

Key Points
Deficiency of TPP2 is associated with Evans syndrome and viral infection susceptibility.
TPP2 deficiency links premature immunosenescence of T and B cells with severe autoimmunity.
Abstract
Autoimmune cytopenia is a frequent manifestation of primary immunodeficiencies. Two siblings presented with Evans syndrome, viral infections, and progressive leukopenia. DNA available from one patient showed a homozygous frameshift mutation in tripeptidyl peptidase II (TPP2) abolishing protein expression. TPP2 is a serine exopeptidase involved in extralysosomal peptide degradation. Its deficiency in mice activates cell death programs and premature senescence. Similar to cells from naïve, uninfected TPP2-deficient mice, patient cells showed increased major histocompatibility complex I expression and most CD8+ T-cells had a senescent CCR7-CD127−CD28−CD57+ phenotype with poor proliferative responses and enhanced staurosporine-induced apoptosis. T-cells showed increased expression of the effector molecules perforin and interferon-γ with high expression of the transcription factor T-bet. Age-associated B-cells with a CD21− CD11c+ phenotype expressing T-bet were increased in humans and mice, combined with antinuclear antibodies. Moreover, markers of senescence were also present in human and murine TPP2-deficient fibroblasts. Telomere lengths were normal in patient fibroblasts and granulocytes, and low normal in lymphocytes, which were compatible with activation of stress-induced rather than replicative senescence programs. TPP2 deficiency is the first primary immunodeficiency linking premature immunosenescence to severe autoimmunity. Determination of senescent lymphocytes should be part of the diagnostic evaluation of children with refractory multilineage cytopenias.
Introduction
Evans syndrome is defined by the simultaneous or sequential development of immune thrombocytopenic purpura and autoimmune hemolytic anemia.1 In about 50% of cases, it is associated with systemic autoimmune disease, such as systemic lupus erythematosus, lymphoproliferative disease, or primary immunodeficiencies.2 In this latter group of diseases, the variety of predisposing genetic defects illustrates the multiple checkpoints that can be affected in the loss of immunologic tolerance.3 However, despite the increased molecular understanding, the question whether a genetic predisposition contributes to the autoimmune cytopenia remains unresolved for most patients.4
Immunosenescence is one pathomechanism that has been associated with autoimmunity.5 For T cells, age-associated skewing of the antigen-receptor repertoire related to decreased thymic output and homeostatic proliferation of potentially autoreactive clones,6 and age-associated alterations in the antigen-receptor signaling network,7 have been put forward as potential explanations. For B cells, a decline of B-cell generation in bone marrow with age and shifts in naïve and antigen-experienced peripheral B-cell subsets could be linked to autoimmunity.8 Premature immunosenescence can occur as a consequence of chronic immune stimulation, such as persistent viral infections.9 In addition, genetic factors favoring premature differentiation and/or persistence of senescent immune cells could be a predisposing factor for autoimmunity, even in the absence of persistent infections.
Tripeptidylpeptidase II (TPP2) is a molecule that has been previously linked to immunosenescence. TPP2 is a cellular protease that operates mostly downstream of proteasomes in cytosolic proteolysis.10-12 It is important for cell proliferation and survival, in particular under conditions of cellular stress,13,14 and may contribute to an antiapoptotic phenotype.14 In mice, lack of TPP2 activates cell death programs leading to proliferative apoptosis in T cells and premature senescence, particularly of CD8+ T cells. In addition, murine TPP2 deficiency also causes premature senescence in fibroblasts and degenerative alterations at the level of the entire organism.15 However, despite their immunologic alterations, no autoimmunity or immunodeficiency phenotype been described to date in TPP2-deficient mice.
Here, we report 2 siblings with early-onset Evans syndrome, variable lymphoproliferation, and mild infection susceptibility, who both had loss-of-function mutations in the gene encoding TPP2. Immunologic studies in 1 of the patients were compared with those obtained in naïve uninfected TPP2-deficient mice in an attempt to differentiate primary consequences of TPP2 deficiency from those of the infections. Our results document that premature senescence in human TPP2 deficiency also affects B cells in addition to CD8+ T cells and fibroblasts, and it is associated with autoimmunity and immunodeficiency.
