-RgnefΔFAK associated in a complex with endogenous Gα13. Taken together, these results show that the Rgnef-interactive (and dominant-negative acting) domain for Gα13-induced signaling occurs independently of FAK binding to Rgnef.
Domain in the Coiled Coil of p190RhoGEF C Terminus
To determine potential residues responsible for Rgnef-Ct association with Gα13, we performed sequence alignments with other RhoGEFs known to interact with Gα proteins. In particular, we identified a pattern of sequence similarity with the RH domain-containing subfamily of RhoGEFs: p115, Lsc, PDZ, and LARG (Fig. 6). Alignment is based upon the crystal structure of the RH domain of p115GEF (41, 42). Despite low 32% sequence identities shared between the RH domains of p115RhoGEF and PDZ-RhoGEF, these domains share nearly identical three-dimensional structures, a root mean square deviation of 0.608 Å on Cα values (41, 42).
FIGURE 6.

Sequence alignment between the RH domains of RH-RhoGEF protein family and Rgnef. Amino acids are colored based on conservation according to the Clustal scheme. Gold boxes over the alignment indicate the localization of helices based on the crystal structures. Numbers indicate the start and end of the RH-like domain in the respective sequence as follows: p115(h):Q92888, Lsc(m):Q61210, Lsc(r)Q9Z1I6, PDZ(h):O15085, PDZ(r):Q9ES67, LARG(h):Q9NZN5, LARG(m):Q8R4H2, Rgnef(h): Q8N1W1, Rgnef(m):P97433, Rgnef(r):P0C6P5. Five conserved Rgnef human (h) residues subjected to alanine mutagenesis are boxed. m, mouse.
By performing computer modeling using the coordinates of the p115 RhoGEF and PDZ RhoGEF RH domains, we were able to generate a Rgnef-Ct three-dimensional model (Fig. 7A, yellow) that contains similar secondary structure elements with p115 RhoGEF (Fig. 7A, blue) and PDZ RhoGEF (Fig. 7A, salmon). In particular, the organization of Rgnef residues in the arrangement of helices α1, α3, α8, and α9 suggests that a similar binding interface may form with Gα13 (Fig. 7B). Computer modeling reveals potential sites of Gα13 interaction mediated by both hydrophobic or ionic amino acid residues within the putative Rgnef-Ct α3, α8, and α9 modeled structure (Fig. 7B).
FIGURE 7.

Models of putative Rgnef RH-like domain and the complex with Gα13. A, structural superposition of the RH domains of human PDZ-RhoGEF (Protein Data Bank code 3CX8; salmon), p115 RhoGEF (Protein Data Bank code 3AB3; blue) and our computer model of the domain of Rgnef (yellow). B, computer model of the domain of Rgnef (yellow) in complex with Gα13 (blue). Putative residues of charged or hydrophobic side chain interactions are shown in blue. Residues mutated in the Rgnef-5 M are shown in black (corresponding to human His-1396, Glu-1478, Glu-1484, Leu-1486, and Arg-1490). C and D, HEK293 cells were transfected with vectors encoding for HA-Rgnef (full length), HA-Rgnef 1M (C), or HA-Rgnef 5M (D) with or without Gα13Q226L. HA-tagged Rgnef constructs were co-immunoprecipitated with antibodies to Gα13 and detected by Li-COR immunoblotting. Graphical results show immunoblotting band intensities from three independent experiments expressed as fold induction with respect to immunoprecipitated Rgnef wild type and normalized by the total expression level of Rgnef. Values are means ± S.D. (*, p < 0.05; **, p < 0.005; two-tailed t test). E, HEK293 cells expressing HA-Rgnef or HA-Rgnef5M with or without GαqR183C or Gα13Q226L were co-immunoprecipitated with HA tag antibodies and detected by Li-COR immunoblotting. The arrowhead points to an unspecific band detected with the anti-Gq monoclonal antibody. F, HEK293 cells expressing HA-Rgnef or HA-Rgnef 5M were incubated with RhoAG17A-GST beads and visualized by anti-HA tag immunoblotting. G, HEK293 cells were transfected with pSRE.L and pRL-TK, together with either empty vector (Ctl), vector encoding Rgnef or Rgnef5M and SRF activities were measured. Data are means ± S.E. of three independent experiments, each conducted in duplicate (***, p < 0.001).
