

Background: Cancer Osaka thyroid (COT) kinase plays a crucial role in inflammatory diseases and cancer.
Results: Production of catalytically competent COT kinase yielded protein suitable for structure guided drug discovery.
Conclusion: COT kinase has a unique and structurally versatile active site.
Significance: The discovery of a novel variation of the protein kinase fold will impact drug discovery for COT kinase.
Keywords: cancer, drug discovery, immunology, mitogen-activated protein kinase (MAPK), structural biology
Abstract
Macrophages are important cellular effectors in innate immune responses and play a major role in autoimmune diseases such as rheumatoid arthritis. Cancer Osaka thyroid (COT) kinase, also known as mitogen-activated protein kinase kinase kinase 8 (MAP3K8) and tumor progression locus 2 (Tpl-2), is a serine-threonine (ST) kinase and is a key regulator in the production of pro-inflammatory cytokines in macrophages. Due to its pivotal role in immune biology, COT kinase has been identified as an attractive target for pharmaceutical research that is directed at the discovery of orally available, selective, and potent inhibitors for the treatment of autoimmune disorders and cancer. The production of monomeric, recombinant COT kinase has proven to be very difficult, and issues with solubility and stability of the enzyme have hampered the discovery and optimization of potent and selective inhibitors. We developed a protocol for the production of recombinant human COT kinase that yields pure and highly active enzyme in sufficient yields for biochemical and structural studies. The quality of the enzyme allowed us to establish a robust in vitro phosphorylation assay for the efficient biochemical characterization of COT kinase inhibitors and to determine the x-ray co-crystal structures of the COT kinase domain in complex with two ATP-binding site inhibitors. The structures presented in this study reveal two distinct ligand binding modes and a unique kinase domain architecture that has not been observed previously. The structurally versatile active site significantly impacts the design of potent, low molecular weight COT kinase inhibitors.
Introduction
The activation of the MAPK pathways p38, JNK, and ERK plays a crucial role in inflammatory diseases and cancer. COT2 kinase is the single kinase that mediates the activation of the Mek/Erk pathway downstream of the pro-inflammatory receptors of the IL-1/TLR/TNF/IL-17 receptor superfamilies, whereas NF-κB, p38, and JNK are turned on by the MAP3K Tak1. In cells, COT kinase forms a heterotrimeric complex together with NF-κB transcription factor p105 and A20-binding inhibitor of NF-κB (ABIN-2). COT kinase is activated by IκB kinase β (IKKβ), which triggers proteasome-dependent degradation of p105 to p50 and disassembly of the ternary complex (1). Furthermore, IκB kinase β phosphorylates COT kinase at position Ser400, which mediates binding of 14-3-3 protein (2, 3). In addition, the full activation of COT kinase requires phosphorylation of Thr290 in the activation loop by an unknown kinase. Dissociation of activated COT kinase from p105 and ABIN-2 allows substrate loop phosphorylation of the downstream substrate MEK. A number of other downstream targets of COT kinase have been described, and recent data show that as yet uncharacterized substrates required for posttranslational TNF processing are activated while COT kinase is residing in the ternary p105·ABIN-2 complex (4).
COT kinase is widely expressed in the spleen, thymus, liver, brain, testis, intestine, kidney, skeletal muscles, lungs, and pancreas (5). COT kinase has a function in hematopoietic lineages, with the best described role in pro-inflammatory cytokine production in macrophages. Its catalytic activity is required for both TNFα and IL-1β production (6). Single cytokine neutralization of either TNFα or IL-1β by antibody is highly efficient in the clinic for multiple indications. The recently discovered role of COT kinase in IL-17 signaling (7, 8) further adds weight to its therapeutic importance in IL-17-driven diseases such as multiple sclerosis and psoriasis and may explain its function in stromal cells that has been found in inflammatory disease models (9). In cancer, COT kinase seems to have ambivalent roles by being able to act as a tumor suppressor (10) or as a driver of tumorigenesis (11). A COT kinase knock-out background has been shown to be beneficial in prostate cancer and breast cancer (12, 13). In addition, COT kinase was described as an important resistance gene being able to drive MEK-ERK activation by circumventing rapidly accelerated kinase as upstream MAP3K (14).
Although the discovery of COT inhibitors has been reported in the literature (15–22), difficulties in obtaining monodisperse protein and the lack of structural information have hampered the discovery of chemical compounds suitable for clinical studies. Careful selection of protein constructs and optimizations in the protein production procedures allowed us to obtain pure enzyme of exquisite biochemical activity. In this study, we additionally report on two new COT kinase inhibitors, 5-(5-(1H-indol-3-yl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3,4-oxadiazol-2-amine (2) and 5-(2-amino-5-(quinolin-3-yl)pyridin-3-yl)-1,3,4-oxadiazole-2(3H)-thione (3), that display nanomolar inhibitory activity of substrate phosphorylation in our biochemical assay. We also present the first atomic resolution x-ray crystal structures of the human COT kinase domain as complexes with the two inhibitors. These structures reveal that COT kinase adopts a unique and unexpected kinase domain fold. The P-loop (also known as glycine-rich loop) is preceded by a 15-amino acid insert, which turned out to be a central modulator of the size and chemical characteristics of the active site. In addition, the active site of COT kinase is structurally more flexible than previously anticipated. The results presented in this study provide a basis for enzyme production and the design of novel, potent, and selective COT kinase inhibitors.
Experimental Procedures
For protein production and crystallization, all chemicals, unless stated otherwise, were purchased from Sigma-Aldrich.
Protein Expression
The DNA sequence, containing codons optimized for insect cell expression, for full-length human COT kinase (UniProt P41279) was ordered from GeneArt and used as a template for creating truncated variants encompassing the kinase domain. Constructs coding for amino acids 66–395 and 30–404, having an N-terminal hexahistidine tag followed by a PreScission protease cleavage site, were designed and subcloned into a transfer plasmid containing a proximal polyhedrin promotor. Sf21 cells were co-transfected with pFlashbac virus DNA (Oxford Expression Technologies) for baculovirus generation according to the vendor's recommendation. Protein expression was carried out in 1-liter cell cultures of Sf21 cells with plaque-purified viruses. To improve the yields of overexpressed protein, compound 1 was added to the culture at a final concentration of 10 μm at 24 h after infection. Cells were harvested 72 h after infection, and the pellets were frozen and stored at −80 °C.