Patients and methods
Two siblings with early onset Evans syndrome and variable infection susceptibility
The index patient (P1), a boy, who is the second child of consanguineous Palestinian parents, presented at the age of 21 months with Coombs-positive autoimmune hemolytic anemia and immune thrombocytopenia, cervical and axillary lymphadenopathy, and mild-to-moderate intermittent splenomegaly (supplemental Table 1, available on the Blood Web site). He was initially responsive to steroids and IVIG, but remained steroid-dependent and developed recurrent episodes of severe cytopenia, despite treatment with cyclosporine, mycophenolate mofetil, several courses of rituximab, and more than 6 months on sirolimus. Although on immunosuppressive therapy, P1 developed disseminated and prolonged cutaneous chickenpox. He had repeated low-level cytomegalovirus (CMV) viremia without clinical sequelae, which could be controlled with ganciclovir and foscavir. At 10 years of age, he developed numerous flat hypopigmented 1 to 2 mm papular lesions on the face (supplemental Figure 1). Polymerase chain reaction was positive for human papillomavirus (HPV) type 15. Otherwise, his infection history was unremarkable. Mild developmental delay with significant verbal and comprehension impairment was diagnosed and education for children with special needs was recommended. Due to the refractory course of Evans syndrome, he recently underwent hematopoietic stem cell transplantation (HSCT) from an HLA-identical healthy sibling donor after conditioning with fludarabine, targeted low levels of busulfan, thiotepa, and antithymocyte globuline. In October 2014, 6 months after HSCT, the child remains well with 97% donor cell chimerism and no infectious, autoimmune or lymphoproliferative manifestations. He is no longer prescribed any immunosuppressive medication. Thus, at this early time point, the clinical features that led to the decision to transplant have resolved.
An older sibling (S1), a girl, had been followed in another hospital from the age of 18 months when she presented with immune thrombocytopenia followed by Coombs-positive hemolytic anemia 5 months later. She had some lymphadenopathy, but no splenomegaly. She was treated with IVIG and steroids, but became increasingly unresponsive to therapy that included splenectomy at 33 months of age. She developed a left hemispheric ischemic event leading to right-sided hemiparesis at 30 months. At 37 months, she died in the context of an acute hemolytic crisis. She had normal psychomotor development and no increased infection susceptibility. Limited laboratory information and no DNA were available from this sibling.
P1 intermittently had significantly increased IgG and IgM and increased IgG was also documented in S1. IgA and IgE were normal (supplemental Table 1). At age 11 years, P1 had seroconverted to CMV, Epstein-Barr virus, and Varicella zoster virus, and had detectable tetanus antibodies. P1 and S1 were intermittently positive for antinuclear antibodies (scored 1-2 on a scale from 0-4). P1 had normal lymphocyte counts and subpopulations at 21 months. At 10 years, P1 had mild leukopenia and lymphopenia with a reduction in the fraction of naïve CD4 T-cells and low B-cell counts (3 years after rituximab treatment). He had normal levels of serum vitamin B12 and soluble Fas Ligand, no elevation of CD3+ T-cell receptor αβ+CD4−CD8−-double negative T-cells, a moderate elevation of γδT-cells (18%), and a normal proportion of CD4+CD25+FOXP3+ regulatory T-cells.
Five patients with FAS mutations (autoimmune lymphoproliferative syndrome [ALPS-FAS]; ages 6-32 years) and 9 patients with autoimmune cytopenia and lymphoproliferation in the context of the clinical diagnosis common variable immunodeficiency (“CVID”) (http://esid.org/Working-Parties/Registry/Diagnosis-criteria), aged 7 to 17 years were recruited as disease control groups for analysis of senescent T- and B-cells. Two of the CVID patients had biallelic LRBA mutations, 2 had activating mutations in PIK3CD, and 1 had a heterozygous CTLA4 mutation, whereas the remaining 4 patients had defined genetic diagnoses that have not been established yet. Two siblings with autosomal-dominant TERC mutation and clinical dyskeratosis congenita (DKC) served as controls for telomere lengths determination.
Patient and parental consent were given for this study. The study was performed with the approvals of the ethical committees of Hadassah Medical Center and the Ministry of Health and of Freiburg University Medical Center, and in accordance with the Declaration of Helsinki.
Mice
Mice rendered deficient in TPP2 by gene targeting have been previously described.15,16 Mice were analyzed between 6 and 12 months of age. All mouse experiments were approved by the Regierungspräsidium Freiburg (G-06/34).
All experimental procedures are described in the supplemental Methods.