Based on the modeled Rgnef-Gα13 complex, we designed two mutants (Rgnef-1M and Rgnef-5M) that replaced residues predicted to lie in the putative interface with Gα13 (Fig. 7, A and B). In Rgnef-1M, Arg-1490 was mutated to alanine (R1490A), and in Rgnef-5M, four additional Rgnef residues were mutated to alanine (E1484A, H1396A, E1478A, and L1486A) plus Rgnef R1490A. Upon co-expression in HEK293 cells with activated Gα13, Rgnef association was determined by co-immunoprecipitation with antibodies to Rgnef (Fig. 7C) or with Gα13 (Fig. 7D). Rgnef-1M and Rgnef-5M exhibited significantly decreased association with Gα13 compared with Rgnef-WT.
Because both mutationally activated Gα13 and Gαq could activate Rgnef (Fig. 3), we determined whether Rgnef-5M mutations disrupted binding to both Gα13 and Gαq by HEK co-expression and co-immunoprecipitation analyses (Fig. 7E). Surprisingly, Gαq immunoprecipitated equally well with Rgnef-WT and Rgnef-5M, whereas Gα13 showed selectively decreased association with Rgnef-5M. To determine whether alterations in global protein folding may underlie binding differences between Rgnef-WT and Rgenf-5M, RhoGEF (RhoA G17A) and SRE activity assays were performed (Fig. 7F). Notably, Rgnef-5M exhibited equivalent binding to RhoA G17A and significantly enhanced SRE activity in an equivalent manner compared with Rgnef-WT.
Taken together, these results support the notion that Rgnef residues Glu-1484, His-1396, Glu-1478, Leu-1486, and Arg-1490 (mutated in Rgnef-5M) facilitate a binding and signaling linkage with Gα13. Of course, Rgnef and Gα13 association may also be indirect, and these residues within the Rgnef-Ct coiled-coil domain are important in mediating this linkage. Nevertheless, these results strongly support a role for Rgnef as a downstream effector for Gα13, and we have identified a new domain within Rgnef that is important for Gα13 signaling.
Discussion
Although the trophic properties of gastrin are well established (45, 46), the intracellular and molecular mechanisms by which gastrin modulates cell growth in the gastrointestinal tract have yet to be fully elucidated. It has been reported that stimulation of CCK2R by gastrin activates several signal transduction pathways involved in cell proliferation and migration, such as the mitogen-activated protein kinase (MAPK) that includes ERK, JNK, p38 kinase, and RhoA activation (47). Here, we show that Gα13 activates Rgnef, which in turn activates RhoA in human colon cancer DLD-1 cells. Knockdown of Gα13 prevented CCK2R-induced gastrin activation of RhoA, FAK-Tyr(P)-397, and paxillin-Tyr(P)-31 tyrosine phosphorylation. Taken together, the results suggest that the signaling pathway between gastrin-induced CCK2R and RhoA downstream activity involves the activation of Rgnef by Gα13.
Lacking a recognized RH-like domain, Rgnef was not considered a putative effector of Gα13/12. Rgnef family members AKAP-lbc and p114RhoGEF have been previously shown to be activated by G proteins (18, 19). Our studies place Rgnef as the downstream activator of both Gα13 and Gαq. However, our data are only suggestive of Rgnef as a direct effector, and biochemical experiments with purified proteins have been problematic to date. Our results show that both the N terminus with the DH-PH domain and the C-terminal domains of Rgnef are each sufficient to immunoprecipitate with Gα13. A multiple sequence alignment led us to identify a region at the C terminus of Rgnef with weak homology to the RH domain. RH domains of RH-RhoGEFs substantially differ among the family (48) indicating that structural adaptations in Gα13 could allow interaction with several RH domains, despite their considerable divergence. In this study, we showed that this domain is necessary for Gα13 effects. The fact that the mutation of residues located at this region reduce its binding to Gα13 validates the proposed model for the Rgnef/Gα13. A similar region located at the C-terminal region of AKAP-lbc was suggested to be involved in Gα12 interaction (18). However, similar coiled-coil structures in other RhoGEFs proteins have been associated with hetero- and homodimerization (49, 50). Heterodimerization of Rgnef with known RH-RhoGEFs could also explain the effects obtained in this work. Nevertheless, Rgnef is the first RH-GEF demonstrated to be downstream of CCK2R. We also found that Gα13 immunoprecipitated with the N terminus of Rgnef containing the DH and PH domain, which should not dimerize. Previous reports on LARG showed simultaneously interaction with the RH domain (at the N-terminal domain of LARG) and DH-PH domain (51). Similarly, it has been shown that activated Gα13 can interact with the RH domain and the DH-PH domains of p115RhoGEF (45, 46, 52, 53). Further investigations will examine the functional role of the interaction through the region containing the DH-PH domains, if the association of Rgnef with Gα13 is direct, and determine whether Rgnef mediates GTPase-activating protein activity toward Gα13.