Protein Purification
The pellets of 50 g of cells containing overexpressed COT(aa66–395) or COT(aa30–404) were resuspended in 300 ml of lysis buffer (0.05 m Tris-HCl, pH 8, 0.003 m tris(2-carboxyethyl)phosphine (TCEP), 10% (v/v) glycerol, 0.4 m NaCl, 10 mm imidazole, 0.05% (v/v) Triton X-100, and Complete, EDTA-free (Roche Applied Science)). Compound 1 (dissolved in DMSO) was added to a final concentration of 20 μm followed by homogenization of the lysate. Cleared lysates were applied on a Ni-NTA column (Qiagen) equilibrated in buffer A (0.025 m Tris-HCl, pH 8, 0.003 m tris(2-carboxyethyl)phosphine, 10% (v/v) glycerol, 0.4 m NaCl, 0.01 m imidazole). The column was washed extensively with buffer A, followed by another wash step with modified buffer A containing 0.2 m NaCl. The protein was eluted by applying an imidazole gradient from 20 to 290 mm imidazole, and fractions containing the kinase were pooled. Exchange of compound 1 and cleavage of the His6 tag were carried out simultaneously overnight in the presence of 1 mm EDTA, PreScission protease and an excess of new ligand. The final purification step was carried out by size exclusion chromatography using a Superdex 75 HiLoad 16/60 column (GE Healthcare). Fractions containing the protein·ligand complex were pooled and concentrated to a final concentration of about 4 mg/ml.
Crystallization
Both COT·ligand complexes were concentrated to 3.5 mg/ml using an Amicon Ultra centrifugal filter with 10,000 molecular weight cut-off (Millipore) before crystallization by vapor diffusion. Hanging drops consisting of an equal ratio of complex to reservoir solution (1 μl) were set up and equilibrated against 500 μl of reservoir solution containing either 0.1 m HEPES, pH 7.2, 24% (w/v) PEG monomethyl ether 500 (PEGMME500), and 0.05 m MgCl2 (COT·compound 2) or 8% (w/v) PEG4000, 12% (v/v) ethylene glycol and 7% (v/v) isopropyl alcohol (COT·compound 3) in a 24-well VDX plate (Hampton Research) at room temperature. Crystals grew within 2 days and were flash-frozen in liquid nitrogen, either directly after harvest from the mother liquor (COT·compound 2) or after a quick dip into a reservoir solution containing 10% (v/v) glycerol (COT·compound 3).
Diffraction data were collected at 100 K at the Swiss Light Source beamline X10SA at 1.0 Å wavelength. Diffraction images were recorded on a MAR225 CCD detector (MAR Research) and processed and scaled with XDS and XSCALE (23), respectively (Table 1). The COT kinase·compound 2 structure was solved by molecular replacement. A BLAST search was carried out against the Protein Data Bank (PDB) using the human COT kinase amino acid sequence (UniProt P41279) including residues 66–395, and coordinates of the top 99 search results were downloaded. Sequence alignments of the downloaded hits with COT kinase residues 66–395 were created using FASTA (24). To create the search models for molecular replacement, the coordinate files were prepared with Chainsaw, which is part of the CCP4 software suite (25), using the corresponding sequence alignments obtained from FASTA. The models obtained from Chainsaw were aligned in PyMOL (26), and large, protruding loops were removed. Molecular replacement searches with each input model were carried out with Phaser (27) and returned 85 solutions in space group C21 with three copies in the asymmetric unit. Three solutions with the highest log likelihood gain were used as a starting point for initial model building. The COT kinase·compound 2 structure was built using COOT (28) and refined with Phenix (29) and AutoBuster (30) to Rwork = 17.7% and Rfree = 20.7% at a high resolution limit of 2.3 Å. 96.4% of all the residues in the structure are in the favored outlier regions of the Ramachandran plot, and 0.3% are in the outlier regions of the Ramachandran plot. The COT kinase·compound 3 structure was solved by molecular replacement with Phaser using protein coordinates of the structure with compound 2 as input model. The solution from molecular replacement (space group P31 with three copies in the asymmetric unit) was rebuilt in COOT and refined with Phenix and AutoBuster to Rwork = 20.1% and Rfree = 24.0% at a high resolution limit of 2.9 Å. 94.8% of all the residues in the structure are in the favored regions of the Ramachandran plot, and 1.5% are in the outlier regions of the Ramachandran plot. PyMOL (26) was used to prepare all structural figures.
TABLE 1.
Data collection and refinement statistics
| COT/compound 2 | COT/compound 3 | |
|---|---|---|
| Data collection | ||
| Space group | C2 | P3 |
| Unit cell | ||
| a, b, c (Å) | 147.978, 85.113, 85.964 | 101.952, 101.952, 108.315 |
| α, β, γ (°) | 90.0 91.06 90.0 | 90.0 90.0 120.0 |
| Resolution (Å) | 73.8-2.3 | 88.3-2.9 |
| Wavelength (Å) | 1.0000 | 1.0000 |
| Unique reflections | 45,307 | 28,213 |
| Completeness (%) | 99.0 (94.0)a | 100.0 (100.0)a |
| 〈I〉/σ(I) | 10.6 (3.3)a | 14.7 (3.7)a |
| Rsym | 0.10 (0.39)a | 0.09 (0.45)a |
| Refinement | ||
| Resolution (Å) | 34.1-2.3 | 88.3-2.9 |
| Reflections | 45,293 | 28,212 |
| Rwork/Rfree | 0.177/0.207 | 0.201/0.240 |
| No. of waters | 361 | 167 |
| No. of protein atoms | 6939 | 6124 |
| No. of ligand atoms | 452 | 268 |
| Wilson B (Å2) | 36.37 | 76.04 |
a Values in parentheses are for highest-resolution shells.
