

Background: Dyrk1A regulates alternative splicing of exon 10 and phosphorylation of Tau.
Results: Calpain I proteolyzes Dyrk1A and enhances its kinase activity, which promotes exon 10 exclusion and hyperphosphorylation of Tau.
Conclusion: Truncation and activation of Dyrk1A may be responsible for Tau pathology in AD brains.
Significance: These findings indicate a new mechanism linked to Tau pathology in AD.
Keywords: calpain I, Dyrk1A truncation, neurofibrillary pathology, Tau alternative splicing, Tau phosphorylation
Abstract
Hyperphosphorylation and dysregulation of exon 10 splicing of Tau are pivotally involved in pathogenesis of Alzheimer disease (AD) and/or other tauopathies. Alternative splicing of Tau exon 10, which encodes the second microtubule-binding repeat, generates Tau isoforms containing three and four microtubule-binding repeats, termed 3R-Taus and 4R-Taus, respectively. Dual specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A) lies at the Down syndrome critical region of chromosome 21. Overexpression of this kinase may contribute to the early Tau pathology in Down syndrome via phosphorylation of Tau and dysregulation of Tau exon 10. Here, we report that Dyrk1A was truncated at the C terminus and was associated with overactivation of calpain I in AD brain. Calpain I proteolyzed Dyrk1A in vitro first at the C terminus and further at the N terminus and enhanced its kinase activity toward Tau via increased Vmax but not Km. C-terminal truncation of Dyrk1A resulted in stronger activity than its full-length protein in promotion of exon 10 exclusion and phosphorylation of Tau. Dyrk1A was truncated in kainic acid-induced excitotoxic mouse brains and coincided with an increase in 3R-Tau expression and phosphorylation of Tau via calpain activation. Moreover, truncation of Dyrk1A was correlated with an increase in the ratio of 3R-Tau/4R-Tau and Tau hyperphosphorylation in AD brain. Collectively, these findings suggest that truncation/activation of Dyrk1A by Ca2+/calpain I might contribute to Tau pathology via promotion of exon 10 exclusion and hyperphosphorylation of Tau in AD brain.
Introduction
Alzheimer disease (AD)3 is a chronic progressive neurodegenerative disorder characterized by extracellular deposits of β-amyloid as plaques (1), intracellular neurofibrillary tangles (NFTs) consisting of abnormally hyperphosphorylated aggregates of the microtubule-associated protein Tau (2–4), and selective neuronal loss. Tau is a cytosolic phosphoprotein, the major function of which is to stimulate microtubule assembly from tubulin subunits and stabilize the microtubule structure. Normally, Tau contains 2–3 phosphate groups/molecule. In AD brain, Tau is abnormally hyperphosphorylated with 9–10 mol of phosphate/mol of the protein (5, 6). Tau phosphorylation affects its axonal transport and degradation (7). Abnormal hyperphosphorylation of Tau is believed to be responsible for its loss of biological activity, for its gain of toxic activity, and for its aggregation into paired helical filaments (8–16). The correlation between the number of NFTs in brain and the severity of dementia symptoms in the affected individuals (17–19) indicates the pivotal role of Tau pathology in AD and other related neurodegenerative disorders called tauopathies.
Adult human brain expresses six isoforms of Tau from a single gene by alternative splicing of its pre-mRNA (20–22). Alternative splicing of Tau exon 10, which encodes the second microtubule-binding repeat, generates Tau isoforms into two groups, 3R-Taus and 4R-Taus, that contain three and four microtubule-binding repeats, respectively (23). 3R-Taus and 4R-Taus are different in their interactions with Tau kinases and in their biological function in polymerization and stabilization of neuronal microtubules (11, 24–26). In adult human brain, 3R-Taus and 4R-Taus are expressed at similar levels (21, 27). Several specific mutations of the tau gene associated with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) cause dysregulation of Tau exon 10 splicing and lead to a selective increase in either 3R- or 4R-Taus. Therefore, equal levels of 3R-Taus and 4R-Taus may be critical for maintaining optimal neuronal physiology in adult human brain. Dysregulation of Tau exon 10 splicing is sufficient to cause neurofibrillary degeneration (28). However, the exact mechanisms that lead to Tau pathology in AD brain are still unknown.
Down syndrome (DS) is caused by a partial or a complete trisomy of chromosome 21. Individuals with DS develop Alzheimer-type Tau pathology in the 4th decade of life (29). Dyrk1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) lies at the DS critical region of chromosome 21 and encodes a proline/arginine-directed serine/threonine protein kinase (30). Dyrk1A phosphorylates Tau at many sites in vitro and in cultured cells with the most favored at Thr212 (31, 32). Phosphorylation of Tau by Dyrk1A inhibits its function to stimulate microtubule assembly, promotes its self-aggregation into filaments, and primes it for further phosphorylation by glycogen synthase kinase-3β (GSK-3β) (32). Moreover, Dyrk1A phosphorylates splicing factors, including SC35 and ASF/SF2 (alternative splicing factor/splicing factor 2) and suppresses their function in the promotion of Tau exon 10 inclusion (33, 34). Overexpression of Dyrk1A in DS brain due to an extra copy of the gene may contribute to the early Tau pathology via dysregulation of both Tau exon 10 splicing and Tau phosphorylation (32, 33, 35). Several studies have shown that Dyrk1A may be involved in AD pathogenesis (36–39). However, the exact mechanisms by which Dyrk1A promotes Tau pathology remain elusive.
Calpains are a family of calcium-activated cysteine proteases that catalyze limited proteolysis of a variety of cellular proteins in eukaryotes (40). Calpain I, the major calpain isoform in neurons, is present principally as an inactive precursor in the cells. It is activated by autoproteolytic cleavage of the N terminus stimulated by a low micromolar concentration of calcium (μ-calpain). Altered calcium homeostasis, truncation, and activation of calpain I have been reported in brains with early stage AD (41–45). Hence, it is well known that calpain proteolyzes many substrates, including Cdk5 (cyclin-dependent protein kinase 5), regulatory subunit II of protein kinase A (PKA-RII), calcineurin, and Tau, and alters their activities (45–48), indicating that overactivation of calpain I in AD brain may be involved in the pathogenesis of this disease.
In the present study, we found that the truncation of Dyrk1A was increased and was positively correlated with calpain I activation in AD brain. Calpain I proteolyzed Dyrk1A into the AD-like truncated form and elevated its kinase activity in vitro. This truncation enhanced the biological activity of Dyrk1A in the regulation of Tau exon 10 splicing and Tau phosphorylation. Thus, these findings provide a novel possible mechanism of Alzheimer neurofibrillary pathology.
Experimental Procedures
Human Brain Tissues
Medial temporal and frontal cortices from histopathologically confirmed 17 AD and 16 age-matched normal human brains used for this study (Table 1) were obtained from the Sun Health Research Institute Donation Program (Sun City, AZ). Tissues were stored in a −75 °C ultrafreezer until used. The use of frozen human brain tissue was in accordance with the National Institutes of Health guidelines and was approved by our Institutional Review Committee.
TABLE 1.
Human brain tissue of AD and control (Con) tissues used in this study
| Case | Age at death | Gender | PMIa | Braak stageb | Tangle scoresc |
|---|---|---|---|---|---|
| years | h | ||||
| AD 1d | 89 | F | 3.0 | V | 14.5 |
| AD 2d | 80 | F | 2.25 | VI | 14.5 |
| AD 3 | 85 | F | 1.66 | V | 12.0 |
| AD 4d | 78 | F | 1.83 | VI | 15.0 |
| AD 5d | 95 | F | 3.16 | VI | 10.0 |
| AD 6d | 86 | M | 2.25 | VI | 13.5 |
| AD 7d | 91 | F | 3.0 | V | 8.50 |
| AD 8 | 83 | F | 3.0 | VI | 12.4 |
| AD 9 | 74 | M | 2.75 | VI | 14.66 |
| AD 10 | 79 | F | 1.5 | VI | 14.66 |
| AD 11 | 73 | F | 2.0 | V | 15.00 |
| AD 12 | 81 | M | 3.0 | V | 11.00 |
| AD 13 | 76 | M | 2.33 | VI | 15.00 |
| AD 14 | 72 | M | 2.5 | VI | 15.00 |
| AD 15 | 74 | F | 2.83 | VI | 15.00 |
| AD 16 | 76 | M | 4.0 | V | 15.00 |
| AD 17 | 78 | M | 1.83 | VI | 15.00 |
| Mean ± S.D. | 80.59 ± 6.70 | 2.52 ± 0.65 | 13.57 ± 2.05 | ||
| Con 1 | 85 | F | 2.75 | II | 5.0 |
| Con 2 | 82 | F | 2.0 | II | 4.25 |
| Con 3 | 70 | F | 2.0 | I | 0.00 |
| Con 4 | 73 | M | 2.0 | III | 2.75 |
| Con 5 | 78 | M | 1.66 | I | 0.00 |
| Con 6 | 80 | M | 3.25 | I | 2.75 |
| Con 7 | 80 | M | 2.16 | II | 1.00 |
| Con 8 | 83 | F | 3.25 | I | 0.75 |
| Con 9 | 82 | F | 2.25 | II | 3.50 |
| Con 10d | 85 | M | 2.5 | II | 4.25 |
| Con 11d | 86 | F | 2.5 | III | 5.00 |
| Con 12d | 81 | M | 2.75 | III | 6.41 |
| Con 13d | 88 | F | 3.0 | II | 2.00 |
| Con 14d | 90 | F | 3.0 | III | 4.50 |
| Con 15d | 88 | F | 3.5 | III | 2.50 |
| Con 16d | 88 | F | 3.0 | IV | 4.50 |
| Mean ± S.D. | 82.44 ± 5.50 | 2.60 ± 0.55 | 3.07 ± 1.93 |
a Post-mortem interval.
b Neurofibrillary pathology was staged according to Braak and Braak (88).
c Tangle score was a density estimate and was designated as none, sparse, moderate, or frequent (0, 1, 2, or 3 for statistics), as defined according to CERAD Alzheimer disease criteria (89). Five areas (frontal, temporal, parietal, hippocampal, and entorhinal) were examined, and the scores were combined for a maximum of 15.
d Both frontal and temporal cortex.
Animals
Male FVB mice were purchased from Charles River Laboratory and Nanjin animal model center. The animals were housed in a 12-h light/dark schedule with free access to food and water. Animal use was in full compliance with National Institutes of Health guidelines and was approved by our institutional animal care and use committees.
Plasmids, Proteins, and Antibodies
Mammalian expression vectors pcDNA3/Dyrk1A and pCI/Dyrk1A and their deletion mutants tagged with HA at the N terminus and Myc or FLAG at the C terminus were constructed, and their sequences were confirmed. pCI/SI9-LI10 containing a Tau minigene, SI9-LI10, comprising Tau exons 9, 10, and 11 and part of intron 9 and the full length of intron 10 (49) was a gift from Dr. Jianhua Zhou (University of Massachusetts Medical School). Monoclonal antibody 8D9 was raised against a histidine-tagged protein containing the first 160 residues of rat Dyrk1A (35). The monoclonal anti-HA, anti-α-tubulin, anti-His, anti-Dyrk1A (D1694), and anti-β-actin were bought from Sigma. The polyclonal anti-GAPDH was from Santa Cruz Biotechnology, Inc. Monoclonal anti-3R-Tau and anti-4R-Tau were from Upstate Biotechnology, Inc. (Lake Placid, NY). Other Dyrk1A antibodies used in this study were 2771 (Cell Signaling Technology, Danvers, MA), 7D10 (Merck KGaA, Darmstadt, Germany), sc-130741 (Santa Cruz Biotechnology, Inc.), and AF5407 (R&D Systems, Inc., Minneapolis, MN). Monoclonal anti-human Tau (43D) and polyclonal anti-Tau (R134d and 92e) were described previously (50). Peroxidase-conjugated anti-mouse and anti-rabbit IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). The ECL kit was from Thermo Scientific (Rockford, IL), and [γ-32P]ATP was from MP Biomedicals (Irvine, CA).