Results
A homozygous frameshift mutation in TPP2 segregates with disease phenotype
To elucidate the presumed genetic basis of the disease in the 2 siblings with early-onset Evans syndrome and variable susceptibility to viral infections, whole exome sequencing was performed using DNA extracted from the whole blood of P1, a healthy sister and both parents. The patient exome was well covered with an average depth of ×326 (94.5% of the exons were covered > ×10 and 92.8% were covered > ×20). Aligning the patient, 196.94 million reads to the reference human genome revealed 261 516 variants. We removed variants, which were low in depth (< ×8), deep intronic, heterozygous, and those present in the database of single nucleotide polymorphisms build 132 (dbSNP132) or in the in-house database of single nucleotide polymorphisms. Seven variants survived this filtering (supplemental Table 3) but only 1 was predicted to be pathogenic by MutationTaster evaluation.17 This was a homozygous frameshift mutation, chr13: 10326878 c.432delG (p.Ala145Profs×25) in the gene encoding TPP2 (Figure 1A). The frameshift started at Ala145, generating a premature stop codon 25 residues later. The mutation segregated with the disease in the family and was absent from the 6503 healthy individuals whose exome analysis results were available through the Exome Variant Server, National Heart Lung and Blood Institute Exome Sequencing Project (Seattle, WA) (http://evs.gs.washington.edu/EVS/; EVS-v.0.0.21). No mutations were detected in the genes encoding PRKCD, PIK3CD, STK4, CD95, or CD95L. Western blot analysis using an anti-TPP2 antibody recognizing amino acids 165 to 303 failed to detect protein (Figure 1B). In the course of the study, 22 children with autoimmune cytopenia and moderate lymphoproliferation in the absence of FAS mutations were screened and no additional patients with mutations in TPP2 were identified.
Figure 1.
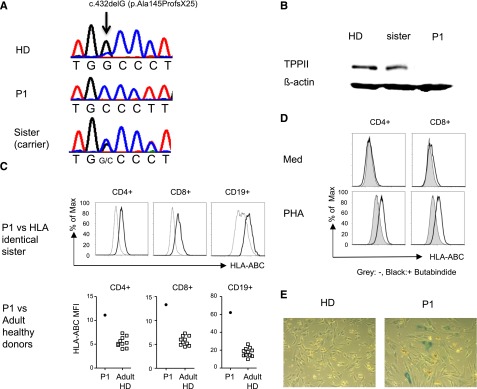
Human TPP2 deficiency reproduces features of the murine knockout phenotype. (A) Illustration of the single nucleotide deletion at position c.432delG of the TPP2 coding sequence in the index patient (P1) and his asymptomatic sister. (B) Western blot analysis of lysates from Epstein Barr virus (EBV) lines of P1, the healthy heterozygous sister, and a healthy donor (HD) incubated with anti-TPP2 antibodies (upper panel) and anti-actin antibodies as a loading control (lower panel). (C) MHC class I expression on fresh T and B cells of P1 compared with his HLA identical healthy heterozygous sister or 10 adult controls (adult HD). (D) Upregulation of MHC class I expression on control CD4 or CD8 T cells after phytohemagglutinin (PHA) stimulation in the absence (gray) or presence (black) of the TPP2 inhibitor butabindide. (E) β-galactosidase activity of primary fibroblasts isolated from skin biopsies of the patient (P1) or a healthy donor (HD). Experiments (C-E) were performed 3 times with similar results. ABCs, age-associated B cells; Max, maximum; Med, medium; TPPII, tripeptidyl peptidase II.
TPP2-deficient mice and humans share features of immune dysregulation and premature fibroblast senescence
Because we only had access to blood from a single patient, we asked whether the main clinical features of the 2 siblings could also be observed in mice with an inactivated TPP2 gene.15,16 Lymphadenopathy and/or splenomegaly were inconsistently observed in about 50% of the mice used in this study (data not shown). Two of 16 mice developed antinuclear antibodies, and 8 had anti-nucleolar or anti-cytoplasmatic antibodies (supplemental Figure 3A). Hypergammaglobulinemia was not observed. Older mice had leukopenia (supplemental Figure 3B), as previously reported. Additional characteristic features previously reported in these mice included increased major histocompatibility complex (MHC) class I expression and premature senescence of fibroblasts.15,16 Similar to the murine findings,16 expression of MHC class I on T and B cells was higher in the patient compared with his HLA-identical healthy heterozygous sister (Figure 1C) and a group of 10 healthy adult controls. Pretreatment of control T cells with the TPP2 inhibitor butabindide18,19 enhanced MHC I upregulation upon phytohemagglutinin stimulation (Figure 1D). Finally, as described in murine cells, fibroblasts of P1 also showed increased staining with β-galactosidase indicating cellular senescence (Figure 1E).