In Vitro Phosphorylation Assay with COT
All assays were performed in 384-well microtiter plates using automated liquid dispensing. The assay plates were prepared by the addition of 50 nl/well of compound solution in 90% (v/v) DMSO. The kinase reactions were started by the stepwise addition of 4.5 μl/well of peptide/ATP solution (50 mm HEPES, pH 7.5, 1 mm DTT, 0.02% (w/v) Tween 20, 0.02% (w/v) BSA, 10 mm β-glycerophosphate, 10 μm sodium orthovanadate, 14 mm MgCl2, 1 mm MnCl2, 306 μm ATP, 4 μm peptide (5-fluorescein-aminohexanoic acid-AGAGSGQLIDSNANSFVGTR-NH2, Biosyntan GmbH)) and 4.5 μl/well of enzyme solution (50 mm HEPES, pH 7.5, 1 mm DTT, 0.02% (w/v) Tween 20, 0.02% (w/v) BSA, 10 mm β-glycerophosphate, 10 μm sodium orthovanadate, 14 mm MgCl2, 1 mm MnCl2, and 17 nm COT kinase (encompassing amino acids 30–404)). Kinase reactions were incubated at 30 °C for 60 min and subsequently terminated by the addition of 16 μl/well of stop solution (100 mm HEPES, pH 7.5, 5% (v/v) DMSO, 0.1% (v/v) Caliper coating reagent, 10 mm EDTA, and 0.015% (w/v) Brij35). Plates with terminated kinase reactions were transferred to the Caliper LC3000 workstations for reading. Phosphorylated and unphosphorylated peptides were separated and quantified using the Caliper microfluidic mobility shift technology.
Chemical Synthesis of Compounds 1, 2, and 3
A detailed protocol for the syntheses of (E)-3-(2-amino-5-(naphthalen-2-yl)pyridin-3-yl)acrylic acid (1), 5-(5-(1H-indol-3-yl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3,4-oxadiazol-2-amine (2) and 5-(2-amino-5-(quinolin-3-yl)pyridin-3-yl)-1,3,4-oxadiazole-2(3H)-thione (3) (see Fig. 1) can be found in the supplemental information.
FIGURE 1.
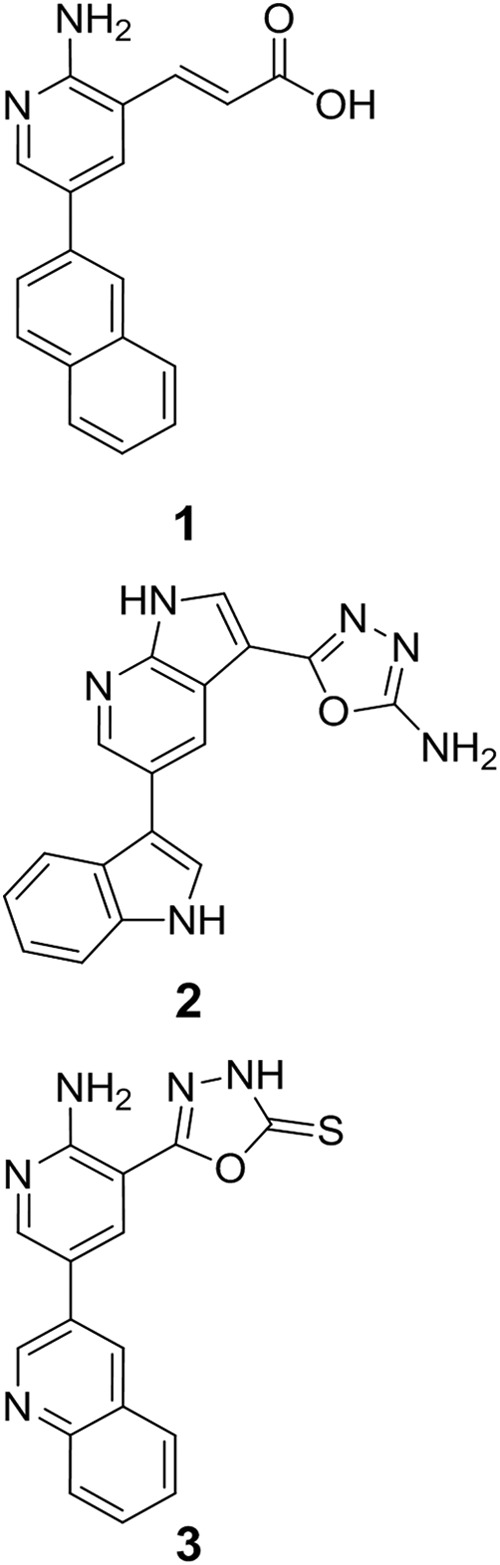
Chemical structures of the COT inhibitors (E)-3-(2-amino-5-(naphthalen-2-yl)pyridin-3-yl)acrylic acid (compound 1), 5-(5-(1H-indol-3-yl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3,4-oxadiazol-2-amine (compound 2) 5-(2-amino-5-(quinolin-3-yl)pyridin-3-yl)-1,3,4-oxadiazole-2(3H)-thione (compound 3).
Results
Recombinant Protein Expression and Purification
A series of DNA plasmids encoding hexahistidine-tagged human COT kinase domain with varying N and C termini was subjected to small scale expression tests in baculovirus-infected Sf21 insect cells. Two constructs, one encompassing amino acid residues 30–404 (aa30–404) and the other one encompassing amino acid residues 66–395 (aa66–395), were identified in a first round of expression experiments to yield soluble but heterogeneously phosphorylated protein. It has been reported previously that expression yields of kinases in baculovirus-infected insect cells can be improved by the addition of specific small molecular weight kinase inhibitors to the cell cultivation medium (31). For both constructs, the addition of the ATP-competitive inhibitor 1 during fermentation and initial purification was essential to increase the overall yield as well as to reduce heterogeneous phosphorylation. In the absence of compound 1, about 0.2 mg of 65% non-phosphorylated and soluble COT kinase (aa30–404) was isolated per liter of cell culture volume after a nickel affinity purification step. The addition of compound 1 greatly enhanced this yield to 4–6 mg of protein per liter of cell culture. Purified COT protein was found 90% non-phosphorylated after the addition of compound 1, suggesting that compound 1 blocks COT kinase autophosphorylation (Table 2). A second construct (aa66–395) was identified using a similar approach and yielded about 2 mg/liter of cell culture after the first purification step. It was possible to quantitatively exchange compound 1 by other ATP site binders by chromatography or to remove it completely to prepare apo-form enzyme (see “Experimental Procedures”).
TABLE 2.