Our results show that in addition to Gα13, Gαq can also act as an upstream activator of Rgnef. Interestingly, previous reports show that both Gα13 and Gαq linked CCK stimulation to the activation of Rac in pancreatic acini (54). For many years, the role of Gαq in RhoA stimulation was under discussion, but investigations supported that Gαq can induce Rho-dependent responses via cooperation with several RH-RhoGEFs, including Lbc-RhoGEF and LARG (20, 37, 55). Later on, it was shown that p63RhoGEF interacts and is activated by Gαq (56, 57). Resolution of the crystal structure of the Gαi/q-p63RhoGEF complex revealed that Gαq interacts with the DH and PH domains of p63RhoGEF (57). Consistently, the functional interaction between Gαq and LARG requires only the DH-PH domains of LARG. Thus, the interaction between Rgnef and Gαq and the fact that Gαq promotes RhoA activation through Rgnef are not surprising. Moreover, the results with the Rgnef-5M point to a clear difference in the mechanism of activation of Rgnef between Gαq and Gα13. Further work will be necessary to address which domain in Rgnef is involved in the activation by Gαq and/or whether p63RhoGEF is also part of this pathway.
Previous studies demonstrated that Rgnef is a key regulator of Rho reactivation, focal adhesion establishment (5), and cell migration downstream of integrins (6). Rgnef can directly bind FAK through a motif in the Rgnef C-terminal region (64), and Rgnef-null cells showed fewer early adhesions upon fibronectin replating (6). Rgnef binding to membrane lipids is needed for activation of FAK at the initial steps of early adhesion, but this effect is independent of its RhoGEF activity (58). Rgnef-GEF activity and Rho activation are required later in cell migration for the process of stability and maturation of adhesion. Interestingly, it has been shown that integrins are noncanonical Gα13-coupled receptors that provide a mechanism for dynamic regulation of RhoA (59), which poses the question whether the link between Gα13 and Rgnef could also be active downstream of integrin adhesions.
In DLD-1 cells, gastrin enhances FAK and paxillin phosphorylation and increases focal adhesion formation linked to the dissolution of cell-cell contacts associated with the loss of E-cadherin surface expression, a process that requires Rgnef (7). Importantly, our results show that activation of Gα13 downstream of gastrin leads to FAK and paxillin phosphorylation and is required for Rgnef activation and Rho activity. Taking into account those results and published records, we propose a model (Fig. 8) whereby, upon gastrin stimulation, Gα13 directs Rgnef to the plasma membrane where it associates with and activates FAK. Next, Gα13GTP promotes Rgnef-GEF activity, leading to RhoA activation, which in turn can induce downstream paxillin phosphorylation and, as result of those processes, cell motility (which may involve FAK dissociation). Previous results demonstrated that RgnefΔFAK did not block the phosphorylation of paxillin downstream of gastrin (7). However, Gα13-induced-SRE activity was inhibited by RgnefΔFAK indicating that analyses of SRE luciferase activity may not always reflect changes in paxillin tyrosine phosphorylation despite both being enhanced by gastrin or activated Gα13 construct expression.
FIGURE 8.
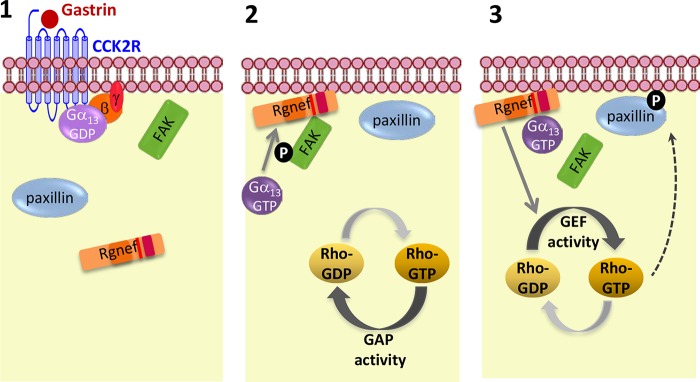
Model of Gα13-dependent activation of Rgnef downstream CCK2R. 1, gastrin stimulation of CCK2 will lead to the activation of Gα13 (GTP-bound form) and dissociation from Gγβ. 2, Gα13 protein activation will help to recruit Rgnef to the plasma membrane where it can bind and facilitate FAK-Tyr-397 phosphorylation. 3, at later points, Gα13 can facilitate activation of the Rgnef-GEF activity leading to the activation of RhoA and signaling cascade that enhances paxillin tyrosine phosphorylation and SRE promoter activity.