TPP2 deficiency leads to premature T-cell senescence
Encouraged by these phenotypic similarities, we performed a parallel immunologic analysis of patient and TPP2-deficient murine lymphocytes to further understand the basis of this genetic disorder. Because TPP2-deficient mice have increased CD44hi memory CD8+ T cells, we carefully analyzed T-cell differentiation using established surface markers for human (CCR7 and CD45RA20) and murine T cells (CD62L and CD44). More than 90% of CD8+ T-cells from P1 had an effector memory or T-effector memory cells reexpressing CD45RA phenotype (Figure 2A-B). A similar effector skewing was observed in naïve TPP2-deficient mice (Figure 2B). Advanced differentiation was also observed among patient and murine CD4+ T-cells (Figure 2A-B). Further analysis of patient CD8+ T cells revealed a predominant CD27−CD28−CD127− phenotype that was not present among CD4+ T cells (Figure 2C). Terminal differentiation of the majority of CD8+ T cells was further reflected by expression of CD57 (Figure 2C-D). Unexpectedly, these cells were largely negative for killer cell lectinlike receptor G1 (Figure 2C), which is usually expressed on CD57+CD8+ T cells. The proportion of CD57+CD8+ T cells in P1 was higher than in ALPS patients, whereas pediatric patients with autoimmune cytopenia and lymphoproliferation fulfilling the clinical criteria for CVID, including patients with activated PI3K δ syndrome, LRBA, and cytotoxic T-lymphocyte–associated protein 4 deficiency showed variable results (Figure 2D). There was also an increased fraction of CD127− CD8+ T cells in naïve TPP2-deficient mice (Figure 2E), indicating that this enhanced differentiation state was not only a consequence of the infection history of the patient. However, in contrast to the patient, most of these murine cells showed increased KLRG1 expression as usually observed for senescent T cells (Figure 2E).
Figure 2.

Advanced differentiation of TPP2-deficient T cells. (A) Left panel: peripheral blood mononuclear cells (PBMC) of the patient (P1, age 11 years) and his asymptomatic heterozygous sister (age 7 years) were analyzed for expression of the differentiation markers CCR7 and CD45RA (gated on CD3+CD4+ and CD3+CD8+ T cells, respectively). Right panel (human PBMC): Proportions of naïve (CCR7+CD45RA+, naive), central memory (CM) (CCR7+CD45RA−), effector memory (EM) (CCR7−CD45RA−), and terminal differentiated T-effector memory cells reexpressing CD45RA (TEMRA) (CCR7−CD45RA+) among CD4+ or CD8+ T cells of the patient (P1) compared with adult healthy donors (HD) (n = 20). (B) Left panel: Splenocytes of wild-type (WT) and TTP2-deficient knockout mice (KO) were analyzed for CD62L and CD44 expression (gated on CD3+CD4+ and CD3+CD8+ T cells). Right panel (mouse splenocytes): Proportions of naïve (CD44−CD62L+), CM (CD44+CD62L+), EM (CD44+CD62L−), and double-negative (DN) (CD44−CD62L−) cells were determined among CD4+ or CD8+ T cells of 6 to 12 months old TPP2 deficient knockout mice (KO) (n = 16) compared with age-matched WT mice (n = 7). (C) Expression of the indicated activation/differentiation markers on CD4 and CD8 T cells of the patient (P1) and his healthy heterozygous sister. (D) Percentage of CD57 expressing CD8 T cells in the patient (P1) compared with adult healthy controls, FAS mutant patients with ALPS (pediatric and adult), and pediatric patients with autoimmune cytopenia and lymphoproliferation in the context of CVID. Of these, CVID patients, 2 had LRBA deficiency (diamonds), 2 had activating PIK3CD mutations (squares), and 1 had a CTLA4 mutation (circle). (E) Expression of KLRG1 and CD127 on CD8 T cells of TPP2-deficient (KO) and age-matched WT mice.
While T-bet expression in CD57+CD8+ T cells of P1 was high as expected for terminally differentiated T cells, the expression of Eomesodermin (Eomes) was reduced and lower compared with CD57+CD8+ T cells from control donors (Figure 3A). The advanced T-cell differentiation was associated with an enhanced effector state. Thus, in contrast to controls, almost all CD8+ T cells and also a relevant fraction of CD4+ T cells expressed perforin (Figure 3B). Furthermore, the fraction of cells responding to phorbol 12-myristate 13-acetate/ionomycin stimulation with expression of interferon (IFN)-γ was enhanced among both patient and murine TPP2-deficient CD4+ and CD8+ T cells (Figure 3C-D).
Figure 3.
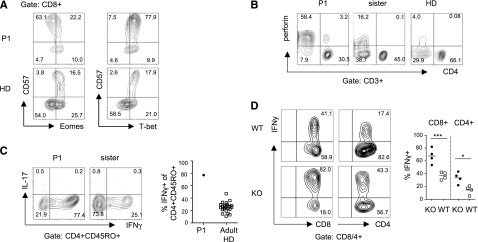
Enhanced effector functions in TPP2 deficient T cells. (A) Expression of the transcription factors Eomesodermin (Eomes) and T-bet in CD8+ T cells of the patient (P1) and a healthy adult control (healthy donor [HD]). (B) Perforin expression in CD3+CD4+ and CD3+CD4− T cells of the patient (P1), his healthy heterozygous sister, and an adult HD control. (C) Intracellular IFN-γ and IL-17 expression in CD4+CD45RO+ T cells after stimulation with phorbol 12-myristate 13-acetate/ionomycin. The percentage of IFN-γ positive T cells in the patient was compared with a group of adult HDs (n = 24). (D) Intracellular IFN-γ of splenic CD3+CD4+ and CD3+CD8+ T cells from TPP2-deficient knockout (KO) (n = 4) and control WT (n = 4) mice. The experiment was repeated twice with similar results.