Optimization of human COT kinase (aa30–404) expression in Sf21 insect cells
The addition of compound 1 during fermentation improved the yield and homogeneity of COT(30–404) obtained after the first purification step (Ni-NTA yield). Fermentation volume was 0.9 l. NA, not applicable.
| Compound 1 | Form of addition | Time of addition | Cultivation time [h] | Ni-NTA yield | Phosphorylation in % |
||
|---|---|---|---|---|---|---|---|
| 0× | 1× | 2× | |||||
| μm | mg | ||||||
| 0 | NA | NA | 64 | 0.2–0.3 | 65 | 25 | 10 |
| 30 | Diluted into medium, sterile filtered | At infection | 63 | 2.4 | 84 | 16 | 0 |
| 10 | DMSO stock diluted into culture | At infection | 64 | 4.2 | 89 | 11 | 0 |
| 10 | DMSO stock diluted into culture | 24 h after infection | 64 | 7 | 90 | 10 | 0 |
Kinase Assays
The use of radioactive filter binding and homogeneous time-resolved fluorescence assays to measure COT kinase activity has been described before (32). For this study, we developed an assay that measures in vitro phosphorylation of a peptide substrate. Enzyme reactions were carried out in 384-well plates. We used the Caliper microfluidic mobility shift technology to quantitatively separate phosphorylated and non-phosphorylated substrate peptides (supplemental Fig. 1). With this experimental setup, the half-maximal inhibitory concentrations (IC50 values) of a large variety of compounds were measured at compound concentrations of up to 10 μm. Chemical derivation of a hit obtained from high throughput screening resulted in the discovery of the two COT inhibitors 2 and 3 discussed in this study (Fig. 1). Both compounds show low nanomolar inhibitory activity of COT kinase in the enzymatic Caliper assay with IC50 values of 13 nm for compound 2 and 17 nm for compound 3.
Overall Structure
Both protein variants mentioned above were subjected to extensive crystallization experiments, and COT kinase (aa66–396) yielded well diffracting crystals. We determined the x-ray co-crystal structures of the human COT kinase domain in complex with compound 2 at 2.3 Å resolution. The crystal structure of this complex has a refined model that includes residues 73–93, 103–139, and 143–392 (Table 1). Another co-crystal of the same human COT kinase domain was solved in complex with compound 3 at a resolution of 2.9 Å. The model of this co-crystal structure contains residues 73–92, 102–331, and 343–389 (Table 1).
Both structures revealed a bilobed kinase domain fold (Fig. 2) with an N-terminal extension that is similar to the one observed in the crystal structure of nuclear factor κB-inducing kinase (PDB entry 4DN5) (33). The N-terminal extension consists of an α-helix (αN2) and a short, parallel β-sheet (βN1 and βN2). Helix αN2 stacks against the central β-sheet and helix αC. βN1 and βN2 pack against and extend the central β-sheet of the N-lobe (Fig. 2A). The loop connecting the N-terminal extension with the N-lobe is disordered in both crystal structures as judged by the absence of continuous electron density for this part of the structure.
FIGURE 2.

Overview of the COT kinase structures. A, the overall structure of the COT·compound 2 structure is shown as graphic representation (Cα trace of the protein, blue). Compound 2 (orange, stick representation) binds to the active site of the COT kinase domain. N-extension, αC-helix, P-loop insert, and the activation segment are highlighted in yellow, cyan, magenta, and red, respectively. B, structure alignment of the COT·compound 2 (blue) with the COT·compound 3 (green) crystal structure. The Cα atoms of both structures were superimposed to generate this alignment (root mean square deviation = 0.454 Å2) Considerable conformational differences occur in the N-lobe of the COT kinase domain, in particular in the P-loop end P-loop insert segments.
The P-loop is a stretch of amino acids that covers the ATP-binding site of kinases and stabilizes the positions of the α and β phosphates of ATP bound to the active site of a kinase. The P-loop contains a conserved glycine-rich sequence termed the GXXGXG motif. Unexpectedly, the GXXGXG motif of the P-loop is preceded by a stretch of 15 amino acids (residues 132–146) that partially covers the active site cleft (Fig. 2). The structures reveal two different conformations of the P-loop and its insert. In the complex with compound 2, truncated electron density for amino acid residues 140–142 indicates that the insert forms a mobile loop. In the COT·compound 3 complex, the P-loop insert forms a short β-hairpin (Fig. 2B). A short stretch of amino acids, Met207 to Gly212, connects the N- and the C-terminal lobes of the kinase and is termed the hinge. The gatekeeper methionine (Met207) side chain, which separates the active site from a hydrophobic pocket in the back, has an extended conformation in both structures. A conserved sequence of Asp-Phe-Gly (DFG motif) has been identified to be involved in the correct positioning of the ATP γ-phosphate in kinases during substrate phosphorylation. The conformation of DFG motif in the C-terminal part of the COT kinase active site is reminiscent of an active conformation of the kinase (DFG-in conformation) in both structures (34).
COT Kinase·Compound 2 Protein-Ligand Interactions
Compound 2 binds to the active site, between the N- and C-terminal lobes, of the COT kinase domain. The P-loop and its insert are collapsed onto the ATP-binding pocket and the bound ligand, thereby shielding compound 2 from the bulk solvent (Fig. 2A). The conformation of the P-loop is further stabilized by an H-bond interaction between the amide NH of Ala148 and the side chain of Asp270 of the DFG motif at the N-terminal end of the activation segment.
The pyrrolopyridine core of compound 2 forms two direct H-bonds with the hinge, whereas Glu208 main chain carboxyl oxygen acts as an H-bond acceptor and Gly210 main chain amide NH acts as a donor (Fig. 3A). The extended side chain of gatekeeper Met207 separates the ATP site from the backpocket of the kinase domain and contacts the pyrrolopyridine core via van der Waals interactions. The indole group of compound 2, which is oriented toward the active site rim, is nested in a small pocket composed of side chain residues of the P-loop insert and the C-terminal lobe. It forms π-π interactions with Trp132 and van der Waals interactions with side chains of Leu134, Phe143, and Pro145 (P-loop insert) and with Gly213, Ser214, and Glu217 (C-terminal lobe). The oxadiazole group of compound 2 engages in direct as well as water-mediated H-bonds with the protein. Direct interactions are built with the side chain of Asp270 (DFG motif) and with the terminal amine of the catalytic lysine (Lys167), which in turn interacts with Asp178 of the C-helix. Water-mediated H-bonds are built with the side chain of Gln182 in the C-helix and with the main chain carbonyl oxygen of Ser257 in the C-terminal lobe.