Cell Culture and Transfection
HEK-293FT cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C. All transfections were performed in triplicate with FuGENE 6 (Roche Applied Science) according to the manufacturer's manual.
Analyses of Levels of Dyrk1A, 3R-Tau, and 4R-Tau and Phosphorylation of Tau
Levels of 3R-Tau, 4R-Tau, and Tau phosphorylation were determined by using quantitative immuno-dot blots or Western blots, as described (32). Bands or dots in immunoblots were visualized by enhanced chemiluminescence (Thermo Scientific) and quantified by densitometry with AIDA 2.0 beta (Raytest, Straubenhardt, Germany). Levels of Tau phosphorylation at each specific site were determined by using phosphorylation-dependent and site-specific Tau antibodies. 3R-Tau and 4R-Tau levels were determined by using antibodies against 3R-Tau and 4R-Tau. Total Tau level was determined by using total Tau antibody R134d, 92e, or 43D. Dyrk1A level in human brains was measured by Western blots developed with monoclonal antibody 8D9 to Dyrk1A. The correlation between human brain Dyrk1A truncation and phosphorylation of Tau or the ratio 3R-Tau/4R-Tau was analyzed by Spearman correlation and linear regression methods.
Immunoprecipitation
HEK-293FT cells were transfected with pcDNA/Dyrk1A, pcDNA3/HA-Dyrk1A-Myc or pCI/HA-Dyrk1A-FLAG, and its deletion mutants tagged with both HA and Myc or FLAG for 48 h as described above. The cells were washed twice with PBS and lysed with lysate buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 50 mm NaF, 1 mm Na3VO4, 0.1% Nonidet P-40, 0.1% Triton X-100, 0.2% sodium deoxycholate, 2 mm EDTA, 1 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin). Insoluble materials were removed by centrifugation; the supernatant was incubated with anti-HA preconjugated onto protein G beads for 4 h at 4 °C. The beads were washed with lysate buffer twice and with TBS twice, and bound proteins were subjected to proteolysis or kinase activity assay.
In Vitro Proteolysis of Dyrk1A
Human brain tissue was homogenized in 9 volumes of buffer consisting of 50 mm Tris-HCl (pH 7.4), 8.5% sucrose, 10 mm β-mercaptoethanol, 2.0 mm EDTA, followed by centrifugation at 16,000 × g at 4 °C for 10 min. The supernatants were incubated in the presence or absence of various concentrations of Ca2+ and/or protease inhibitors for 10 min at 30 °C. The reactions were terminated by the addition of 4-fold concentrated SDS-PAGE sample buffer, followed by heating in boiling water for 5 min. The products of proteolysis were analyzed by Western blots developed with antibodies to calpain I and Dyrk1A.
Immunopurified Dyrk1A was proteolyzed in vitro by incubating with calpain I (Sigma) in proteolysis buffer (50 mm Tris-HCl, pH 7.4, 1 mm CaCl2, 10 mm β-mercaptoethanol) for 10 min at 30 °C. After termination of proteolysis by adding 50 μm N-acetyl-Leu-Leu-Nle-CHO (ALLN; a calpain inhibitor), the proteolyzed products were used for kinase activity assay and for site-specific phosphorylation of Tau by Western blots.
Immunodepletion of Dyrk1A by Anti-His Antibody
Brain extracts were incubated with anti-His precoupled to protein G-agarose beads overnight at 4°C in the presence of 5 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin. The original brain extracts and those after immunodepletion were examined by Western blots with anti-Dyrk1A and anti-GAPDH.
Dyrk1A Kinase Activity Assays
To study the effect of proteolysis of Dyrk1A by calpain I on its kinase activity, Tau441 (0.2 mg/ml) was incubated with the Dyrk1A proteolyzed as above in a reaction buffer consisting of 50 mm Tris-HCl (pH 7.4), 10 mm β-mercaptoethanol, 0.1 mm EGTA, 10 mm MgCl2, and 0.2 mm [γ-32P]ATP (500 cpm/pmol). After incubation at 30 °C for 30 min, the reaction was stopped by applying the reaction mixture on a chromatography paper strip prespotted with 10% trichloroacetic acid; the 32P-labeled Tau was separated from free [γ-32P]ATP by paper chromatography, and the radioactivity of Tau was determined by Cerenkov counting as described previously (51).
To compare the activity of Dyrk1A deletion mutants, Tau441 was incubated with immunoprecipitated wild type Dyrk1A or its deletion mutants in the above reaction buffer for 30 min at 30 °C. The reaction was then stopped by heating with an equal volume of 2× Laemmli sample buffer. The reaction products were separated by SDS-PAGE. Incorporation of 32P was detected in the dried gel by a phosphorimaging system.
To determine the Km and Vmax, various concentrations (2–90 μm) of recombinant Tau441 were incubated with proteolyzed or control-treated Dyrk1A 30 °C for 10 min. Phosphorylation of Tau at Thr212 was then determined by using immuno-dot blot assays developed with anti-Thr(P)212-Tau and quantitated by densitometry. The level of Tau phosphorylation at Thr212 was plotted against Tau concentration with GraphPad Prism version 5 by using a Michaelis-Menten program and also plotted by the Lineweaver-Burk double-reciprocal method. Km value and Vmax value were calculated using the Lineweaver-Burk double-reciprocal method.
Site-specific Phosphorylation of Tau by Proteolyzed Dyrk1A
Immunopurified Dyrk1A with anti-His as described above was proteolyzed in vitro by incubating with 2 μg/ml calpain I (Sigma) in proteolysis buffer (50 mm Tris-HCl, pH 7.4, 1 mm CaCl2, 10 mm β-mercaptoethanol) for 10 min at 30 °C. After termination of proteolysis by adding 50 μm ALLN, the proteolyzed Dyrk1A was incubated with Tau441 (0.2 mg/ml) in a reaction buffer consisting of 50 mm Tris-HCl (pH 7.4), 10 mm β-mercaptoethanol, 0.1 mm EGTA, 10 mm MgCl2, and 0.2 mm ATP. After incubation at 30 °C for various periods, the reaction was stopped by adding acetic acid (2% final concentration). The reaction products were subjected to immuno-dot blot analysis for the site-specific phosphorylation of Tau. The phosphorylation level of Tau at each site was plotted against the reaction times, and curves were fitted using GraphPad Prism version 5 by a Boltzmann sigmoidal program.
Mass Spectrum Analysis
Immunopurified Dyrk1A was proteolyzed in vitro by calpain I, as described above, and separated by SDS-PAGE. After Coomassie Blue staining, the full-length, the 70-kDa, and the 42-kDa Dyrk1A bands were dissected, reduced with 10 mm DTT at 56 °C for 30 min, and alkylated with 50 mm iodoacetamide for 30 min. The gel bands were destained with 50% acetonitrile in 50 mm NH4HCO3 and dehydrated with 100% acetonitrile. Trypsin digestion (13 μg/ml) was performed overnight prior to peptide extraction in 5% formic acid, acetonitrile (1:2, v/v). The extracted peptides were analyzed on a quadrupole Orbitrap (Q-Exactive, Thermo Scientific) mass spectrometer equipped with a nanoflow HPLC instrument (Easy nLC, Thermo Fisher Scientific) and a nanoelectrospray ion source (Proxeon Biosystems). Peptide samples were loaded onto a C18 reversed phase column (15-cm length, 75-μm inner diameter; Michrom Bioresources) and eluted with a linear gradient from 5 to 35% acetonitrile containing 0.5% formic acid in 30 min. The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS2 acquisition. The 10 most intense ions of survey full scan (m/z 400–1600) were sequentially isolated and fragmented by higher energy C-trap dissociation. Fragment spectra were acquired in the Orbitrap mass analyzer. An ion threshold of 10,000 counts was used. The raw files were processed using Proteome Discoverer workflow (version 1.3; Thermo Scientific). The MS data were searched against the human IPI database (version 3.68). Carabamidomethlyation of cysteine was set as a fixed modification, and oxidation of methionine was chosen as variable modifications for database searching. Both peptide and protein identification were filtered at 1% false discovery rate.
Effect of Post-mortem Delay on the Truncation of Dyrk1A
Adult C57B6 mice (5 months old) were killed by CO2 chamber. The dead bodies were kept at room temperature or 4 °C for various times. The forebrains were analyzed by Western blots.
Kainic Acid Injection of Mice
Male FVB mice (25–30 g body weight, 12 weeks old) were housed individually in a 12-h light/dark schedule with free access to food and water. A single dose of kainic acid (20 mg/kg body weight) was injected intraperitoneally (48). The mice were then sacrificed at 2. 5, 6, 10, 24, and 36 h after injection, and forebrains were immediately removed and stored at −80 °C.
For calpain inhibition, the FVB mice above were injected intracerebroventricularly with calpeptin. Specifically, the mice were first anesthetized by intraperitoneal injection of Avertin (Sigma) and placed on a stereotactic instrument. After the scalp was incised and retracted, a 10-μl syringe (Hamilton) was stereotactically placed into the lateral ventricle of the cerebrum according to stereotaxic coordinates (bregma and dura of anterior-posterior 0.35 mm, left lateral 1.0 mm and dorsal-ventral 2.5 mm). A total of 2 μl of 2 μm calpeptin dissolved in DMSO was injected into the left ventricle of the brain. The same volume of DMSO was injected into the left ventricle for control animals. Mice were treated with kainic acid (KA) 3 h after the injection.
Quantitation of Tau Exon 10 Splicing by Reverse Transcription-PCR (RT-PCR)
Total cellular RNA was isolated from cultured cells by using the RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany). One microgram of total RNA was used for first-strand cDNA synthesis with oligo(dT)15–18 by using the Omniscript reverse transcription kit (Qiagen). To measure the alternative splicing of Tau exon 10, PCR was performed by using PrimeSTARTM HS DNA polymerase (Takara Bio Inc., Otsu, Shiga, Japan) with primers 5′-GGTGTCCACTCCCAGTTCAA-3′ (forward) and 5′-CCCTGGTTTATGATGGATGTTGCCTAATGAG-3′ (reverse) at 98 °C for 3 min, 98 °C for 10 s, and 68 °C for 40 s for 30 cycles and then 68 °C for 10 min for extension. The PCR products were resolved on 1.5% agarose gels and quantitated using the Molecular Imager system (Bio-Rad).
Statistical Analyses
Where appropriate, the data are presented as mean ± S.D. Data points were compared by one-way analysis of variance for multiple-group analysis and Student's t test (for the data with normal distribution) or Mann-Whitney test (for the data with non-normal distribution) for two-group comparison. For the analyses of the correlation between Dyrk1A truncation with calpain I activation or the ratio of 3R-Tau/4R-Tau or Tau phosphorylation, the linear regression was performed, and the Spearman correlation coefficient r was calculated and indicated in each panel.