To assess whether the increased CD8 T-cell differentiation was linked to the enhanced MHC I expression associated with TPP2 deficiency, we compared EBV lines or PBMC from the patient and his heterozygous healthy HLA-identical sister in their ability to induce proliferation of allogeneic healthy donor responder T cells. However, no differences could be detected (supplemental Figure 4). Analysis of the T-cell repertoire revealed significant skewing among CD8+, but not among CD4+ T cells in the patient (supplemental Figure 2A). In contrast, TPP2-deficient mice had a normal polyclonal T-cell repertoire, suggesting that the oligoclonal expansions in the patient were not a direct consequence of the genetic defect (supplemental Figure 2B).
TPP2 deficiency reduces T-cell proliferation and lymphocyte survival
Previous murine experiments had indicated reduced proliferation associated with enhanced activation induced cell death in TPP2-deficient T cells.15 The proliferative response of patient cells to stimulation with anti-CD3/28 coated beads or with phytohemagglutinin was reduced, but it was present for CD4+ T cells, whereas no response could be induced in CD8+ T cells (Figure 4A). Moreover, proliferation of control T cells was diminished after pretreatment with butabindide (Figure 4B). Due to the poor proliferative response, we could not generate enough activated human T cells to study restimulation-induced cell death. Therefore, we analyzed apoptosis induction in EBV lines from the patient, his heterozygous sister, a healthy donor, and a patient with autoimmune lymphoproliferative syndrome caused by a germline FAS mutation (ALPS-FAS). Although irradiation or stimulation with anti-Fas antibody or etoposide induced apoptosis in patient cells to a similar extent as in controls, patient EBV lines were more susceptible to staurosporine-induced cell death (Figure 4C). Pretreatment of control EBV lines with butabindide also enhanced the apoptosis response to staurosporine (Figure 4D). In combination with previous reports in mice, these results suggest that TPP2 deficiency confers a lymphocyte proliferation and survival defect.
Figure 4.

Defective proliferation and enhanced susceptibility to apoptosis. (A) PBMC of the patient (P1) or his healthy heterozygous sister were left untreated or stimulated with PHA or anti-CD3/CD28 beads and carboxyfluorescein diacetate succinimidyl ester (CSFE) dilution of CD4+ or CD8+ T cells was determined after 5 days incubation (unstimulated: gray; stimulated: black). The experiment was performed 3 times with similar results. (B) Control PBMC were labeled with CFSE and stimulated with phytohemagglutinin (PHA) or anti-CD3/CD28 beads in the absence (gray line) or presence (black line) of the TPP2 inhibitor butabindide. CFSE dilution of CD4+ and CD8+ T cells was analyzed after 5 days. The experiment was repeated 3 times with similar results. (C) EBV lines of the patient (P1), his healthy heterozygous sister, a healthy donor (HD) control and a FAS-mutant ALPS patient were stimulated with increasing concentrations of anti-Fas antibody cross-linked with protein A, the topoisomerase inhibitor etoposide (eto), the protein kinase inhibitor staurosporine (sts), or were sublethally irradiated (30 Gy). Viable Annexin V/PI negative cells were determined by flow cytometry after 24 hours or on day 0, 2, and 4, respectively. Results are representative of 3 independent experiments with similar results. (D) Cells of an EBV line from a healthy donor were stimulated with increasing concentrations of staurosporine in the absence (filled squares) or presence (open squares) of the TPP2 inhibitor butabindide and viable cells were determined after 24 hours. The experiment was repeated twice with similar results.
TPP2 deficiency leads to premature senescence of B cells
Analysis of the B cell compartment was performed 3 years after rituximab treatment. There was a mild relative reduction of IgD+CD27+ marginal-zone like (5.3%) and IgD-CD27+ class-switched memory B cells (2.7%), and a moderate increase in the percentage of CD38+ IgM+ transitional B cells (18%). Most prominent was an elevated fraction of CD21low cells, which have been characterized as a preactivated, functionally attenuated, and autoreactive population in patients with CVID or autoimmune diseases21,22 (Figure 5A). Recently, a related population of CD11c+ age-associated B cells (ABCs) has been described that accumulate in aged humans and mice23,24 and has been associated with autoimmunity.24,25 Indeed, most CD21low B cells of P1 expressed CD11c (Figure 5B) and the transcription factor T-bet. This population was not observed in healthy adult donors or patients with ALPS, but in a proportion of pediatric patients with autoimmunity and lymphoproliferation fulfilling the clinical criteria for CVID. Of note, among the few disease control patients with a molecular diagnosis including activating PI3K δ syndrome, LRBA, or CLTA-4 deficiency, some had elevated ABCs and some had elevated CD57+ CD8+ T cells without obvious correlation. Importantly, we also observed a significant increase in the fraction of CD11c+CD11b+ B cells in TPP2-deficient mice, suggesting that the relative abundance of this B-cell population resulted from the genetic defect and was not induced by the particular medical history of the patient (Figure 5C).