FIGURE 3.

Details of the active site pocket of COT kinase. A, compound 2 (sticks representation; carbon in light blue) binds into the active site of COT (Cα carbon atoms in brown). The blue surface represents the shape of the binding pocket including protein residues within 5 Å around the ligand. B, binding of compound 3 to the active site of COT. (Cα carbon trace in gray). The orange surface represents the shape of the binding pocket including protein residues within 5 Å around the ligand. Nitrogen, oxygen, and sulfur atoms are highlighted in dark blue, red, and yellow, respectively. Polar interactions between ligand and/or protein atoms within 3.2 Å distance are marked with dashed green lines. Water molecules are represented as red spheres.
COT Kinase·Compound 3 Protein-Ligand Interactions
Compound 3 binds to the active site of COT and its amino-pyridine core group forms two H-bonds with the main chain of Glu208 and Gly210 in the hinge (Fig. 3B). The thiocarbonyl group at the oxadiazole interacts with the catalytic Lys167 side chain amine. The quinoline group of compound 3 binds into a small pocket formed by residues of the P-loop insert (Trp132, Leu134, and Ile144) and the C-terminal lobe (Gly213, Ser214, and Glu217). The P-loop undergoes an unprecedented frameshift of the IPRG motif so that Arg146 assumes the position of Glu145 and its side chain points down toward the active site, rather than up. The frameshift is accompanied by a partial disordering of the P-loop β sheet and is likely driven by the formation of a salt bridge between the Arg146 side chain and the acidic oxadizolthione moiety of 3 (Fig. 3B). Consequently, Pro145 is flipped out, and the side chain of Ile144 is oriented such that it stacks onto the quinoline of compound 3. The P-loop insert opens up and adopts a β-hairpin structure (Fig. 4). At the back of the active site, the side chain of gatekeeper Met207 adopts an extended conformation and interacts via van der Waals contacts with the oxadiazole group of compound 3. The side chain of Asp270 (DFG motif) is rotated away and does not interact with either Lys167 or compound 3. The salt bridge between Lys167 and Asp178 in the C-helix is broken, as the side chain of Asp178 is rotated away from Lys164 (Fig. 3, A and B).
FIGURE 4.
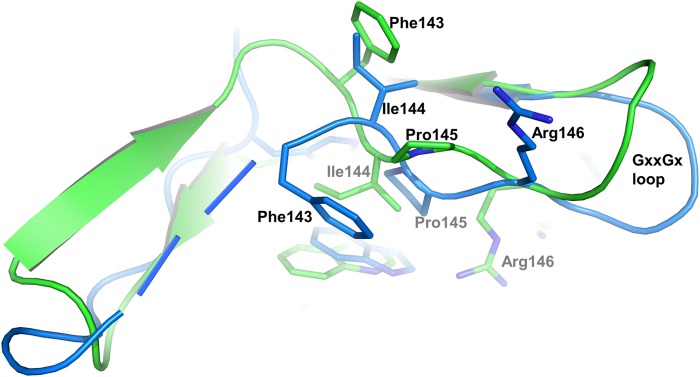
Details of the P-loop conformational differences. Differences in the P-loop and P-loop insert are depicted as an overlay of Cα atoms of COT·compound 2 and COT·compound 3 in graphic representation. Structure alignment and color coding from Fig. 2B are preserved.
Discussion
Although the overall folds are similar in both COT kinase co-crystal structures, the conformations of the N-terminal lobe, and in particular of the P-loop and its insert, differ considerably in the two structures (Fig. 2B). In contrast, the C-lobes in both structures appear to be more rigid and adopt an active conformation (35). The activation segment encompassing residues Asp270 to Glu297 is in an extended conformation (Figs. 2B and 5), similar to the structure of cAMP-dependent protein kinase bound to a peptide inhibitor (PDB ID 1ATP) (36). The DFG phenylalanine (Phe271) side chain points toward the back of the active site and intercalates between Ile181 and Gln182 of helix αC. When compared with the canonical kinase fold, helix αC of COT is shortened by two residues with only eight amino acids between Asp178 and the HEN hairpin motif (His187-Glu188-Asn189). This deletion is reflected in a 310 helix around Asp178. The highly conserved catalytic Glu residue is replaced by Asp178 in COT kinase, a replacement that is only observed in MEK4 (PDB entry 3ALN) (37), MEK6 (PDB entry 3ENM) (37), and MEK7 (PDB entry 2DYL). The relatively short helix αC, on the other hand, is further fixed by a zipper-like motif of interlocked hydrophobic amino acids, involving Val179 of helix αC and residues of the central β-sheet (Leu193 and Leu205) and helix αN2 (Leu84 and Ala88). The activation segment is further stabilized in its observed conformation by the bifurcated H-bond interaction of Thr290 with Asp253 and Lys255 of the catalytic loop. This structural motif, also termed C-terminal anchor of the activation segment, is commonly found in active state serine-threonine kinase structures and is crucial for the correct positioning of the substrate with respect to the ATP γ-phosphate (34). Together with Ser400, Thr290 has been identified as a phosphorylation site required for COT kinase activation (38, 39). Phosphorylation of Thr290 introduces a negative charge that must lead to an electrostatic repulsion from Asp178 in the catalytic loop and therefore destabilize the C-terminal anchor of the activation segment. Given the structural diversity observed in several other kinase structures, it is likely that a destabilization of one of the anchor points in the activation segment would lead to a change of its conformation (34). Such a change in structure may in turn facilitate further structural rearrangements in the COT kinase·ABIN-2·p105 complex that, along with IκB kinase β-dependent phosphorylation of COT kinase Ser400 and subsequent partial degradation of p105, eventually would lead to macrophage activation (3).
FIGURE 5.