Results
Truncation of Dyrk1A Is Elevated in AD Brain and Correlates with the Activation of Calpain I
To learn the role of Dyrk1A in AD brain, we first analyzed the level of Dyrk1A in frontal cortices from seven AD and seven age- and post-mortem interval-matched control brains obtained <3.5 h after death (Table 1; cases with Footnote d) by Western blots. Dyrk1A has multiple splicing forms (52) and post-translational modifications (53). Thus, Dyrk1A in human brain homogenates showed as a smear polypeptide ladder in Western blots developed by 8D9, an antibody against amino acids (aa) 91–151 of Dyrk1A (Fig. 1A). The total level of Dyrk1A in AD brains was similar to that in control brains (Fig. 1, A and B). However, the level of full-length Dyrk1A was reduced (Fig. 1, A and B), and truncation of Dyrk1A seen by the ratio of truncated/full-length forms of this protein was increased in AD brains (Fig. 1, A and C). The decreased level of the full-length form and increased truncation of Dyrk1A in AD brains were confirmed by antibodies against N-terminal and C-terminal regions of Dyrk1A (Fig. 2, A and B). The antibody against the C terminus of Dyrk1A only immunoreacted with its full length (Fig. 2, A and C), which suggests that truncated forms may lack the C terminus.
FIGURE 1.

Truncation of Dyrk1A is increased and correlated with calpain I activation in AD brain. A, expression of Dyrk1A and calpain I in AD and control (Con) frontal cortices. Western blots of frontal cortical homogenates from seven AD and seven control cases were developed with anti-Dyrk1A (8D9, against aa 91–151), anti-calpain I, and GAPDH. The positions of the full-length and the truncated forms of Dyrk1A are indicated on the right of the blot. The GAPDH blot was included as a loading control. B, similar level of Dyrk1A in AD and control brains. Western blots as shown in A were quantitated by densitometry, and the levels of full-length, truncated, and total Dyrk1A, after being normalized by GAPDH levels, are presented as mean ± S.D. (error bars); the difference was analyzed by Mann-Whitney test; **, p < 0.01. C, increased truncation of Dyrk1A in AD brain. Blots in A were quantitated by densitometry, and the relative ratios of the truncated over the full-length Dyrk1A are presented as mean ± S.D.; the difference was analyzed by Mann-Whitney test. ***, p < 0.001. D, overactivation of calpain I in AD brain. The blots were quantitated by densitometry, and the ratios of the truncated calpain I over the total calpain I, including both the full-length and the truncated forms, are presented as activation of calpain I as mean ± S.D.; ***, p < 0.001. E, correlation between calpain I activation and Dyrk1A truncation in human brain; there was no significant difference in the slop between AD and control brains, and therefore we employed AD and control cases together for this analysis. The relative activation of calpain I (truncated/(full-length + truncated)) (y axis) was plotted against the truncation of Dyrk1A (x axis). The correlation between them was analyzed by Spearman correlation analysis.
FIGURE 2.

Truncation of Dyrk1A is correlated with calpain I activation. A, Western blots of frontal cortical homogenates from seven AD and seven control cases (Con) developed with anti-N-terminal (D1694, against aa 32–51) and anti-C-terminal (7D10, against aa 664–754) regions of Dyrk1A, respectively. B and C, increased truncation of Dyrk1A or decreased full-length Dyrk1A in AD brain. The blots in A were quantitated by densitometry. The relative truncation (truncated/total) (B) and levels of Dyrk1A are shown as mean ± S.E. (error bars) (D). The Mann-Whitney test was used for statistical analyses. D and E, correlation of truncated or full-length Dyrk1A with calpain I activation. The truncation (D) or level (E) of Dyrk1A was plotted against the calpain I activation. The correlation was determined by Spearman correlation analysis. F, an up to 3-h post-mortem interval did not increase the truncation of Dyrk1A in mouse brains. Adult mice were killed by CO2 chamber. The dead bodies were kept at room temperature or 4 °C for various times. The forebrains were analyzed by Western blots.
To learn whether the truncation of Dyrk1A was caused by post-mortem delay, we compared the post-mortem delay. We found that there was no significant difference in post-mortem interval between AD and control cases used for the above study (Table 1). Dyrk1A truncation was not correlated with the post-mortem interval (Data not shown). Moreover, we kept the mouse bodies up to 3 h at room temperature or 4 °C after death to mimic post-mortem delay, and then removed the brains and studied Dyrk1A by Western blots. We did not observe any increase in truncation of Dyrk1A during 3 h post-mortem interval (Fig. 2F). These findings demonstrated that an up to 3-h post-mortem interval does not cause extra truncation of Dyrk1A. Thus, increased truncation of Dyrk1A in AD brains that we observed probably represented pathology and not post-mortem changes.
In agreement with previous reports (44, 45), we found that calpain I was truncated from 80 kDa into 78- and 76-kDa forms and activated by autoproteolysis in AD brain (Fig. 1, A and D). Activation of calpain I is known to lead to proteolysis/truncation of several brain proteins (45, 46, 48, 54). To learn whether Dyrk1A truncation is associated with activation of calpain I, we determined the correlation between Dyrk1A truncation and activation of calpain I in the frontal cortices of human brains. We found a strong positive correlation (r = 0. 8808, p < 0.0001) between calpain I activation and Dyrk1A truncation (Fig. 1E). This correlation was also observed by using the antibodies against the N terminus or C terminus of Dyrk1A, respectively (Fig. 2, D and E).
Dyrk1A Is Proteolyzed by Calpain I
Calpain I is a calcium-dependent protease, and it is activated by micromolar Ca2+. To investigate whether activated calpain I is responsible for the truncation of Dyrk1A, we performed in vitro proteolysis assays. We first added various concentrations of calcium together with 2.0 mm EDTA to normal human brain extract and incubated it at 30 °C for 10 min and then determined the truncation/activation of calpain I and the truncation of Dyrk1A by Western blots. We found that calpain I was proteolyzed from 80 kDa to 78- and 76-kDa active forms in a Ca2+ dose-dependent manner, which coincided with an increase in the truncated forms of Dyrk1A (Fig. 3A). The proteolysis was carried out in the presence of 2.0 mm EDTA to chelate endogenous divalent metals. Most of the added Ca2+ that allowed only free Ca2+ above the 2.0 mm chelating capacity of EDTA amounted to micromolar levels of free Ca2+ in the reaction mixture during incubation of the brain extracts. Therefore, these results suggest that the Dyrk1A proteolysis in the human brain extract could have resulted from activation of calpain I, rather than calpain II, which requires millimolar concentrations of free Ca2+ for activation.
FIGURE 3.

Dyrk1A is proteolyzed by calcium-mediated truncation/activation of calpain I. A, calcium-activated proteolysis of calpain I and Dyrk1A in the human brain extract. Normal human brain frontal cortical extract was incubated at 30 °C for 10 min in the presence of 2.0 mm EDTA and various concentrations (0.00–2.14 mm) of CaCl2. Then the incubated tissue extract was analyzed by Western blots developed with anti-calpain I or anti-Dyrk1A. B, selective inhibition of calcium-activated proteolysis of calpain I and Dyrk1A. Normal human frontal cortical extract was incubated at 30 °C for 10 min in the presence of 2.0 mm each of EDTA and CaCl2 plus various selective protease inhibitors, as indicated in the figure, followed by Western blots probed with anti-calpain I or anti-Dyrk1A (8D9, against aa 91–151) to detect the proteolysis. An arrow indicates the full-length calpain I (top) or Dyrk1A (bottom); vertical lines indicate the truncated calpain I or Dyrk1A. Apr, aprotinin; Pep, pepstatin; Leu, leupeptin; Calp, calpestatin peptide. C, proteolysis of Dyrk1A by calpain I. Dyrk1A was immunoprecipitated by anti-His from HEK-293FT cells that overexpressed Dyrk1A and incubated with various concentration of calpain I in the presence of CaCl2 for 10 min at 30 °C, followed by Western blot analyses. AD, AD brain; Con, control brain; HC, heavy chain of antibody; LC, light chain of antibody. D, depletion of the full-length but not truncated Dyrk1A from human brain extract by anti-His antibody. Human brain frontal cortical extracts from AD and control cases were incubated with anti-His precross-linked protein G-agarose beads overnight at 4 °C. The same volumes of the original brain extracts and of those after immunodepletion were subjected to Western blots developed with anti-Dyrk1A (8D9) and anti-GAPDH. The depletion of full-length Dyrk1A by anti-His from AD brain indicated cleavage of Dyrk1A upstream of the histidine domain.
To confirm that Dyrk1A in human brain extract is proteolyzed by calpain, we studied the inhibition of Ca2+-stimulated proteolysis of calpain I and Dyrk1A with various selective protease inhibitors. When normal human brain extract was incubated in the presence of aprotinin, a serine protease inhibitor, or pepstatin, an aspartic protease inhibitor, no significant inhibition of the proteolysis of either calpain I or Dyrk1A was observed (Fig. 3B). These results excluded the involvement of serine and aspartic proteases in the proteolysis of calpain I and Dyrk1A in the brain. In contrast, when leupeptin, a selective inhibitor of cysteine and serine proteases, and ALLN, a calpain and cysteine protease inhibitor, were included in the incubation mixture, a marked inhibition of calpain I proteolysis and an almost complete blockage of Ca2+-induced Dyrk1A proteolysis were observed (Fig. 3B). A specific calpain inhibitor, calpepstatin peptide, also inhibited the autoproteolysis of calpain I and prevented Dyrk1A from proteolysis. Taken together, these results indicated that most probably the elevated Ca2+ induces autoproteolysis and activation of calpain I, which in turn cleaves Dyrk1A into the truncated forms in AD brain.
To further confirm the proteolysis of Dyrk1A by calpain I, we overexpressed Dyrk1A in HEK-293FT cells and immunoprecipitated it with anti-His antibody. Dyrk1A contains a His repeat at its C-terminal region and thus can be immunoprecipitated by anti-His antibody (Fig. 3C). The immunopurified Dyrk1A was proteolyzed with purified calpain I, and then the reaction products were analyzed by Western blots. We observed that purified Dyrk1A was proteolyzed by calpain I in a dose-dependent manner (Fig. 3C). Similar to the pattern of Dyrk1A in AD brain homogenate, the proteolyzed products showed smear and were detected by antibody 8D9 against aa 91–150 of Dyrk1A, but not by anti-His (Fig. 3C), suggesting that the proteolyzed products did not have a His repeat. The majority of the proteolyzed products were around 50 kDa. These results suggest that Dyrk1A is proteolyzed by calpain I to the AD-like pattern and that the cleavage sites may locate at the N-terminal side of the His repeat, the PEST (a peptide sequence that is rich in proline, glutamic acid, serine, and threonine) domain (Fig. 4A), which can be easily attacked by calpain I (55).
FIGURE 4.