Figure 5.

Abnormal B-cell differentiation in TPP2 deficiency. (A-B) Human CD19+ B cells were analyzed for expression of differentiation and senescence markers. The proportion of CD11c+CD21− ABCs was compared between the patient (P1), 8 adult controls (HD), 5 untreated ALPS-FAS patients, and pediatric patients with autoimmune cytopenia and lymphoproliferation in the context of CVID. Patient B cells were analyzed for T-bet expression. (C) The proportions of CD11b+ and CD11b/c+ double-positive splenic CD19+ B cells were determined in 6- to 12-month-old TPP2 knockout (KO) and WT mice (n = 4). The experiment was repeated twice with similar results.
TPP2 deficiency does not affect telomere integrity
To assess whether TPP2 deficiency affects telomere integrity, we analyzed telomere lengths using flow fluorescence in situ hybridization technology in PBMC from P1, his healthy sister, and 2 patients with DKC as a positive control. Although lymphocyte telomere length of the sibling was at the 50th percentile of healthy children, patient lymphocytes showed a telomere length below the 25th percentile (Figure 6A-C). This is much higher than what can be observed in DKC patients. Considering that patient lymphocytes contained 40% of CD57+CD8+ T cells, this is an expected result and does not indicate impaired telomere maintenance. In addition, both siblings had similar telomere lengths between the 25th and 50th percentile in granulocytes (Figure 6D), and further analysis of fibroblasts revealed even longer telomere lengths (data not shown). These data suggest that TPP2 deficiency is not associated with a general enhancement of replicative senescence.
Figure 6.
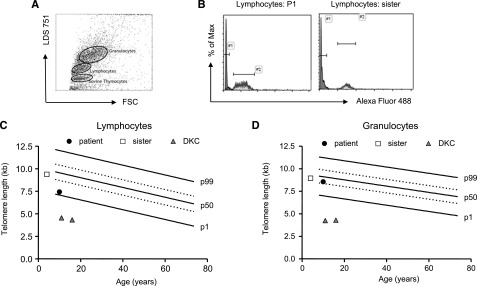
Telomere lengths of TPP2-deficient lymphocytes and granuloyctes. (A) Exemplary fluorescence-activated cell sorter plot of the flow- fluorescence in situ hybridization analysis: human lymphocytes, granulocytes, and cow thymocytes were discriminated by forward scatter (FSC) and LDS 751 staining. (B) Telomere lengths were determined by subtracting telomere-Alexa-Fluor 488 intensity of stained from the unstained lymphocytes of the patient (P1) or the healthy carrier sister. Cow thymocytes with known telomere length were used as an internal control and to calculate telomere lengths in kilobases. Absolute telomere lengths of lymphocytes (C) and granulocytes (D) of the patient, his sister, and 2 patients with DKC are shown in the context of age-dependent percentiles (black lines: 99th, 50th, and 1st percentile, dotted lines: 75th and 25th percentile).
Discussion
We detected biallelic TPP2 mutations leading to absent protein expression in a child presenting with early-onset Evans syndrome. The following arguments suggest a causal relationship between this genetic defect and the clinical phenotype: (1) Features of cellular immunosenescence in patient T and B cells including phenotypic alterations, defects in apoptosis, and proliferation and functional effector skewing were also observed in TPP2-deficient mice. In addition to published reports, this was supported by parallel experimentation with human and murine cells in this study. (2) The reduced proliferation, increased apoptosis, and increased MHC class I expression of patient cells could be reproduced by treating control cells with the TPP2 inhibitor butabindine. (3) Similar to P1 and the possibly affected sibling, TPP2-deficient mice show an increased incidence of autoantibodies. (4) TPP2-deficient mouse and patient fibroblasts show premature senescence. (5) A recent congress abstract described a similar phenotype of autoimmune cytopenia, infection susceptibility and developmental delay in 2 siblings with TPP2 deficiency and premature fibroblast and CD8 T-cell senescence.26
Current understanding of the function of TPP2 in vivo is mainly based on observations in TPP2-deficient mice. Previous studies have documented an immunosenescence phenotype associated with enhanced cellular death programs. The mice showed age-related lymphopenia associated with enhanced apoptosis of immature thymocytes and accelerated thymic involution. In addition, proliferative apoptosis of peripheral T cells was increased.15 The T-cell phenotype was skewed toward memory T cells and anti-CD3 stimulated T cells stained positive for acidic β-galactosidase, further indicating premature senescence. An increased basal activity of nuclear factor κB, attenuated nuclear factor κB activation, and increased p53 expression in TPP2-deficient T cells further supported cellular senescence. Moreover, MHC class I expression was increased,16 as is observed in age-associated inflammation.27 Clinically, TPP2-deficient mice showed variable splenomegaly and/or lymphadenopathy associated with extramedullary hematopoiesis.