RhoA activation is implicated in colorectal tumor progression (2), and the levels of Rgnef mRNA and protein increase with colon carcinoma tumor progression (7). Although CCK2R is frequently overexpressed in cancers and the oncogenic properties of gastrin in the colonic mucosa of mouse are well established (60), its role in humans is still controversial (61). Nevertheless, the presence of CCK2R is recognized to provide a growth advantage to tumors where it is expressed (60). Together our results suggest a mechanism whereby gastrin stimulation of Gα13 activates Rgnef implicating a role in human colon carcinoma. Little is known about the specific functions of Gα13 in cancer, and those studies have focused on Gα12 (29). It is documented that Gα13 expression is markedly increased during prostate cancer progression and that micro RNAs regulate its expression post-transcriptionally (62). Gα13 also mediates lysophosphatidic acid-stimulated invasive migration of pancreatic cancer cells (63). The future analysis of Gα13 levels will help to elucidate whether its expression varies during colon carcinoma progression and further elucidate its function in colon cancer.
Acknowledgment
We thank Monica Pons for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant CA180769 (to D. S.). This work was also supported by Spanish Ministerio de Economia y Competitividad Grant BFU2011-30080.
- GEF
- guanine nucleotide exchange factor
- FAK
- focal adhesion kinase
- GPCR
- G protein-coupled receptor
- RH
- regulator of G protein signaling homology
- Rgnef-CT
- Rgnef C-terminal
- SRE
- serum-response element
- SRF
- serum-response factor
- RBD
- Rho binding domain
- IP
- immunoprecipitation
- CCK2R
- type 2 cholecystokinin receptor
- Scr
- scrambled
- PH
- pleckstrin homology
- DOC
- deoxycholate.
References
- 1. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 2. Sahai E., Marshall C. J. (2002) RHO-GTPases and cancer. Nat. Rev. Cancer 2, 133–142 [DOI] [PubMed] [Google Scholar]
- 3. Leve F., Morgado-Díaz J. A. (2012) Rho GTPase signaling in the development of colorectal cancer. J. Cell. Biochem. 113, 2549–2559 [DOI] [PubMed] [Google Scholar]
- 4. van Horck F. P., Ahmadian M. R., Haeusler L. C., Moolenaar W. H., Kranenburg O. (2001) Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 276, 4948–4956 [DOI] [PubMed] [Google Scholar]
- 5. Lim Y., Lim S.-T., Tomar A., Gardel M., Bernard-Trifilo J. A., Chen X. L., Uryu S. A., Canete-Soler R., Zhai J., Lin H., Schlaepfer W. W., Nalbant P., Bokoch G., Ilic D., Waterman-Storer C., Schlaepfer D. D. (2008) PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J. Cell Biol. 180, 187–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller N. L., Lawson C., Chen X. L., Lim S.-T., Schlaepfer D. D. (2012) Rgnef (p190RhoGEF) knockout inhibits RhoA activity, focal adhesion establishment, and cell motility downstream of integrins. PLoS One 7, e37830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu H.-G., Nam J.-O., Miller N. L., Tanjoni I., Walsh C., Shi L., Kim L., Chen X. L., Tomar A., Lim S.-T., Schlaepfer D. D. (2011) p190RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 71, 360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu J., Zhai J., Lin H., Nie Z., Ge W. W., García-Bermejo L., Muschel R. J., Schlaepfer W. W., Cañete-Soler R. (2003) Cytoplasmic retention sites in p190RhoGEF confer anti-apoptotic activity to an EGFP-tagged protein. Brain Res. Mol. Brain Res. 117, 27–38 [DOI] [PubMed] [Google Scholar]
- 9. Lin H., Zhai J., Schlaepfer W. W. (2005) RNA-binding protein is involved in aggregation of light neurofilament protein and is implicated in the pathogenesis of motor neuron degeneration. Hum. Mol. Genet. 14, 3643–3659 [DOI] [PubMed] [Google Scholar]