Proteolysis of Dyrk1A by calpain I at the C terminus precedes that at the N terminus. A, diagram of recombinant Dyrk1A and epitopes of the Dyrk1A antibodies used in this study. B, Western blots of proteolyzed Dyrk1A. Dyrk1A was immunoprecipitated by anti-HA from HEK-293FT cells that overexpressed Dyrk1A and was incubated with various concentrations of calpain I in the presence of CaCl2 for 10 min at 30 °C, followed by Western blots developed with antibodies indicated below the blots. C, cleavage of Dyrk1A by calpain I in vitro at both the C terminus and N terminus. Dyrk1A tagged with HA at the N terminus and with FLAG at the C terminus was immunoprecipitated by anti-HA or anti-His and then incubated with calpain I for 10 min. The proteolyzed products were analyzed by Western blots developed with antibodies indicated below the blots. D, mass spectrometry of truncated Dyrk1A. Truncated Dyrk1A by calpain I was separated by SDS-PAGE, and the full-length, ∼70-kDa, and ∼42-kDa Dyrk1A bands were subjected to MS analysis. The MS-detected peptides are marked by green color. E, diagram of the consensus sequences for calpain cleavage. F, predicted cleavage sites by calpain. Arrows indicate possible cleavage sites of Dyrk1A by calpain I according to its consensus cleavage sites. The framed sequence is the PEST region.
To identify whether the His repeat of Dyrk1A in human brain are also truncated, we immunodepleted the human brain extracts with anti-His antibody. The immunodepleted extracts were then detected by Western blots developed with anti-Dyrk1A (8D9). We found that the full-length Dyrk1A was successfully depleted, but the truncated Dyrk1A bands were identical to those of the extracts without immunodepletion (Fig. 3D), indicating that most truncated Dyrk1A in human brains lacks the His repeats. Consistent with these results, Western blots of human brain homogenates developed with the C-terminal antibody did not recognize any of the truncated Dyrk1A (Fig. 2A).
To study the kinetics of proteolysis, we incubated immunopurified Dyrk1A with various concentrations of calpain I and analyzed the proteolytic products by Western blots using several antibodies that recognize different epitopes of Dyrk1A (Fig. 4A). We observed that with increasing concentrations of calpain I, Dyrk1A was first truncated to the ∼70-kDa and further to the ∼42-kDa fragments (Fig. 4B). The large Dyrk1A fragments (∼70 kDa) truncated by low concentration of calpain I were labeled by antibodies D1694, 8D9, and AF5407, which recognize the N-terminal and middle regions of Dyrk1A (aa 429–502), but not by the C-terminal antibody 7D10 or anti-His (Fig. 4B). These results suggest that calpain I cleaves Dyrk1A first at a site(s) upstream of the His repeat. The ∼42-kDa truncated Dyrk1A fragments generated by higher concentrations of calpain I were not stained by the N-terminal Dyrk1A antibody D1694 (Fig. 4B, left), suggesting that Dyrk1A was further cleaved by calpain I at the N terminus to produce smaller fragments.
To further characterize the cleavage of Dyrk1A by calpain I, we overexpressed Dyrk1A tagged with HA at the N terminus and with FLAG at the C terminus in HEK-293FT cells and immunopurified it by anti-HA or anti-His. After incubation of the purified Dyrk1A with 1 μg/ml calpain I at 30 °C for 10 min, we examined the reaction mixtures using Western blots and found that the large (∼70-kDa) truncated Dyrk1A fragments were labeled by antibodies D1694, 2771, 8D9, sc-130741, and AF5407, which recognize the N-terminal and middle regions of Dyrk1A, but not by the C-terminal antibodies 7D10 or anti-His or by anti-FLAG (Fig. 4C). We also found that the ∼42-kDa Dyrk1A fragments were labeled by antibodies against the middle region of Dyrk1A, 8D9, SC-130741, and AF5407, but not by either the N-terminal antibody D1694 or the C-terminal antibodies (Fig. 4C). These results suggest that the large (∼70-kDa) and small (∼42-kDa) Dyrk1A fragments were truncated by calpain I in vitro at the C terminus and at both the N and the C terminus, respectively.
To analyze the truncation of the fragments by mass spectrometry (MS), we cut out the 70- and 42-kDa Dyrk1A bands as well as the full-length protein from SDS gels and subjected them to trypsin digestion. A total of 12 peptides were identified from the 42-kDa band, but the first and the last peptides identified from full-length Dyrk1A were absent from the truncated Dyrk1A (Fig. 4D). The first and the last amino acids detected from the 42-kDa band were Leu87 and Lys472, respectively. Therefore, we speculated that calpain I cleaved Dyrk1A outside of Leu87–Lys472. Moreover, the 42 kDa band was not labeled by antibody D1694 (against aa 32–51) or anti-His (against aa 590–610) but by antibody AF5407 (against aa 429–502). According to consensus motifs for calpain cleavage (56) (Fig. 4E), we analyzed potential calpain cleavage sites within aa 51–86 and 472–589 and predicted four potential cleavage sites within aa 51–86 and 14 potential cleavage sites within aa 472–589, which contains the PEST domain (Fig. 4F).
Proteolysis of Dyrk1A by Calpain I Increases Its Kinase Activity
To determine the impact of calpain I-induced proteolysis on Dyrk1A, we determined its kinase activity toward Tau441. We proteolyzed Dyrk1A with various concentration of calpain I in vitro for 10 min at 30 °C. The proteolyzed Dyrk1A was used to phosphorylate Tau441 with [γ-32P]ATP for 30 min in vitro. The incorporation of 32Pi into Tau was calculated and plotted against the calpain I concentration. We found that the proteolysis of Dyrk1A elevated its kinase activity in a manner dependent on calpain I concentration, reaching ∼3-fold increase at 2 μg/ml calpain I (Fig. 5A).
FIGURE 5.

Proteolysis of Dyrk1A by calpain I enhances its kinase activity. A, increase of Dyrk1A activity by proteolysis with calpain I. Immunopurified Dyrk1A with anti-His from Dyrk1A-overexpressing HEK-293FT cells was proteolyzed with various concentrations of calpain I as described in C. After adding ALLN to inhibit proteolysis, the kinase activity of proteolyzed Dyrk1A was assayed toward Tau441 as a substrate in the presence of a calpain inhibitor, ALLN. B and C, increased site-specific phosphorylation of Tau by proteolyzed Dyrk1A. Immunopurified Dyrk1A was proteolyzed with 2.0 μg/ml calpain 1 for 10 min at 30 °C. After adding 50 μm ALLN to terminate the proteolysis reaction, the proteolyzed Dyrk1A and control-treated Dyrk1A (no calpain I) were incubated with Tau (0.2 mg/ml) for various periods at 30 °C in the reaction buffer to phosphorylate Tau. The phosphorylated Tau products were subjected to immuno-dot blots developed with phosphorylation-dependent and site-specific Tau antibodies and total Tau antibody (B). Tau phosphorylation at each individual site was then quantified densitometrically, and the relative levels after being normalized with the total Tau levels were plotted against reaction times. Each curve was fitted with GraphPad Prism version 5 by a Boltzmann sigmoidal program (C). D, enzyme kinetics of Dyrk1A-catalyzed Tau phosphorylation. Various concentrations of Tau441 were incubated with either the proteolyzed Dyrk1A or the control-treated Dyrk1A (no calpain I) at 30 °C for 10 min, and Tau phosphorylation at Thr212 in the reaction mixtures was determined by immuno-dot blots. E and F, the blots shown in C were quantified densitometrically, and the data were plotted against Tau concentration and fitted with GraphPad Prism version 5 by a Michaelis-Menten program (E) or as a Lineweaver-Burk plot (F), and Km and Vmax were determined from the latter.
To learn the effect of Dyrk1A truncation on Tau phosphorylation at various sites, we used calpain I-proteolyzed Dyrk1A to phosphorylate Tau441 for various time and then employed immuno-dot blots developed with site-specific antibodies to various Tau phosphorylation sites. We observed that Tau was more effectively phosphorylated by proteolyzed Dyrk1A than the control-treated enzyme at Ser199, Thr205, Thr212, Thr217, Ser396, and Ser404 (Fig. 5, B and C). These results revealed that proteolysis of Dyrk1A enhances its kinase activity and increases the ability to phosphorylate Tau in a site-specific manner.
Thr212 of Tau is the most efficient phosphorylation site of Dyrk1A (31, 32). The smaller difference in Thr212 phosphorylation by the truncated Dyrk1A and the control-treated Dyrk1A, as shown in Fig. 4, B and C, might result from a rapid saturation of the phosphorylation reaction. Thus, we determined the Km and Vmax for phosphorylation of Tau at Thr212 by Dyrk1A. We found that, as compared with the non-proteolyzed Dyrk1A, the proteolyzed Dyrk1A phosphorylated Thr212 of Tau severalfold more efficiently than the control-treated kinase (Vmax of 654.6 versus 140.8) (Fig. 5, D and E). However, the truncation did not change the Km of Dyrk1A significantly, suggesting that the truncation does not affect its affinity to Tau as a substrate.
C-terminally Truncated Forms of Dyrk1A Have Higher Kinase Activity
The above studies demonstrated that Dyrk1A was truncated in the PEST domain in AD brain. We constructed mutants, progressively deleted from the C terminus, Dyrk1A(1–474), Dyrk1A(1–588), and Dyrk1A(1–616) (Fig. 6A). These deletion mutants of Dyrk1A were tagged with HA at the N terminus. We overexpressed the full-length Dyrk1AFL and its deletion mutants in HEK-293FT cells and immunoprecipitated them with anti-HA antibody. The kinase activity of the immunopurified Dyrk1A mutants was assayed toward Tau441 as a substrate and using [γ-32P]ATP. We found that Dyrk1A(1–474) had the highest activity toward Tau441 (Fig. 6B), even when the amount of protein immunoprecipitated was the lowest of all of the constructs studied (Fig. 6B).
FIGURE 6.

C-terminal truncation of Dyrk1A increases its kinase activity. A, schematics of Dyrk1A domain structures and deletion mutations of Dyrk1A studied. NLS, nuclear localization sequence. B, increased phosphorylation of Tau by deletion mutants of Dyrk1A. Dyrk1A and its deletion mutants, Dyrk1A(1–616), Dyrk1A(1–588), and Dyrk1A(1–474), tagged with HA at the N terminus were overexpressed in HEK-293FT cells and immunoprecipitated (IP) with anti-HA antibody (mouse). The immunocomplexes were incubated with Tau441 and [γ-32P]ATP for 30 min at 30 °C and separated by SDS-PAGE and visualized with Coomassie Blue staining. After drying the gel, the 32P incorporated into Tau441 was measured by using phosphorimaging (BAS-1500, Fuji). The level of 32P incorporated into Tau was normalized by the protein level detected by Coomassie Blue staining. Results are shown as mean ± S.D. (error bars) (n = 3). The level of immunoprecipitated Dyrk1A or its deletion mutants was determined by Western blots (Wb) with anti-HA (rabbit) antibody. C, overexpression of Dyrk1A deletion mutants suppressed Tau exon 10 inclusion. HEK-293FT cells were co-transfected with Tau minigene pCI/SI9-LI10 and pcDNA3/Dyrk1AFL or its deletion mutants tagged with HA at a ratio of 1:8 for 48 h. The cell lysates were analyzed by Western blots developed with anti-HA for the expression of Dyrk1A and its deletion mutants. The GAPDH blot was included as a loading control. RT-PCR was employed to determine the splicing products of exon 10. The ratio of inclusion and exclusion of Tau exon 10 was calculated after densitometric quantification. Results are shown as the mean ± S.D. (n = 3). D, site-specific phosphorylation of Tau by Dyrk1A and its deletion mutants. The lysates of HEK-293FT cells double-transfected with pCI/Tau441 and pcDNA3/Dyrk1AFL or its deletion mutants were subjected to Western blots developed with site-specific and phosphorylation-dependent anti-Tau, as indicated, and total Tau with anti-total Tau (R134d). The relative levels of Tau phosphorylation, after densitometric quantification of the blots and normalization by the total Tau level, are shown as mean ± S.D. (n = 3); the level of Dyrk1AFL was defined as 100. *, p < 0. 05; **, p < 0. 01; ***, p < 0. 001.