The knockout mice also indicated that TPP2 is not only relevant in the hematopoietic system.15,16 Thus, premature senescence of fibroblasts was associated with aged appearance, reduced body weight, and premature death of elderly mice.15 Despite the immunologic alterations, there was only a minor phenotype after infection with lymphocytic choriomeningitis virus. The mice showed a minimal reduction in the number of virus-specific T cells, but cytotoxicity was normal and virus elimination was unimpaired (Firat et al16 and Peter Aichele, Institute of Immunology, University of Freiburg, oral communication).
Based on these previous findings, it was highly surprising that human TPP2 deficiency was observed in a patient with a life-threatening immunodeficiency. Nevertheless, the underlying cellular phenotype of our patient well reflected the immunosenescence observed in mice. In CD8+ T cells, we observed a highly differentiated CCR7−CD45RA+CD27−CD28− and CD57+ phenotype.28 Such characteristics of senescent cells can also be induced by repeated proliferation induced by persistent viruses such as CMV.29 However, even though our patient had intermittent low-level CMV reactivation, the extent of accumulation of senescent CD8 T cells was very unusual for his age. Moreover, we confirmed this phenotype in the absence of infection in TPP2-deficient mice, extending previous observations.15 The dominance of the transcription factor T-bet relative to the expression of Eomes could be related to the advanced T-cell differentiation. T-bet drives T-cell differentiation with enhanced effector activity, whereas Eomes expression is linked to memory cell formation with high proliferative capacity and self-renewal.30 Both patient and murine cells showed an excessive IFN-γ response and patient T cells highly expressed perforin. It is conceivable that this enhanced IFN-γ production contributes to the increased MHC class I expression that was observed in the patient as in the TPP2-deficient mice,16 and this can also be seen in aged individuals.27 Reduced proliferation is also in line with T-cell senescence.15
Confirming murine observations, patient fibroblasts also showed features of premature senescence. Moreover, the mild developmental delay observed in our patient was also reported by Hambleton et al.26 TPP2 deficiency thus appears to be a syndromic disease that is not restricted to the immune system. However, although the further course remains to be evaluated, in our patient, at the age of 12 years, none of the potential extrahematopoietic manifestations were considered severe enough to represent a contraindication for HSCT.
It is a new finding that TPP2 deficiency causes increased infection susceptibility. Although a not further specified broad susceptibility to infections from early childhood was noted in the patients reported by Hambleton et al,26 by the age of 12 years, our patient had a history of widespread cutaneous varicella infection, intermittent CMV viremia, and cutaneous HPV infection. In part, these infections may be explained by immunosuppressive therapy. However, extensive and recalcitrant HPV infection is unusual. In particular, β-HPVs, such as genotype 15 detected in our patient. are weakly virulent and unusual in healthy individuals,31 even under immunosuppressive therapy, although they are characteristic for epidermodysplasia verruciformis ([EV] OMIM 226400), a rare genodermatosis. Infection by EV-specific β-HPV genotypes has also been demonstrated in 2 autosomal recessive disorders affecting T-cell immunity; RHOH deficiency,32 and MST1 deficiency.33 TPP2 deficiency thus represents a further T-cell deficiency predisposing to EV-specific HPVs.
It was even more unexpected, that TPP2 deficiency is associated with autoantibody-mediated autoimmunity, manifesting as severe Evans syndrome leading to a transplant indication in P1 and an early death in S1. Autoimmune cytopenia in 2 patients and autoimmune hepatitis in 1 was also reported by Hambleton et al.26 We could further document that most TPP2-deficient mice developed antinucleolar, anticytoplasmic, or antinuclear antibodies, which were also detected in P1 and S1. Aging is associated with increased autoantibody formation.34 A subset of B cells termed ABCs accumulating in the elderly has recently been characterized in humans and mice by the expression of CD11b, CD11c, and T-bet.23,24 Interestingly, these ABCs appear earlier in autoimmune prone mice and are found in humans suffering from autoimmunity,24 including patients with common variable immunodeficiency.21,22,25 Their depletion in mice leads to a reduction in autoantibodies. We found ABCs in the 12-year-old patient and in TPP2-deficient mice, illustrating premature immunosenescence also in the B-cell compartment and providing a potential link to the observed autoantibody-mediated disease in TPP2 deficiency.