- 10. Rico B., Beggs H. E., Schahin-Reed D., Kimes N., Schmidt A., Reichardt L. F. (2004) Control of axonal branching and synapse formation by focal adhesion kinase. Nat. Neurosci. 7, 1059–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim J.-S., Bareiss S., Kim K. K., Tatum R., Han J.-R., Jin Y. H., Kim H., Lu Q., Kim K. (2006) Presenilin-1 inhibits δ-catenin-induced cellular branching and promotes δ-catenin processing and turnover. Biochem. Biophys. Res. Commun. 351, 903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim H., Oh M., Lu Q., Kim K. (2008) E-Cadherin negatively modulates δ-catenin-induced morphological changes and RhoA activity reduction by competing with p190RhoGEF for δ-catenin. Biochem. Biophys. Res. Commun. 377, 636–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gebbink M. F., Kranenburg O., Poland M., van Horck F. P., Houssa B., Moolenaar W. H. (1997) Identification of a novel, putative Rho-specific GDP/GTP exchange factor and a RhoA-binding protein: control of neuronal morphology. J. Cell Biol. 137, 1603–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt A., Hall A. (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 15. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 16. Sternweis P. C., Carter A. M., Chen Z., Danesh S. M., Hsiung Y.-F., Singer W. D. (2007) Regulation of Rho guanine nucleotide exchange factors by G proteins. Adv. Protein Chem. 74, 189–228 [DOI] [PubMed] [Google Scholar]
- 17. Aittaleb M., Boguth C. A., Tesmer J. J. (2010) Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diviani D., Soderling J., Scott J. D. (2001) AKAP-Lbc anchors protein kinase A and nucleates Gα12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 276, 44247–44257 [DOI] [PubMed] [Google Scholar]
- 19. Niu J., Profirovic J., Pan H., Vaiskunaite R., Voyno-Yasenetskaya T. (2003) G protein βγ subunits stimulate p114RhoGEF, a guanine nucleotide exchange factor for RhoA and Rac1: regulation of cell shape and reactive oxygen species production. Circ. Res. 93, 848–856 [DOI] [PubMed] [Google Scholar]
- 20. Dutt P., Kjoller L., Giel M., Hall A., Toksoz D. (2002) Activated Gαq family members induce Rho GTPase activation and Rho-dependent actin filament assembly. FEBS Lett. 531, 565–569 [DOI] [PubMed] [Google Scholar]
- 21. Siehler S. (2009) Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 158, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang H., Wu D., Simon M. I. (1993) The transforming activity of activated Gα12. FEBS Lett. 330, 319–322 [DOI] [PubMed] [Google Scholar]
- 23. Jones T. L., Gutkind J. S. (1998) Gα12 requires acylation for its transforming activity. Biochemistry 37, 3196–3202 [DOI] [PubMed] [Google Scholar]
- 24. Kumar R. N., Shore S. K., Dhanasekaran N. (2006) Neoplastic transformation by the gep oncogene, Gα12, involves signaling by STAT3. Oncogene 25, 899–906 [DOI] [PubMed] [Google Scholar]
- 25. Juneja J., Cushman I., Casey P. J. (2011) G12 Signaling through c-Jun NH2-terminal kinase promotes breast cancer cell invasion. PLoS One 6, e26085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meigs T. E., Fedor-Chaiken M., Kaplan D. D., Brackenbury R., Casey P. J. (2002) Gα12 and Gα13 negatively regulate the adhesive functions of cadherin. J. Biol. Chem. 277, 24594–24600 [DOI] [PubMed] [Google Scholar]
- 27. Krakstad B. F., Ardawatia V. V., Aragay A. M. (2004) A role for Gα12/Gα13 in p120ctn regulation. Proc. Natl. Acad. Sci. U.S.A. 101, 10314–10319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ardawatia V. V., Masià-Balagué M., Krakstad B. F., Johansson B. B., Kreitzburg K. M., Spriet E., Lewis A. E., Meigs T. E., Aragay A. M. (2011) Gα(12) binds to the N-terminal regulatory domain of p120(ctn), and downregulates p120(ctn) tyrosine phosphorylation induced by Src family kinases via a RhoA independent mechanism. Exp. Cell Res. 317, 293–306 [DOI] [PubMed] [Google Scholar]