To investigate the effect of truncated Dyrk1As on the regulation of Tau exon 10 splicing, we co-transfected them individually with Tau minigene pCI/SI9-LI10 into HEK-293FT cells. After 48-h transfection, Tau exon 10 splicing was determined by RT-PCR. We observed that the expression levels of Dyrk1AFL and Dyrk1A(1–474) were similar, but those of Dyrk1A(1–616) and Dyrk1A(1–588) were slightly higher than the expression level of Dyrk1AFL (Fig. 6C). Overexpression of Dyrk1AFL and its deletion mutants inhibited Tau exon 10 inclusion significantly (Fig. 6C). Dyrk1A(1–616) and Dyrk1A(1–588) showed slightly higher activity than that of Dyrk1AFL in the suppression of Tau exon 10 inclusion, but Dyrk1A(1–474) showed considerably more inhibition of exon 10 inclusion (Fig. 6C).
Dyrk1A phosphorylates Tau in vitro and in vivo (32). Overexpression of Dyrk1A and its mutants significantly elevated Tau phosphorylation at Ser199, Thr205, Thr212, Thr217, Ser396, Ser404, and Ser422 site-specifically (Fig. 6D). Compared with Dyrk1AFL, increased Tau phosphorylation at Ser199, Thr205, Thr212, Thr217, Ser396, Ser404, and Ser422 was observed by overexpression of the deletion mutants (Fig. 6D). Overexpression of Dyrk1A(1–588) or Dyrk1A(1–522) elevated Tau phosphorylation at Ser217 and Ser422 the most effectively (Fig. 6D). Dyrk1A(1–474) expression showed the highest ability in the phosphorylation of Tau at Ser199, Thr205, and Ser404 (Fig. 6D). These data suggest that truncated forms of Dyrk1A, especially Dyrk1A(1–474), are more active than Dyrk1AFL, and the C terminus may play an inhibitory role in the kinase activity of Dyrk1A toward Tau.
Excitotoxicity Induces Truncation of Dyrk1A and Increases the Ratio of 3R-Tau/4R-Tau and Tau Phosphorylation in Mice
Excitotoxicity induces calcium influx and calpain I activation (48, 57). To study if Dyrk1A is truncated in the excitotoxic brain, we injected mice with KA intraperitoneally, which is a commonly used method to induce excitotoxicity, and measured Dyrk1A truncation. Spectrin IIα is specifically cleaved by activated calpain to 145-kDa truncated product. In the excitotoxic mouse brain, we found a 145-kDa spectrin IIα 24 h after KA injection (Fig. 7A), indicating calpain activation. In these mouse brains, we also observed a time-dependent truncation of Dyrk1A after KA injection (Fig. 7, A and B). Rodent brain mainly expresses 4R-Tau and only very little 3R-Tau (58). We investigated the levels of Tau in the KA-induced excitotoxic brain. We found that the levels of total Tau, 3R-Tau, and 4R-Tau were first slightly increased and then were followed by a dramatic decrease (Fig. 7C). The changes of 4R-Tau were more marked than 3R-Tau (Fig. 7C). Thus, the ratio of 3R-Tau/4R-Tau was dramatically increased during excitotoxicity (Fig. 7D).
FIGURE 7.

KA-induced excitotoxicity causes Dyrk1A truncation and increase in 3R-Tau expression and Tau phosphorylation in mice. A and B, KA-induced excitotoxicity caused the truncation of Dyrk1A. Homogenates of forebrains of mice sacrificed at the indicated time points after KA injection were analyzed by Western blots developed with Dyrk1A antibody (8D9, against aa 91–151) and anti-spectrin IIα and quantified densitometrically. The ratio of truncated/total was calculated and plotted against the time after KA injection. C–E, excitotoxicity induced by KA injection led to increased expression of 3R-Tau and phosphorylation of Tau at Thr212. Forebrain homogenates were analyzed by Western blots developed with anti-3R-Tau (RD3), anti-4R-Tau (RD4), anti-Thr(P)212-Tau, anti-total Tau, and, as loading control, anti-actin (C). The ratio of 3R-Tau/4R-Tau (D) and phosphorylated Tau at Thr212 normalized with total Tau level (E) were plotted against the time after KA injection and fitted by a Boltzmann sigmoidal program.
To learn the effect of truncation and activation of Dyrk1A by excitotoxicity on Tau phosphorylation, we determined the level of Tau phosphorylation at Thr212, the most favored site of Dyrk1A, and found that Thr212 phosphorylation was dramatically increased (Fig. 7, C and E). Thus, we found that truncation of Dyrk1A in excitotoxic brain coincides with increase in 3R-Tau expression and Tau Thr212 phosphorylation.
Inhibition of Calpain Prevents Truncation of Dyrk1A, 3R-Tau Expression, and Tau Phosphorylation at Thr212
To study the involvement of calpain I activation in truncation of Dyrk1A that increases 3R-Tau expression and Tau phosphorylation, we injected calpeptin, a calpain inhibitor, intracerebroventricularly and then determined the levels of Dyrk1A and Tau. We found that inhibition of calpain by calpeptin significantly abolished the KA-induced Dyrk1A truncation (Fig. 8, A and B). Moreover, the KA-induced increases in the ratio of 3R-Tau/4R-Tau and Tau phosphorylation at Thr212 were markedly reduced by calpeptin treatment (Fig. 8, C–E). These results suggested that truncation and activation of Dyrk1A were caused by calpain activation, and inhibition of calpain prevented the Dyrk1A truncation and increase in 3R-Tau expression and Tau phosphorylation.
FIGURE 8.
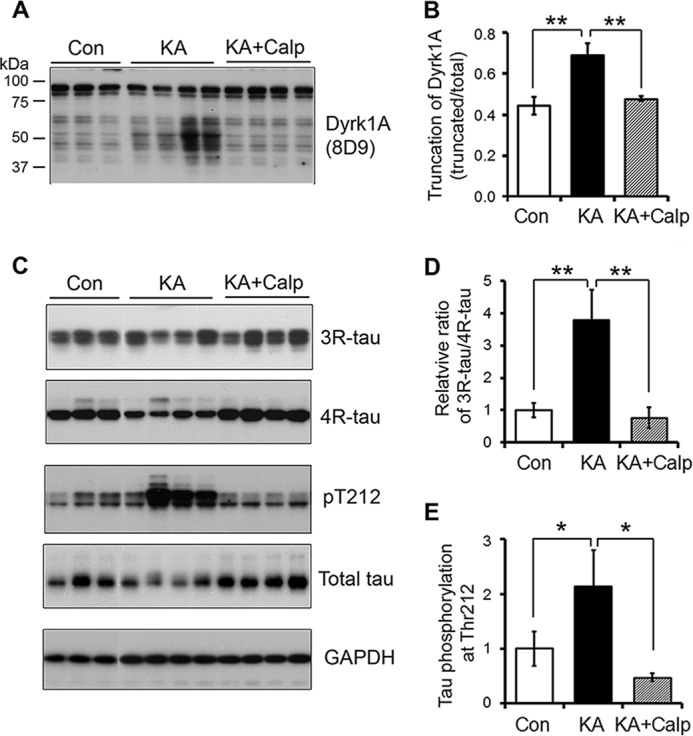
Inhibition of calpain prevents KA-induced truncation of Dyrk1A, 3R-Tau expression, and Tau Thr212 phosphorylation. A and B, inhibition of calpain prevented KA-induced truncation of Dyrk1A. The calpain inhibitor calpeptin was intracerebroventricularly injected 3 h before KA intraperitoneal injection. Homogenates of forebrains of mice sacrificed after 12 h of KA injection were analyzed by Western blots developed with Dyrk1A antibody (8D9, against aa 91–151) and quantified densitometrically. The ratio of truncated/total Dyrk1A was calculated and is shown as mean ± S.D. (error bars) (n = 4–6); **, p < 0.01. C–E, inhibition of calpain suppressed 3R-Tau expression and Tau phosphorylation at Thr212 caused by KA. 3R-Tau, 4R-Tau, total Tau, and phosphorylated Tau in the above samples were analyzed by Western blots (C) and quantified as described above. The ratio of 3R-Tau/4R-Tau (D) and level of phosphorylated Tau at Thr212 normalized with total Tau (E) were calculated and shown as mean ± S.D. (n = 4–6); *, p < 0.05.
Truncation of Dyrk1A Correlates with the Increased 3R-Tau Expression and Tau Phosphorylation in AD Brain
Alterations of Tau exon 10 alternative splicing leading to imbalanced expression of 3R-Tau and 4R-Tau are seen in several Tauopathies (28). Dyrk1A suppresses Tau exon 10 inclusion, resulting in an increase of 3R-Tau expression (33). To determine whether the truncation and activation of Dyrk1A disrupts the ratio of 3R-Tau/4R-Tau in AD brain, we measured 3R-Tau and 4R-Tau levels in the homogenates of the frontal cortices by immunoblots and calculated the ratio of 3R-Tau/4R-Tau. Then we performed Spearman correlation and linear regression analyses. We observed that Dyrk1A truncation was positively correlated with the ratio of 3R-Tau to 4R-Tau in AD brain (Fig. 9A), suggesting that truncation and activation of Dyrk1A might contribute to the imbalance of 3R-Tau and 4R-Tau expression in AD brain.
FIGURE 9.

Levels of 3R-Tau and Tau phosphorylation directly correlate with truncation of Dyrk1A in AD brain. A, correlation of 3R-Tau/4R-Tau ratio (y axis) with Dyrk1A truncation (x axis) in AD brain. The level of Dyrk1A truncation in frontal cortices from 12 AD cases was plotted against the 3R-Tau/4R-Tau ratio. The correlation was analyzed by Spearman correlation analysis. B, immunohistochemical staining of sections from AD temporal cortex with anti-3R-Tau and anti-4R-Tau. Anti-3R-Tau (RD3) labels neurofibrillary tangles more than anti-4R-Tau (RD4). C, correlation between Dyrk1A truncation and site-specific phosphorylation of Tau. Levels of Dyrk1A truncation and Tau phosphorylation at individual phosphorylation sites in the frontal cortical homogenates from 12 AD cases were determined by Western blots (not shown). The levels of Tau phosphorylation at individual phosphorylation sites (y axis) were then plotted against the corresponding Dyrk1A truncation (x axis), and the correlation was analyzed by Spearman correlation analysis. A linear regression line is shown when the correlation reaches statistical significance (p < 0.05).
To determine whether an increased 3R-Tau/4R-Tau ratio is associated with neurofibrillary pathology, we immunostained the temporal cortices from AD and control cases with anti-3R-Tau and anti-4R-Tau. In control brains, 3R-Tau- and 4R-Tau-NFTs were rare. In AD brains, NFTs were observed both with anti-3R-Tau and anti-4R-Tau, but the 3R-Tau-positive NFTs are approximately 2 times more plentiful than 4R-Tau-positive NFTs in the same area (Fig. 9B). These results suggest the association of 3R-Tau with neurofibrillary pathology in the AD brain.