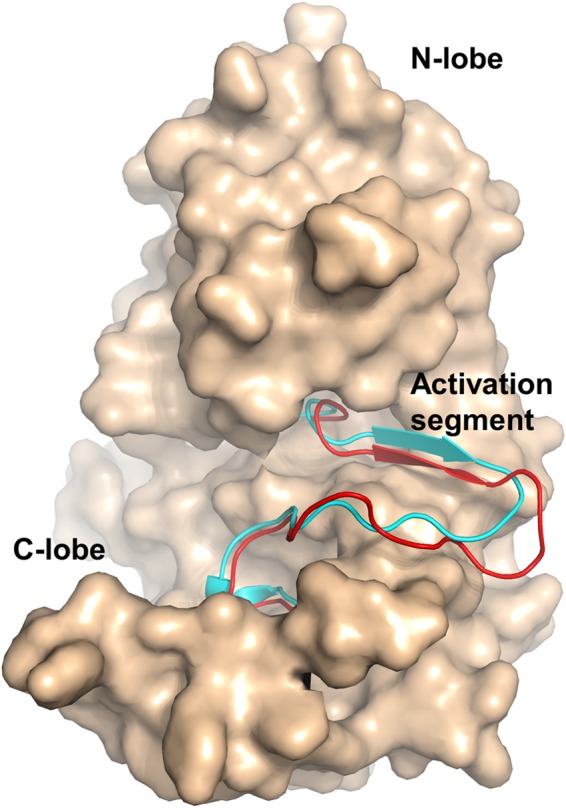
Comparison of the activation segments of COT·compound 2 and cAMP-dependent PKA. Coordinates of the Cα atoms of COT·compound 2 and cAMP-dependent protein kinase in the active state (PDB ID 1ATP) were superimposed. The structure of COT is shown as surface representation with the activation segment (amino acids 270–297) highlighted in red (graphic representation). The superimposed activation segment of PKA is highlighted in cyan (graphic representation).
The discovery of small molecule inhibitors in high throughput screens using COT kinase has been described (15–22). Although some progress in the advancement of these molecules has been made, the low sequence homology of COT kinase to other serine-threonine (ST) kinases and the lack of structural information have hampered rapid progress in the understanding of the structure-activity relationship of the reported compounds. Certainly, the unexpected presence of the 15-amino acid-long P-loop insert revealed by the two crystal structures discussed here, which appears to be rather flexible, prevented the creation of accurate homology models in the past. The crystal structures also revealed a P-loop that is partially collapsed onto the active site cleft, leaving the ligands deeply buried between the N- and C-lobes. Despite their similar structures and binding modes, ligands 2 and 3 induce large conformational shifts of the P-loop that demonstrate conformational flexibility bestowed by the P-loop insert (Fig. 4). The inherent flexibility of COT kinase is also reflected in the low melting point (Tm) of our constructs (Tm ∼310 K) in the absence of compound in the active site. The addition of an ATP-competitive compound increased Tm by about 10 K. We therefore conclude that the flexibility of COT kinase leads to a strong induced fit component to binding, which is one explanation for the steep structure-activity relationship that we encountered in our medicinal chemistry program. The active sites differ not only in their shapes, but also in their electronic properties. Although both compounds form similar H-bond interactions with the backbone of Glu208 and Gly210 in the hinge of the kinase, the different cores (pyrrolopyridine versus aminopyridine) lead to subtle differences in the overall shapes of the bound compounds (Figs. 1 and 3). These small shape differences lead to a substantial conformational change in the N-terminal lobe and to a remodeling of the active site (Figs. 3 and 4). The distance from the quinolone to the thiocarbonyl sulfur in compound 3 is slightly larger than the distance from the indole to the oxadiazole NH2 group in compound 2. This leaves enough space for Arg146 side chain to fit between the thione and the quinoline substituents of compound 3 (Fig. 3B) and allows for a conformational change of the P-loop and its insert. Consequently, Pro145 is flipped out, and the side chain of Ile144 is oriented such that it stacks onto the quinoline of compound 3. The P-loop insert adopts a β-hairpin structure, and the downward position of Arg146 causes the GXXGX loop to open up (Figs. 2B and 4).
The two crystal structures of human COT kinase in complex with ATP-competitive inhibitors described in this study revealed the unique architecture of this kinase, which offers opportunities to improve the potency and selectivity of ATP-competitive inhibitors. Small changes in the chemical structure of an inhibitor can have a large effect on the conformation of the active site, which partially accounts for the steep structure-activity relationship of COT kinase inhibitors (15–22, 40) and reduces the predictability of structure-guided medicinal chemistry approaches, such as scaffold morphing and fragment growing. Although the presented co-crystal structures of COT kinase inhibitor complexes provided a first glimpse on the binding mode of early hits, the high throughput in vitro phosphorylation assay we report above became an invaluable tool to rapidly test newly synthesized derivatives for potency and selectivity. Furthermore, we propose that this assay could also be used to validate experimental procedures that include COT kinase variants from species other than human. An amino acid sequence alignment of human, mouse, and rat COT kinase shows differences in the amino acid compositions of the P-loop and its insert (Fig. 6). As stated above, this part of the COT kinase is susceptible to conformational changes that also depend on the chemical characteristics of the ligand. It is therefore very well conceivable that a potent inhibitor of the human variant may lose potency on the rat or mouse variant and vice versa, a consideration that needs to be taken into account when comparing results from assays that use variants from different species.
FIGURE 6.

Sequence comparison of human, mouse, and rat COT kinase domain. An alignment of the sequences of COT kinase from mouse, rat, and human, encompassing amino acids 66–395, is shown. The green box highlights residues that belong to the P-loop and its insert (position 132–152). Numbering is based on the human sequence (UniProt P41279). Positions with fully conserved residues are marked with an asterisk. Conservation of between groups of strongly similar and weakly similar properties are indicated with a colon or a period, respectively.
In this study, we focused on ATP-competitive inhibitors that bind to the active site of COT kinase. However, it should be noted that drug discovery approaches, which target the ternary COT kinase·ABIN-2·p105 complex or aim at an allosteric modulation of the kinase activity, may also represent attractive strategies for the discovery of selective inhibitors of the COT kinase dependent axis of the MEK/ERK pathway.
Supplementary Material
Acknowledgments
We thank the MX group of the Swiss Light Source at the Paul Scherrer Institute in Villigen, Switzerland for their continuous and outstanding support. We also thank M. Geiser for subcloning the DNA constructs used in this study.
Authors are employees and/or own shares of Novartis Pharma AG.

This article contains supplemental text and Fig. S1.