- 29. Kelly P., Moeller B. J., Juneja J., Booden M. A., Der C. J., Daaka Y., Dewhirst M. W., Fields T. A., Casey P. J. (2006) The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U.S.A. 103, 8173–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kelly P., Stemmle L. N., Madden J. F., Fields T. A., Daaka Y., Casey P. J. (2006) A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J. Biol. Chem. 281, 26483–26490 [DOI] [PubMed] [Google Scholar]
- 31. Kelly P., Casey P. J., Meigs T. E. (2007) Biologic functions of the G12 subfamily of heterotrimeric g proteins: growth, migration, and metastasis. Biochemistry 46, 6677–6687 [DOI] [PubMed] [Google Scholar]
- 32. Aragay A. M., Collins L. R., Post G. R., Watson A. J., Feramisco J. R., Brown J. H., Simon M. I. (1995) G12 requirement for thrombin-stimulated gene expression and DNA synthesis in 1321N1 astrocytoma cells. J. Biol. Chem. 270, 20073–20077 [DOI] [PubMed] [Google Scholar]
- 33. Escrieut C., Gigoux V., Archer E., Verrier S., Maigret B., Behrendt R., Moroder L., Bignon E., Silvente-Poirot S., Pradayrol L., Fourmy D. (2002) The biologically crucial C terminus of cholecystokinin and the nonpeptide agonist SR-146,131 share a common binding site in the human CCK1 receptor. Evidence for a crucial role of Met-121 in the activation process. J. Biol. Chem. 277, 7546–7555 [DOI] [PubMed] [Google Scholar]
- 34. Johansson B. B., Minsaas L., Aragay A. M. (2005) Proteasome involvement in the degradation of the G(q) family of Gα subunits. FEBS J. 272, 5365–5377 [DOI] [PubMed] [Google Scholar]
- 35. Benincá C., Planagumà J., de Freitas Shuck A., Acín-Perez R., Muñoz J. P., de Almeida M. M., Brown J. H., Murphy A. N., Zorzano A., Enríquez J. A., Aragay A. M. (2014) A new noncanonical pathway of Gα(q) protein regulating mitochondrial dynamics and bioenergetics. Cell. Signal. 26, 1135–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. García-Mata R., Wennerberg K., Arthur W. T., Noren N. K., Ellerbroek S. M., Burridge K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 [DOI] [PubMed] [Google Scholar]
- 37. Sagi S. A., Seasholtz T. M., Kobiashvili M., Wilson B. A., Toksoz D., Brown J. H. (2001) Physical and functional interactions of Gαq with Rho and its exchange factors. J. Biol. Chem. 276, 15445–15452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hill C. S., Wynne J., Treisman R. (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 39. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 40. Sali A. (1995) Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today 1, 270–277 [DOI] [PubMed] [Google Scholar]
- 41. Chen Z., Singer W. D., Danesh S. M., Sternweis P. C., Sprang S. R. (2008) Recognition of the activated states of Gα13 by the rgRGS domain of PDZRhoGEF. Structure 16, 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hajicek N., Kukimoto-Niino M., Mishima-Tsumagari C., Chow C. R., Shirouzu M., Terada T., Patel M., Yokoyama S., Kozasa T. (2011) Identification of critical residues in Gα13 for stimulation of p115RhoGEF activity and the structure of the Gα13-p115RhoGEF regulator of G protein signaling homology (RH) domain complex. J. Biol. Chem. 286, 20625–20636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki N., Nakamura S., Mano H., Kozasa T. (2003) Gα12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. U.S.A. 100, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koh T. J., Chen D. (2000) Gastrin as a growth factor in the gastrointestinal tract. Regul. Pept. 93, 37–44 [DOI] [PubMed] [Google Scholar]
- 46. Chu S., Schubert M. L. (2013) Gastric secretion. Curr. Opin. Gastroenterol. 29, 636–641 [DOI] [PubMed] [Google Scholar]
- 47. Taniguchi T., Takaishi K., Murayama T., Ito M., Iwata N., Chihara K., Sasaki T., Takai Y., Matsui T. (1996) Cholecystokinin-B/gastrin receptors mediate rapid formation of actin stress fibers. Oncogene 12, 1357–1360 [PubMed] [Google Scholar]
- 48. Sprang S. R., Chen Z., Du X. (2007) Structural basis of effector regulation and signal termination in heterotrimeric Gα proteins. Adv. Protein Chem. 74, 1–65 [DOI] [PubMed] [Google Scholar]