To study the impact of truncation and activation of Dyrk1A on Tau pathology in AD brain, we measured Tau phosphorylation in the frontal cortices from 12 AD cases at individual sites by Western blots and determined the correlation of Dyrk1A truncation with hyperphosphorylation of Tau at various pathological sites. We found that Dyrk1A truncation had a positive correlation with Tau phosphorylation at Thr181, Ser202, Thr205, Thr212, Ser214, Thr217, Ser262, Ser396, Ser404, and Ser422 (Fig. 9C), suggesting that truncation and activation of Dyrk1A might contribute to Tau hyperphosphorylation in AD brain.
Tau pathology is seen in both temporal and frontal cortices in AD brain. In order to compare Dyrk1A truncation and Tau phosphorylation in these two areas, we determined Dyrk1A truncation and Tau phosphorylation in six AD and six control cases by Western blots and immuno-dot blots, respectively. We found that the truncation of Dyrk1A was increased significantly in both frontal and temporal cortices. Dyrk1A was slightly more truncated in the frontal cortex than the temporal cortex (Fig. 10A). In these two areas, Tau was hyperphosphorylated at many sites, including Ser199, Ser202, Thr205, Thr212, Thr217, Ser262, Ser396, Ser404, and Ser422, in AD cases (Fig. 10, B and C). The phosphorylation of Tau in the frontal cortex also appeared to be greater than in the temporal cortex at all of the sites studied except for Ser214 (Fig. 10, B and C). These results further support the involvement of the truncation and activation of Dyrk1A in the hyperphosphorylation of Tau in AD brain.
FIGURE 10.

Dyrk1A truncation and Tau phosphorylation are greater in frontal cortex than temporal cortex. A, Western blots of frontal (FC) or temporal cortical (TC) homogenates from six AD and six control cases developed with antibody 8D9 to Dyrk1A. The blots were quantitated by densitometry, and the ratio of the truncated over total Dyrk1A is shown as mean ± S.D. (error bars) paired two-tailed Student's t test after log transformation). B, the levels of total Tau and Tau phosphorylated at the indicated individual phosphorylation sites in frontal or temporal cortical extracts of AD and control brains were analyzed by immuno-dot blots developed with a mixture of R134d and 92e as total and several phosphorylation-dependent/site-specific Tau antibodies. C, blots in B were quantified after normalization by the total Tau level, and the relative levels of site-specific Tau phosphorylation are shown as mean ± S.D. (six pairs, paired two-tailed Student's t test after log transformation). The levels of phosphorylation of Tau at all detected sites, except Ser214 (pS214), were higher (p < 0.01) in the frontal cortex than in the temporal cortex.
Discussion
The present study shows that Dyrk1A is truncated and positively correlated with the activation of calpain I as well as with an increased ratio of 3R-Tau to 4R-Tau and hyperphosphorylation of Tau at multiple sites in AD brain. The activation of calpain I in AD brain could be due to calcium overload resulting from excitotoxicity, β-amyloid neurotoxicity, and/or free radical injury (59). In vitro calpain I proteolyzed Dyrk1A at the C terminus first, followed by the N terminus, and produced AD-like truncation, resulting in an increase in its kinase activity. The cleavage of Dyrk1A by calpain I at C terminus was localized between aa 480 and 597, probably in the PEST region. Overexpression of C-terminally truncated Dyrk1A promoted Tau phosphorylation and Tau exon 10 exclusion. Excitotoxicity induced by KA led to AD-like truncation of Dyrk1A, which coincided with increased 3R-Tau expression and Tau phosphorylation at Thr212 in mouse brain. Thus, it appears that truncation and activation of Dyrk1A by activated calpain I in AD brain probably contributes to Tau pathology in AD through (i) an increase of the 3R-Tau/4R-Tau ratio by inhibition of ASF/SF2- and SC35-promoted Tau exon 10 inclusion and (ii) hyperphosphorylation of Tau by direct phosphorylation of Tau and by priming it for further abnormal hyperphosphorylation by GSK-3β (Fig. 11).
FIGURE 11.

Proposed mechanism by which Dyrk1A activation by Ca2+/calpain I contributes to neurofibrillary pathology in AD brain. Intraneuronal calcium is increased due to excitotoxicity, β-amyloid neurotoxicity, and free radical injury and activates calpain I, which proteolyzes Dyrk1A and in turn activates it. Activated Dyrk1A phosphorylates SC35 and ASF/SF2 and suppresses their facilitation in Tau exon 10 inclusion, resulting in an increased ratio of 3R-Tau/4R-Tau. Activated Dyrk1A also phosphorylates Tau and makes it a more favorable substrate for GSK-3β as well as promoting Tau exon 10 exclusion. All of these changes lead to abnormal hyperphosphorylation of Tau and finally neurofibrillary pathology.
Hyperphosphorylation and deposition of Tau in affected neurons is a hallmark of the pathogenesis of Alzheimer neurofibrillary degeneration (60). The exact causes leading to abnormal hyperphosphorylation of Tau in AD remain elusive, although dysregulation of Tau kinases and Tau phosphatases, impaired glucose uptake and utilization via decreased Tau O-GlcNAcylation, and mitochondrial oxidative stress have been implicated (61). Many protein kinases can phosphorylate Tau in vitro. Among these kinases, GSK-3β, Cdk5, protein kinase A (PKA), calcium and calmodulin-dependent protein kinase-II, casein kinase-1, mitogen-activated protein kinases (MAPKs), stress-activated protein kinases (SAPKs), and Dyrk1A have been implicated most often in the abnormal hyperphosphorylation of Tau (60). Dyrk1A phosphorylates Tau predominantly at Thr212 but also at Thr181, Ser199, Ser202, Thr205, Thr212, Thr217, Thr231, Ser396, Ser404, and Ser422 both in vitro and in cultured cells (31, 32). Moreover, prephosphorylation of Tau by Dyrk1A promotes further phosphorylation of Tau by GSK-3β (32). Overexpression of Dyrk1A in DS trisomic mouse models, such as Ts65Dn and Ts1Cje, and in human Dyrk1A transgenic mice leads to hyperphosphorylation of Tau in a site-specific manner (32, 62, 63).
An imbalance in the expressions of 3R-Tau and 4R-Tau generated by alternative splicing of Tau exon 10 causes neurofibrillary pathology (64). ASF/SF2 and SC35 play critical and regulatory roles in Tau exon 10 splicing in that they act on polypurine enhancer and SC35-like enhancer of exon 10, respectively, and promote Tau exon 10 inclusion (34, 65, 66). We previously showed that in addition to phosphorylating Tau, Dyrk1A phosphorylates splicing factors ASF/SF2 and SC35 and prevents them from facilitating Tau exon 10 inclusion (33, 34). Thus, overexpression of Dyrk1A due to trisomy in DS brain leads to an increased ratio of 3R-Tau/4R-Tau, an imbalance that is known to associate with neurofibrillary pathology. The Tau 3R/4R ratio is also known to be increased in Pick disease and some cases of FTDP-17 (28). These observations suggest that neurofibrillary pathology can be caused by up-regulation of Dyrk1A through phosphorylating Tau and disturbing the normal 1:1 ratio of 3R-Tau to 4R-Tau.
Small changes in the protein level of Dyrk1A can result in a disease phenotype in patients (67) and in mouse models (68), suggesting that the dosage and/or activity of Dyrk1A have to be tightly regulated. Autophosphorylation of Dyrk1A at Tyr312 (Tyr321 in the longest isoform) in the activation loop is required to achieve its full kinase activity (69, 70). However, dephosphorylation with protein-tyrosine phosphatases does not inactivate mature Dyrk1A (70), suggesting that Tyr312 phosphorylation is required for the switch to, rather than for maintaining, the active conformation. In addition, Dyrk1A is regulated by interacting proteins. 14-3-3β binds to Ser520-phosphorylated Dyrk1A in the PEST region and stimulates its catalytic activity (71). Binding of RanBPM to the C terminus (aa 550–563) modulates Dyrk1A activity negatively. Other proteins, such as WD40 repeat protein and SPRED 1 and 2, also are reported to interact with Dyrk1A and modulate it to phosphorylate the substrates (72). Here, we reported that proteolysis of Dyrk1A by calpain I increased its kinases activity toward Tau by 3-fold, which did not change the affinity toward Tau but increased Vmax to phosphorylate Tau.
In the present study, we found for the first time that the truncation of Dyrk1A is significantly increased in AD brain, and the truncation enhances its kinase activity. Thus, another way to regulate Dyrk1A activity is through its truncation by calpain I. This truncation was probably caused by the activation of calpain I due to calcium overload in AD brain. Calpains I and II are well known to proteolyze the same substrates but require micromolar and millimolar calcium, respectively. Thus, calcium overload reaching millimolar concentration can also probably cause truncation and activation of Dyrk1A by calpain II. Recent studies reported that besides millimolar calcium, phosphorylation of calpain II by ERK1/2 can activate it (73, 74). Thus, the possibility that, in addition to calpain I, calpain II can also lead to Tau pathology by truncation and an increase in activity of Dyrk1A cannot be ruled out. Because we were able to almost completely block Dyrk1A truncation produced by micromolar concentration of calcium by calpain inhibitor calpepstatin, it appears that calpain I is most likely the primary enzyme involved in this pathology in AD brain. Dyrk1A has a long kinase domain, followed by a PEST region, histidine repeat, and serine/threonine-rich domain (30). Calpain I cleaved Dyrk1A in vitro first at the C terminus and further at the N terminus dose-dependently. Moreover, the proteolysis of Dyrk1A by calpain I increased its kinase activity, but this increase was not elevated accordingly with the proteolysis induced by higher concentration of calpain I, suggesting that its N terminus may not be very critical for its kinase activity.
To dissect the inhibitory region, we generated a series of deletion mutants of Dyrk1A according to its functional domains. We found that deletion of the C terminus, including the Ser/Thr-rich domain of Dyrk1A, increased its activity. Additional deletions up to the His repeats did not further increase Dyrk1A activity. Therefore, downstream of His repeats, Dyrk1A may contain a weak inhibitory domain. Removing the C terminus from the PEST region (the Dyrk1A(1–474) mutant) increased Dyrk1A kinase activity dramatically, indicating another inhibitory domain in this region. Proteolysis of Dyrk1A by calpain I was probably in its PEST domain in vitro, which is known to be easily attacked by calpain in other proteins (55). Truncated Dyrk1A forms in AD brain could not be immunodepleted by anti-His antibody and were around 40–50 kDa, indicating that they were similar to the truncated Dyrk1A forms produced by calpain I in vitro and that the truncation was located upstream of the His repeat.
Many putative etiologic factors of AD, including excitotoxicity, β-amyloid neurotoxicity, and free radical injury, have in common the potential for disrupting intracellular calcium homeostasis (75–77). Although there is a lack of direct evidence of altered calcium homeostasis in AD brain, dysregulation of calcium is one of the major hypotheses that may explain the pathogenic mechanism of the disease (59, 78). Both calpain I and II are present principally as inactive precursors in the cell, and they are activated by calcium-stimulated autoproteolytic cleavage of the N-terminal sequence in response to calcium influx. Calpain I, which is the major calpain isoform in neurons, is fully activated by low micromolar concentrations of calcium (hence, it is also called μ-calpain), whereas calpain II requires low millimolar levels of calcium for optimal activity (hence, it is also called m-calpain). It is reported that extrasynaptic, but not synaptic, stimulation of the NMDA receptor invokes calpain I activation (79). Calpain I/II is thought to play a critical role in the activation of neuronal Cdk5 (46), MAPK pathway (43), PKA (48), protein phosphatase 2B (45), and strial-enriched protein-tyrosine phosphatase (79) as well as truncation of Tau (47). Here, we observed the truncation of Dyrk1A to 50 kDa in KA-induced excitotoxic brain, and inhibition of calpain with calpeptin almost abolished Dyrk1A truncation, which further confirms truncation of Dyrk1A by calpain. Elevated cleavage and activation of calpain has been reported previously in AD brain (44, 45). We recently reported that PKA phosphorylated ASF/SF2 and enhanced its function in Tau exon 10 inclusion, and a down-regulation of PKA induced by calpain I activation in AD brain was correlated with the increase of 3R-Tau level (65). The present study suggests that calpain activation may also play a role in neurodegeneration via truncation and activation of Dyrk1A in AD brain.