The atomic coordinates and structure factors (codes 4Y83 and 4Y85) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- COT
- Cancer Osaka thyroid
- Ni-NTA
- nickel-nitrilotriacetic acid
- aa
- amino acids
- DMSO
- dimethyl sulfoxide.
References
- 1. Beinke S., Robinson M. J., Hugunin M., Ley S. C. (2004) Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IκB kinase-induced proteolysis of NF-κB1 p105. Mol. Cell. Biol. 24, 9658–9667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ben-Addi A., Mambole-Dema A., Brender C., Martin S. R., Janzen J., Kjaer S., Smerdon S. J., Ley S. C. (2014) IκB kinase-induced interaction of TPL-2 kinase with 14-3-3 is essential for Toll-like receptor activation of ERK-1 and -2 MAP kinases. Proc. Natl. Acad. Sci. U.S.A. 111, E2394–E2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roget K., Ben-Addi A., Mambole-Dema A., Gantke T., Yang H.-T., Janzen J., Morrice N., Abbott D., Ley S. C. (2012) IκB kinase 2 regulates TPL-2 activation of extracellular signal-regulated kinases 1 and 2 by direct phosphorylation of TPL-2 serine 400. Mol. Cell. Biol. 32, 4684–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang H. T., Papoutsopoulou S., Belich M., Brender C., Janzen J., Gantke T., Handley M., Ley S. C. (2012) Coordinate regulation of TPL-2 and NF-κB signaling in macrophages by NF-κB1 p105. Mol. Cell. Biol. 32, 3438–3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patriotis C., Makris A., Bear S. E., Tsichlis P. N. (1993) Tumor progression locus 2 (Tpl-2) encodes a protein kinase involved in the progression of rodent T-cell lymphomas and in T-cell activation. Proc. Natl. Acad. Sci. U.S.A. 90, 2251–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dumitru C. D., Ceci J. D., Tsatsanis C., Kontoyiannis D., Stamatakis K., Lin J. H., Patriotis C., Jenkins N. A., Copeland N. G., Kollias G., Tsichlis P. N. (2000) TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 7. Xiao Y., Jin J., Chang M., Nakaya M., Hu H., Zou Q., Zhou X., Brittain G. C., Cheng X., Sun S.-C. (2014) TPL2 mediates autoimmune inflammation through activation of the TAK1 axis of IL-17 signaling. J. Exp. Med. 211, 1689–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sriskantharajah S., Gückel E., Tsakiri N., Kierdorf K., Brender C., Ben-Addi A., Veldhoen M., Tsichlis P. N., Stockinger B., O'Garra A., Prinz M., Kollias G., Ley S. C. (2014) Regulation of experimental autoimmune encephalomyelitis by TPL-2 kinase. J. Immunol. 192, 3518–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Acker G. J., Perides G., Weiss E. R., Das S., Tsichlis P. N., Steer M. L. (2007) Tumor progression locus-2 is a critical regulator of pancreatic and lung inflammation during acute pancreatitis. J. Biol. Chem. 282, 22140–22149 [DOI] [PubMed] [Google Scholar]
- 10. Gkirtzimanaki K., Gkouskou K. K., Oleksiewicz U., Nikolaidis G., Vyrla D., Liontos M., Pelekanou V., Kanellis D. C., Evangelou K., Stathopoulos E. N., Field J. K., Tsichlis P. N., Gorgoulis V., Liloglou T., Eliopoulos A. G. (2013) TPL2 kinase is a suppressor of lung carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 110, E1470–E1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vougioukalaki M., Kanellis D. C., Gkouskou K., Eliopoulos A. G. (2011) Tpl2 kinase signal transduction in inflammation and cancer. Cancer Lett. 304, 80–89 [DOI] [PubMed] [Google Scholar]
- 12. Kim J. Y., Lim S.-C., Kim G., Yun H. J., Ahn S.-G., Choi H. S. (2014) Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene 10.1038/onc.2014.418 [DOI] [PubMed] [Google Scholar]
- 13. Lee H. W., Cho H. J., Lee S. J., Song H. J., Cho H. J., Park M. C., Seol H. J., Lee J.-I., Kim S., Lee H. M., Choi H. Y., Nam D.-H., Joo K. M. (2015) Tpl2 induces castration resistant prostate cancer progression and metastasis. Int. J. Cancer 136, 2065–2077 [DOI] [PubMed] [Google Scholar]
- 14. Johannessen C. M., Boehm J. S., Kim S. Y., Thomas S. R., Wardwell L., Johnson L. A., Emery C. M., Stransky N., Cogdill A. P., Barretina J., Caponigro G., Hieronymus H., Murray R. R., Salehi-Ashtiani K., Hill D. E., Vidal M., Zhao J. J., Yang X., Alkan O., Kim S., Harris J. L., Wilson C. J., Myer V. E., Finan P. M., Root D. E., Roberts T. M., Golub T., Flaherty K. T., Dummer R., Weber B. L., Sellers W. R., Schlegel R., Wargo J. A., Hahn W. C., Garraway L. A. (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Y., Cole D., Denny R. A., Anderson D. R., Ipek M., Ni Y., Wang X., Thaisrivongs S., Chamberlain T., Hall J. P., Liu J., Luong M., Lin L. L., Telliez J. B., Gopalsamy A. (2011) Discovery of indazoles as inhibitors of Tpl2 kinase. Bioorg. Med. Chem. Lett. 21, 4758–4761 [DOI] [PubMed] [Google Scholar]
- 16. Kaila N., Green N., Li H. Q., Hu Y., Janz K., Gavrin L. K., Thomason J., Tam S., Powell D., Cuozzo J., Hall J. P., Telliez J. B., Hsu S., Nickerson-Nutter C., Wang Q., Lin L. L. (2007) Identification of a novel class of selective Tpl2 kinase inhibitors: 4-alkylamino-[1,7]naphthyridine-3-carbonitriles. Bioorg. Med. Chem. 15, 6425–6442 [DOI] [PubMed] [Google Scholar]
- 17. Hu Y., Green N., Gavrin L. K., Janz K., Kaila N., Li H. Q., Thomason J. R., Cuozzo J. W., Hall J. P., Hsu S., Nickerson-Nutter C., Telliez J. B., Lin L. L., Tam S. (2006) Inhibition of Tpl2 kinase and TNF-α production with quinoline-3-carbonitriles for the treatment of rheumatoid arthritis. Bioorg. Med. Chem. Lett. 16, 6067–6072 [DOI] [PubMed] [Google Scholar]