- 49. Grabocka E., Wedegaertner P. B. (2007) Disruption of oligomerization induces nucleocytoplasmic shuttling of leukemia-associated rho Guanine-nucleotide exchange factor. Mol. Pharmacol. 72, 993–1002 [DOI] [PubMed] [Google Scholar]
- 50. Baisamy L., Jurisch N., Diviani D. (2005) Leucine zipper-mediated homo-oligomerization regulates the Rho-GEF activity of AKAP-Lbc. J. Biol. Chem. 280, 15405–15412 [DOI] [PubMed] [Google Scholar]
- 51. Suzuki N., Tsumoto K., Hajicek N., Daigo K., Tokita R., Minami S., Kodama T., Hamakubo T., Kozasa T. (2009) Activation of leukemia-associated RhoGEF by Gα13 with significant conformational rearrangements in the interface. J. Biol. Chem. 284, 5000–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wells C. D., Liu M.-Y., Jackson M., Gutowski S., Sternweis P. M., Rothstein J. D., Kozasa T., Sternweis P. C. (2002) Mechanisms for reversible regulation between G13 and Rho exchange factors. J. Biol. Chem. 277, 1174–1181 [DOI] [PubMed] [Google Scholar]
- 53. Chen Z., Guo L., Hadas J., Gutowski S., Sprang S. R., Sternweis P. C. (2012) Activation of p115-RhoGEF requires direct association of Gα13 and the Dbl homology domain. J. Biol. Chem. 287, 25490–25500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sabbatini M. E., Bi Y., Ji B., Ernst S. A., Williams J. A. (2010) CCK activates RhoA and Rac1 differentially through Gα13 and Gαq in mouse pancreatic acini. Am. J. Physiol. Cell Physiol. 298, C592–C601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Booden M. A., Siderovski D. P., Der C. J. (2002) Leukemia-associated Rho guanine nucleotide exchange factor promotes Gαq-coupled activation of RhoA. Mol. Cell. Biol. 22, 4053–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lutz S., Freichel-Blomquist A., Yang Y., Rümenapp U., Jakobs K. H., Schmidt M., Wieland T. (2005) The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J. Biol. Chem. 280, 11134–11139 [DOI] [PubMed] [Google Scholar]
- 57. Lutz S., Shankaranarayanan A., Coco C., Ridilla M., Nance M. R., Vettel C., Baltus D., Evelyn C. R., Neubig R. R., Wieland T., Tesmer J. J. (2007) Structure of Gαq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923–1927 [DOI] [PubMed] [Google Scholar]
- 58. Miller N. L., Lawson C., Kleinschmidt E. G., Tancioni I., Uryu S., Schlaepfer D. D. (2013) A noncanonical role for Rgnef in promoting integrin-stimulated focal adhesion kinase activation. J. Cell Sci. 126, 5074–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gong H., Shen B., Flevaris P., Chow C., Lam S. C., Voyno-Yasenetskaya T. A., Kozasa T., Du X. (2010) G protein subunit G 13 binds to integrin IIb 3 and mediates integrin “Outside-in” signaling. Science 327, 340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dufresne M., Seva C., Fourmy D. (2006) Cholecystokinin and gastrin receptors. Physiol. Rev. 86, 805–847 [DOI] [PubMed] [Google Scholar]
- 61. Fourmy D., Gigoux V., Reubi J. C. (2011) Gastrin in gastrointestinal diseases. Gastroenterology 141, 814–818 [DOI] [PubMed] [Google Scholar]
- 62. Rasheed S. A., Teo C. R., Beillard E. J., Voorhoeve P. M., Casey P. J. (2013) MicroRNA-182 and microRNA-200a control G protein subunit α-13 (GNA13) expression and cell invasion synergistically in prostate cancer cells. J. Biol. Chem. 288, 7986–7995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gardner J. A., Ha J. H., Jayaraman M., Dhanasekaran D. N. (2013) The gep proto-oncogene Gα13 mediates lysophosphatidic acid-mediated migration of pancreatic cancer cells. Pancreas 42, 819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhai J., Lin H., Nie Z., Wu J., Cañete-Soler R., Schlaepfer W. W., Schlaepfer D. D. (2003) Direct interaction of focal adhesion kinase with p190RhoGEF. J. Biol. Chem. 278, 24865–24873 [DOI] [PubMed] [Google Scholar]