In the present study, we observed that truncation of Dyrk1A enhanced its kinase activity. In KA-induced excitotoxic brain, truncation of Dyrk1A was accompanied by an increase in the ratio of 3R-Tau/4R-Tau and Tau phosphorylation at Thr212, a major Dyrk1A site. Prevention of Dyrk1A truncation by calpain inhibition also abolished the increase in the 3R-Tau/4R-Tau ratio and Thr212 phosphorylation. Interestingly, increased truncation of Dyrk1A in AD brain was associated with increased Tau phosphorylation and 3R-Tau expression. Some previous studies assayed mRNAs of Tau isoforms and found either an increase in 4R-Tau (80, 81) or no change (82, 83) in sporadic AD. It is well known that RNA is quickly degraded in post-mortem tissues, and protein is much more stable than mRNA (84, 85). Here, we did not obtain reliable data on the mRNA level of Tau isoforms, but we observed increased mRNA level of 3R-Tau in DS mouse (Tg65Dn) brains (data not shown). By immunohistochemical staining, Espinoza et al. (86) found that some advanced AD cases had a large amount of 3R-Tau-positive, but not 4R-Tau-positive, NFTs that were thioflavin-S-positive. More severe pathology appeared in association with more abundant 3R-Tau-positive tangles. These findings suggest that aggregation and deposition of 3R-Tau may be associated with more advanced stages and are consistent with our observations of increased 3R-Tau/4R-Tau ratio in AD. Interestingly, the NMDA receptor antagonist memantine has a modest effect in moderate to severe AD (87), suggesting that dysregulation of calcium/calpain may be related to the progression of AD.
In summary, in the present study, we found that activation of calpain I probably led to truncation and activation of Dyrk1A, which phosphorylated Tau and promoted 3R-Tau expression, contributing to Tau pathogenesis and neurofibrillary pathology in AD. Further study of this newly identified mechanism will help in the development of novel therapeutic strategies to prevent or inhibit neurofibrillary pathology in AD and related tauopathies and thus treat these diseases.
Acknowledgments
We thank Dr. F. Chen (Nanjing Medical University) for statistical consultation, M. Marlow for editorial suggestions, and J. Murphy for secretarial assistance. We are also grateful to the Sun Health Research Institute Brain Donation Program (Sun City, AZ) for the provision of post-mortem human brain tissue.
This work was supported in part by Nantong University and the New York State Office of People with Developmental Disabilities and by grants from the National Natural Science Foundation of China (Grant 81030059), the United States Alzheimer's Association (Grant IIRG-10-173154), the Basic Research Program of the Jiangsu Education Department (Grant 10KJA310040), and the Priority Academic Program Development of Jiangsu Higher Education Institutions. The Brain Donation Program is supported in part by National Institutes of Health Grant P30 AG19610 (Arizona Alzheimer's Disease Core Center).
- AD
- Alzheimer disease
- NFT
- neurofibrillary tangle
- ALLN
- N-acetyl-Leu-Leu-Nle-CHO
- 3R- and 4R-Tau
- Tau isoform containing three and four microtubule-binding repeats, respectively
- DS
- Down syndrome
- GSK-3β
- glycogen synthase kinase-3β
- aa
- amino acids
- PEST
- peptide sequence that is rich in proline, glutamic acid, serine, and threonine
- FL
- full-length
- KA
- kainic acid.
References
- 1. Glenner G. G., Wong C. W., Quaranta V., Eanes E. D. (1984) The amyloid deposits in Alzheimer's disease: their nature and pathogenesis. Appl. Pathol. 2, 357–369 [PubMed] [Google Scholar]
- 2. Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y. C., Zaidi M. S., Wisniewski H. M. (1986) Microtubule-associated protein tau: a component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089 [PubMed] [Google Scholar]
- 3. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee V. M., Balin B. J., Otvos L., Jr., Trojanowski J. Q. (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678 [DOI] [PubMed] [Google Scholar]
- 5. Köpke E., Tung Y. C., Shaikh S., Alonso A. C., Iqbal K., Grundke-Iqbal I. (1993) Microtubule-associated protein tau: abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384 [PubMed] [Google Scholar]
- 6. Ksiezak-Reding H., Liu W. K., Yen S. H. (1992) Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 597, 209–219 [DOI] [PubMed] [Google Scholar]
- 7. Rodríguez-Martin T., Cuchillo-Ibáñez I., Noble W., Nyenya F., Anderton B. H., Hanger D. P. (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iqbal K., Grundke-Iqbal I., Zaidi T., Merz P. A., Wen G. Y., Shaikh S. S., Wisniewski H. M., Alafuzoff I., Winblad B. (1986) Defective brain microtubule assembly in Alzheimer's disease. Lancet 2, 421–426 [DOI] [PubMed] [Google Scholar]
- 9. Alonso A. C., Zaidi T., Grundke-Iqbal I., Iqbal K. (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 5562–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alonso A. C., Grundke-Iqbal I., Iqbal K. (1996) Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787 [DOI] [PubMed] [Google Scholar]
- 11. Alonso A. D., Zaidi T., Novak M., Barra H. S., Grundke-Iqbal I., Iqbal K. (2001) Interaction of tau isoforms with Alzheimer's disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J. Biol. Chem. 276, 37967–37973 [DOI] [PubMed] [Google Scholar]
- 12. Alonso A., Zaidi T., Novak M., Grundke-Iqbal I., Iqbal K. (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fath T., Eidenmüller J., Brandt R. (2002) Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J. Neurosci. 22, 9733–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson G. R., Wiedau-Pazos M., Sang T. K., Wagle N., Brown C. A., Massachi S., Geschwind D. H. (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34, 509–519 [DOI] [PubMed] [Google Scholar]
- 15. Lucas J. J., Hernández F., Gómez-Ramos P., Morán M. A., Hen R., Avila J. (2001) Decreased nuclear β-catenin, tau hyperphosphorylation, and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 20, 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pérez M., Hernández F., Gómez-Ramos A., Smith M., Perry G., Avila J. (2002) Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. Eur. J. Biochem. 269, 1484–1489 [DOI] [PubMed] [Google Scholar]
- 17. Alafuzoff I., Iqbal K., Friden H., Adolfsson R., Winblad B. (1987) Histopathological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol. 74, 209–225 [DOI] [PubMed] [Google Scholar]
- 18. Riley K. P., Snowdon D. A., Markesbery W. R. (2002) Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann. Neurol. 51, 567–577 [DOI] [PubMed] [Google Scholar]
- 19. Arriagada P. V., Marzloff K., Hyman B. T. (1992) Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology 42, 1681–1688 [DOI] [PubMed] [Google Scholar]
- 20. Andreadis A., Brown W. M., Kosik K. S. (1992) Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633 [DOI] [PubMed] [Google Scholar]
- 21. Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526 [DOI] [PubMed] [Google Scholar]
- 22. Goedert M., Wischik C. M., Crowther R. A., Walker J. E., Klug A. (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U.S.A. 85, 4051–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goedert M., Spillantini M. G., Potier M. C., Ulrich J., Crowther R. A. (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goode B. L., Chau M., Denis P. E., Feinstein S. C. (2000) Structural and functional differences between 3-repeat and 4-repeat tau isoforms: implications for normal tau function and the onset of neurodegenetative disease. J. Biol. Chem. 275, 38182–38189 [DOI] [PubMed] [Google Scholar]
- 25. Lu M., Kosik K. S. (2001) Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol. Biol. Cell 12, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh T. J., Grundke-Iqbal I., Wu W. Q., Chauhan V., Novak M., Kontzekova E., Iqbal K. (1997) Protein kinase C and calcium/calmodulin-dependent protein kinase II phosphorylate three-repeat and four-repeat tau isoforms at different rates. Mol. Cell. Biochem. 168, 141–148 [DOI] [PubMed] [Google Scholar]
- 27. Kosik K. S., Orecchio L. D., Bakalis S., Neve R. L. (1989) Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 28. Goedert M., Jakes R. (2005) Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250 [DOI] [PubMed] [Google Scholar]
- 29. Wisniewski K. E., Wisniewski H. M., Wen G. Y. (1985) Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann. Neurol. 17, 278–282 [DOI] [PubMed] [Google Scholar]
- 30. Kentrup H., Becker W., Heukelbach J., Wilmes A., Schürmann A., Huppertz C., Kainulainen H., Joost H. G. (1996) Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J. Biol. Chem. 271, 3488–3495 [DOI] [PubMed] [Google Scholar]
- 31. Woods Y. L., Rena G., Morrice N., Barthel A., Becker W., Guo S., Unterman T. G., Cohen P. (2001) The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 355, 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu F., Liang Z., Wegiel J., Hwang Y. W., Iqbal K., Grundke-Iqbal I., Ramakrishna N., Gong C. X. (2008) Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 22, 3224–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi J., Zhang T., Zhou C., Chohan M. O., Gu X., Wegiel J., Zhou J., Hwang Y. W., Iqbal K., Grundke-Iqbal I., Gong C. X., Liu F. (2008) Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 283, 28660–28669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qian W., Liang H., Shi J., Jin N., Grundke-Iqbal I., Iqbal K., Gong C. X., Liu F. (2011) Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 39, 6161–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wegiel J., Kuchna I., Nowicki K., Frackowiak J., Dowjat K., Silverman W. P., Reisberg B., DeLeon M., Wisniewski T., Adayev T., Chen-Hwang M. C., Hwang Y. W. (2004) Cell type- and brain structure-specific patterns of distribution of minibrain kinase in human brain. Brain Res. 1010, 69–80 [DOI] [PubMed] [Google Scholar]
- 36. Kimura R., Kamino K., Yamamoto M., Nuripa A., Kida T., Kazui H., Hashimoto R., Tanaka T., Kudo T., Yamagata H., Tabara Y., Miki T., Akatsu H., Kosaka K., Funakoshi E., Nishitomi K., Sakaguchi G., Kato A., Hattori H., Uema T., Takeda M. (2007) The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15–23 [DOI] [PubMed] [Google Scholar]
- 37. Ferrer I., Barrachina M., Puig B., Martínez de Lagrán M., Martí E., Avila J., Dierssen M. (2005) Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 20, 392–400 [DOI] [PubMed] [Google Scholar]
- 38. Ryu Y. S., Park S. Y., Jung M. S., Yoon S. H., Kwen M. Y., Lee S. Y., Choi S. H., Radnaabazar C., Kim M. K., Kim H., Kim K., Song W. J., Chung S. H. (2010) Dyrk1A-mediated phosphorylation of Presenilin 1: a functional link between Down syndrome and Alzheimer's disease. J. Neurochem. 115, 574–584 [DOI] [PubMed] [Google Scholar]