- 18. Gavrin L. K., Green N., Hu Y., Janz K., Kaila N., Li H. Q., Tam S. Y., Thomason J. R., Gopalsamy A., Ciszewski G., Cuozzo J. W., Hall J. P., Hsu S., Telliez J. B., Lin L. L. (2005) Inhibition of Tpl2 kinase and TNF-α production with 1,7-naphthyridine-3-carbonitriles: synthesis and structure-activity relationships. Bioorg. Med. Chem. Lett. 15, 5288–5292 [DOI] [PubMed] [Google Scholar]
- 19. Wu J., Green N., Hotchandani R., Hu Y., Condon J., Huang A., Kaila N., Li H.-Q., Guler S., Li W., Tam S. Y., Wang Q., Pelker J., Marusic S., Hsu S., Perry Hall J., Telliez J.-B., Cui J., Lin L.-L. (2009) Selective inhibitors of tumor progression loci-2 (Tpl2) kinase with potent inhibition of TNF-α production in human whole blood. Bioorg. Med. Chem. Lett. 19, 3485–3488 [DOI] [PubMed] [Google Scholar]
- 20. Green N., Hu Y., Janz K., Li H. Q., Kaila N., Guler S., Thomason J., Joseph-McCarthy D., Tam S. Y., Hotchandani R., Wu J., Huang A., Wang Q., Leung L., Pelker J., Marusic S., Hsu S., Telliez J. B., Hall J. P., Cuozzo J. W., Lin L. L. (2007) Inhibitors of tumor progression loci-2 (Tpl2) kinase and tumor necrosis factor α (TNF-α) production: selectivity and in vivo antiinflammatory activity of novel 8-substituted-4-anilino-6-aminoquinoline-3-carbonitriles. J. Med. Chem. 50, 4728–4745 [DOI] [PubMed] [Google Scholar]
- 21. George D., Friedman M., Allen H., Argiriadi M., Barberis C., Bischoff A., Clabbers A., Cusack K., Dixon R., Fix-Stenzel S., Gordon T., Janssen B., Jia Y., Moskey M., Quinn C., Salmeron J. A., Wishart N., Woller K., Yu Z. (2008) Discovery of thieno[2,3-c]pyridines as potent COT inhibitors. Bioorg. Med. Chem. Lett. 18, 4952–4955 [DOI] [PubMed] [Google Scholar]
- 22. Cusack K., Allen H., Bischoff A., Clabbers A., Dixon R., Fix-Stenzel S., Friedman M., Gaumont Y., George D., Gordon T., Grongsaard P., Janssen B., Jia Y., Moskey M., Quinn C., Salmeron A., Thomas C., Wallace G., Wishart N., Yu Z. (2009) Identification of a selective thieno[2,3-c]pyridine inhibitor of COT kinase and TNF-α production. Bioorg. Med. Chem. Lett. 19, 1722–1725 [DOI] [PubMed] [Google Scholar]
- 23. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132, 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pearson W. R., Lipman D. J. (1988) Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A. 85, 2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bricogne G., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P, Sharff A., Smart O.S., Vonrhein C., Womack T.O., B. E. (2011) Buster, Version 1.11.4, Global Phasing Ltd., Cambridge, United Kingdom [Google Scholar]
- 31. Strauss A., Fendrich G., Horisberger M. A., Liebetanz J., Meyhack B., Schlaeppi J. M., Schmitz R. (2007) Improved expression of kinases in Baculovirus-infected insect cells upon addition of specific kinase inhibitors to the culture helpful for structural studies. Protein Expr. Purif. 56, 167–176 [DOI] [PubMed] [Google Scholar]
- 32. Jia Y., Quinn C. M., Bump N. J., Clark K. M., Clabbers A., Hardman J., Gagnon A., Kamens J., Tomlinson M. J., Wishart N., Allen H. (2005) Purification and kinetic characterization of recombinant human mitogen-activated protein kinase kinase kinase COT and the complexes with its cellular partner NF-κ B1 p105. Arch. Biochem. Biophys. 441, 64–74 [DOI] [PubMed] [Google Scholar]
- 33. Liu J., Sudom A., Min X., Cao Z., Gao X., Ayres M., Lee F., Cao P., Johnstone S., Plotnikova O., Walker N., Chen G., Wang Z. (2012) Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 287, 27326–27334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nolen B., Taylor S., Ghosh G. (2004) Regulation of protein kinases: controlling activity through activation segment conformation. Mol Cell. 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 35. Zheng J., Trafny E. A., Knighton D. R., Xuong N. H., Taylor S. S., Ten Eyck L. F., Sowadski J. M. (1993) 2.2-˚ refined crystal structure of the catalytic subunit of cAMP-dependent protein-kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. D Biol. Crystallogr. 49, 362–365 [DOI] [PubMed] [Google Scholar]
- 36. Matsumoto T., Kinoshita T., Kirii Y., Yokota K., Hamada K., Tada T. (2010) Crystal structures of MKK4 kinase domain reveal that substrate peptide binds to an allosteric site and induces an auto-inhibition state. Biochem. Biophys. Res. Commun. 400, 369–373 [DOI] [PubMed] [Google Scholar]
- 37. Min X., Akella R., He H., Humphreys J. M., Tsutakawa S. E., Lee S. J., Tainer J. A., Cobb M. H., Goldsmith E. J. (2009) The structure of the MAP2K MEK6 reveals an autoinhibitory dimer. Structure 17, 96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cho J., Tsichlis P. N. (2005) Phosphorylation at Thr-290 regulates Tpl2 binding to NF-κB1/p105 and Tpl2 activation and degradation by lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 102, 2350–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson M. J., Beinke S., Kouroumalis A., Tsichlis P. N., Ley S. C. (2007) Phosphorylation of TPL-2 on serine 400 is essential for lipopolysaccharide activation of extracellular signal-regulated kinase in macrophages. Mol. Cell. Biol. 27, 7355–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. George D., Salmeron A. (2009) Cot/Tpl-2 protein kinase as a target for the treatment of inflammatory disease. Curr. Top. Med Chem. 9, 611–622 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.