- 39. Ryoo S. R., Cho H. J., Lee H. W., Jeong H. K., Radnaabazar C., Kim Y. S., Kim M. J., Son M. Y., Seo H., Chung S. H., Song W. J. (2008) Dual-specificity tyrosine (Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer's disease. J. Neurochem. 104, 1333–1344 [DOI] [PubMed] [Google Scholar]
- 40. Goll D. E., Thompson V. F., Li H., Wei W., Cong J. (2003) The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 41. Grynspan F., Griffin W. B., Mohan P. S., Shea T. B., Nixon R. A. (1997) Calpains and calpastatin in SH-SY5Y neuroblastoma cells during retinoic acid-induced differentiation and neurite outgrowth: comparison with the human brain calpain system. J. Neurosci. Res. 48, 181–191 [PubMed] [Google Scholar]
- 42. Nixon R. A., Saito K. I., Grynspan F., Griffin W. R., Katayama S., Honda T., Mohan P. S., Shea T. B., Beermann M. (1994) Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease. Ann. N.Y. Acad. Sci. 747, 77–91 [DOI] [PubMed] [Google Scholar]
- 43. Veeranna, Kaji T., Boland B., Odrljin T., Mohan P., Basavarajappa B. S., Peterhoff C., Cataldo A., Rudnicki A., Amin N., Li B. S., Pant H. C., Hungund B. L., Arancio O., Nixon R. A. (2004) Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am. J. Pathol. 165, 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saito K., Elce J. S., Hamos J. E., Nixon R. A. (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 90, 2628–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu F., Grundke-Iqbal I., Iqbal K., Oda Y., Tomizawa K., Gong C. X. (2005) Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J. Biol. Chem. 280, 37755–37762 [DOI] [PubMed] [Google Scholar]
- 46. Lee M. S., Kwon Y. T., Li M., Peng J., Friedlander R. M., Tsai L. H. (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364 [DOI] [PubMed] [Google Scholar]
- 47. Yang L. S., Ksiezak-Reding H. (1995) Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur. J. Biochem. 233, 9–17 [DOI] [PubMed] [Google Scholar]
- 48. Liang Z., Liu F., Grundke-Iqbal I., Iqbal K., Gong C. X. (2007) Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J. Neurochem. 103, 2462–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu Q., Guo J., Zhou J. (2004) A minimal length between tau exon 10 and 11 is required for correct splicing of exon 10. J. Neurochem. 90, 164–172 [DOI] [PubMed] [Google Scholar]
- 50. Liu F., Iqbal K., Grundke-Iqbal I., Hart G. W., Gong C. X. (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 10804–10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu F., Iqbal K., Grundke-Iqbal I., Gong C. X. (2002) Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3β. FEBS Lett. 530, 209–214 [DOI] [PubMed] [Google Scholar]
- 52. Bol S. M., Moerland P. D., Limou S., van Remmerden Y., Coulonges C., van Manen D., Herbeck J. T., Fellay J., Sieberer M., Sietzema J. G., van 't Slot R., Martinson J., Zagury J. F., Schuitemaker H., van 't Wout A. B. (2011) Genome-wide association study identifies single nucleotide polymorphism in DYRK1A associated with replication of HIV-1 in monocyte-derived macrophages. PloS One 6, e17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adayev T., Chen-Hwang M. C., Murakami N., Wegiel J., Hwang Y. W. (2006) Kinetic properties of a MNB/DYRK1A mutant suitable for the elucidation of biochemical pathways. Biochemistry 45, 12011–12019 [DOI] [PubMed] [Google Scholar]
- 54. Simpkins K. L., Guttmann R. P., Dong Y., Chen Z., Sokol S., Neumar R. W., Lynch D. R. (2003) Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 23, 11322–11331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rechsteiner M., Rogers S. W. (1996) PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21, 267–271 [PubMed] [Google Scholar]
- 56. Tompa P., Buzder-Lantos P., Tantos A., Farkas A., Szilágyi A., Bánóczi Z., Hudecz F., Friedrich P. (2004) On the sequential determinants of calpain cleavage. J. Biol. Chem. 279, 20775–20785 [DOI] [PubMed] [Google Scholar]
- 57. Wu H. Y., Tomizawa K., Oda Y., Wei F. Y., Lu Y. F., Matsushita M., Li S. T., Moriwaki A., Matsui H. (2004) Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J. Biol. Chem. 279, 4929–4940 [DOI] [PubMed] [Google Scholar]
- 58. Hanes J., Zilka N., Bartkova M., Caletkova M., Dobrota D., Novak M. (2009) Rat tau proteome consists of six tau isoforms: implication for animal models of human tauopathies. J. Neurochem. 108, 1167–1176 [DOI] [PubMed] [Google Scholar]
- 59. LaFerla F. M. (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 3, 862–872 [DOI] [PubMed] [Google Scholar]
- 60. Iqbal K., Liu F., Gong C. X., Alonso Adel C., Grundke-Iqbal I. (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang J. Z., Liu F. (2008) Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 85, 148–175 [DOI] [PubMed] [Google Scholar]
- 62. Shukkur E. A., Shimohata A., Akagi T., Yu W., Yamaguchi M., Murayama M., Chui D., Takeuchi T., Amano K., Subramhanya K. H., Hashikawa T., Sago H., Epstein C. J., Takashima A., Yamakawa K. (2006) Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum. Mol. Genet. 15, 2752–2762 [DOI] [PubMed] [Google Scholar]
- 63. Ryoo S. R., Jeong H. K., Radnaabazar C., Yoo J. J., Cho H. J., Lee H. W., Kim I. S., Cheon Y. H., Ahn Y. S., Chung S. H., Song W. J. (2007) DYRK1A-mediated hyperphosphorylation of Tau: a functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 282, 34850–34857 [DOI] [PubMed] [Google Scholar]
- 64. Goedert M. (2005) Tau gene mutations and their effects. Mov. Disord. 20, S45–S52 [DOI] [PubMed] [Google Scholar]
- 65. Shi J., Qian W., Yin X., Iqbal K., Grundke-Iqbal I., Gu X., Ding F., Gong C. X., Liu F. (2011) Cyclic AMP-dependent protein kinase regulates the alternative splicing of tau exon 10: a mechanism involved in tau pathology of Alzheimer disease. J. Biol. Chem. 286, 14639–14648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. D'Souza I., Schellenberg G. D. (2006) Arginine/serine-rich protein interaction domain-dependent modulation of a tau exon 10 splicing enhancer: altered interactions and mechanisms for functionally antagonistic FTDP-17 mutations Δ280K and N279K. J. Biol. Chem. 281, 2460–2469 [DOI] [PubMed] [Google Scholar]
- 67. Møller R. S., Kübart S., Hoeltzenbein M., Heye B., Vogel I., Hansen C. P., Menzel C., Ullmann R., Tommerup N., Ropers H. H., Tümer Z., Kalscheuer V. M. (2008) Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 82, 1165–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dierssen M., de Lagran M. M. (2006) DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A): a gene with dosage effect during development and neurogenesis. Sci. World J. 6, 1911–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wiechmann S., Czajkowska H., de Graaf K., Grötzinger J., Joost H. G., Becker W. (2003) Unusual function of the activation loop in the protein kinase DYRK1A. Biochem. Biophys. Res. Commun. 302, 403–408 [DOI] [PubMed] [Google Scholar]
- 70. Adayev T., Chen-Hwang M. C., Murakami N., Lee E., Bolton D. C., Hwang Y. W. (2007) Dual-specificity tyrosine phosphorylation-regulated kinase 1A does not require tyrosine phosphorylation for activity in vitro. Biochemistry 46, 7614–7624 [DOI] [PubMed] [Google Scholar]
- 71. Alvarez M., Altafaj X., Aranda S., de la Luna S. (2007) DYRK1A autophosphorylation on serine residue 520 modulates its kinase activity via 14-3-3 binding. Mol. Biol. Cell 18, 1167–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Becker W., Sippl W. (2011) Activation, regulation, and inhibition of DYRK1A. FEBS J. 278, 246–256 [DOI] [PubMed] [Google Scholar]
- 73. Glading A., Bodnar R. J., Reynolds I. J., Shiraha H., Satish L., Potter D. A., Blair H. C., Wells A. (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol. 24, 2499–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zadran S., Jourdi H., Rostamiani K., Qin Q., Bi X., Baudry M. (2010) Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J. Neurosci. 30, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi D. W., Koh J. Y. (1998) Zinc and brain injury. Annu. Rev. Neurosci. 21, 347–375 [DOI] [PubMed] [Google Scholar]
- 76. Khachaturian Z. S. (1989) The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging (Milano) 1, 17–34 [DOI] [PubMed] [Google Scholar]
- 77. Mattson M. P., Rydel R. E. (1992) β-Amyloid precursor protein and Alzheimer's disease: the peptide plot thickens. Neurobiol. Aging 13, 617–621 [DOI] [PubMed] [Google Scholar]
- 78. Bezprozvanny I., Mattson M. P. (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu J., Kurup P., Zhang Y., Goebel-Goody S. M., Wu P. H., Hawasli A. H., Baum M. L., Bibb J. A., Lombroso P. J. (2009) Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 29, 9330–9343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Glatz D. C., Rujescu D., Tang Y., Berendt F. J., Hartmann A. M., Faltraco F., Rosenberg C., Hulette C., Jellinger K., Hampel H., Riederer P., Möller H. J., Andreadis A., Henkel K., Stamm S. (2006) The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2-BETA1 changes in sporadic Alzheimer's disease. J. Neurochem. 96, 635–644 [DOI] [PubMed] [Google Scholar]
- 81. Yasojima K., McGeer E. G., McGeer P. L. (1999) Tangled areas of Alzheimer brain have upregulated levels of exon 10 containing tau mRNA. Brain Res. 831, 301–305 [DOI] [PubMed] [Google Scholar]
- 82. Chambers C. B., Lee J. M., Troncoso J. C., Reich S., Muma N. A. (1999) Overexpression of four-repeat tau mRNA isoforms in progressive supranuclear palsy but not in Alzheimer's disease. Ann. Neurol. 46, 325–332 [DOI] [PubMed] [Google Scholar]
- 83. Boutajangout A., Boom A., Leroy K., Brion J. P. (2004) Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer's disease. FEBS Lett. 576, 183–189 [DOI] [PubMed] [Google Scholar]
- 84. Stan A. D., Ghose S., Gao X. M., Roberts R. C., Lewis-Amezcua K., Hatanpaa K. J., Tamminga C. A. (2006) Human postmortem tissue: what quality markers matter? Brain Res. 1123, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chevyreva I., Faull R. L., Green C. R., Nicholson L. F. (2008) Assessing RNA quality in postmortem human brain tissue. Exp. Mol. Pathol. 84, 71–77 [DOI] [PubMed] [Google Scholar]
- 86. Espinoza M., de Silva R., Dickson D. W., Davies P. (2008) Differential incorporation of tau isoforms in Alzheimer's disease. J. Alzheimers Dis. 14, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Reisberg B., Doody R., Stöffler A., Schmitt F., Ferris S., Möbius H. J., and Memantine Study Group (2003) Memantine in moderate-to-severe Alzheimer's disease. N. Engl. J. Med. 348, 1333–1341 [DOI] [PubMed] [Google Scholar]
- 88. Braak H., Braak E. (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278; discussion 278–284 [DOI] [PubMed] [Google Scholar]
- 89. Mirra S. S., Heyman A., McKeel D., Sumi S. M., Crain B. J., Brownlee L. M., Vogel F. S., Hughes J. P., van Belle G., Berg L. (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41, 479–486 [DOI] [PubMed] [Google Scholar]