What are the molecular mechanisms leading to immunosenescence in the absence of TPP2? In principle, 2 forms of cellular senescence can be differentiated. Whereas replicative senescence is associated with shortened telomeres, stress-induced senescence is associated with DNA damage, activated stress pathways, or a disbalance of proliferation-associated transcription factors.35 The observation of normal telomere length in patient fibroblasts and the expected shorter telomeres in predominantly senescent lymphocytes indicates that TPP2 is not required for general maintenance of telomere integrity such as in dyskeratosis congenita. In fact, previous observations support that TPP2 rather protects cells under conditions of cellular stress.10 As a peptidase, TPP2 may have an impact on the intracellular concentration of short peptides modulating protein-protein interactions or a direct role in signaling by targeting natural substrates.36 One conceivable target is the Akt/mTOR pathway. Its activation has previously been linked to senescence associated β-galactosidase activity in human fibroblasts37 and to CD8 T-cell senescence.38 In fact, recent observations in patients with gain-of-function mutations in the PI3K catalytic subunit or loss of the regulatory subunit p85 suggest that activation of this pathway may be associated with immunodeficiency and autoimmunity in human patients.38-40 We found slightly enhanced S6 phosphorylation in patient T cells (data not shown), but could not reproduce this in murine T cells. The lack of a clinical response to sirolimus in our patient may further suggest that this is not a major pathway linking TPP2 deficiency to autoimmunity and immunodeficiency. Thus, the molecular link from TPP2 deficiency to premature cellular senescence remains elusive.
In summary, in combination with the findings of Hambleton et al, our observations in a single patient and the corresponding gene-inactivated mice identify TPP2 deficiency as a new syndromal primary immunodeficiency leading to an increased susceptibility to viruses and autoimmunity. It is the first primary immunodeficiency clearly linking premature senescence with autoimmunity. The combination of an accumulation of senescent cells, autoimmunity, and immunodeficiency appear to be recurrent themes in pediatric patients with a number of recently described diseases including CTLA4 deficiency,41,42 activated PI3K δ syndrome, PI3K p85 deficiency, or LRBA deficiency.43 Although not specific for a particular genetic condition, the determination of senescent T and B cells should be part of the diagnostic evaluation of any child with refractory multilineage cytopenias.
Acknowledgments
The authors thank Ursula Warthorst for excellent technical assistance; Odeya Ehrilch, Dalia Bassa, and the team at the Pediatric Hematology-Oncology and Bone Marrow Transplant (BMT) Department for their treatment of the patient; and the patient’s family for their trust and support.
This work was supported by the German Federal Ministry of Education and Research (BMBF 01EO1303 grant to the Center for Chronic Immunodeficiency and BMBF 01GM1111B grant to the PID-Net Initiative) and the DFG (TRR130 to H.E. and Eh145-6); and P.S. was supported by a grant from the Joint Research Fund of the Hebrew University and the Hadassah Hebrew University Hospitals.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.S. identified the patient, provided all clinical information, and revised the paper; A.R.-E. performed most human experiments and wrote the paper; R.G., E.F., S.Z., H.E., and G.N. performed mouse experiments; S.R.-V. analyzed skin lesions; U.F. and S.N. performed protein analysis; S.F., M.R., B.K., and M.R. assisted with human lymphocyte analysis; K.W. advised on B-cell analysis and revised the manuscript; O.E., V.M.P., and A.B. performed genetic and bioinformatics analysis; F.B. and T.H.B. analyzed telomere lengths; S.E. coordinated the research; and A.R.-E. and S.E. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stephan Ehl, Center for Chronic Immunodeficiency, Breisacher Strasse 117, Freiburg, 79106 Germany; e-mail: stephan.ehl@uniklinik-freiburg.de.
References
- 1.Wang WC. Evans syndrome in childhood: pathophysiology, clinical course, and treatment. Am J Pediatr Hematol Oncol. 1988;10(4):330–338. doi: 10.1097/00043426-198824000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Price V. Auto-immune lymphoproliferative disorder and other secondary immune thrombocytopenias in childhood. Pediatr Blood Cancer. 2013;60(Suppl 1):S12–S14. doi: 10.1002/pbc.24343. [DOI] [PubMed] [Google Scholar]
- 3.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162. doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rensing-Ehl A, Janda A, Lorenz MR, et al. Sequential decisions on FAS sequencing guided by biomarkers in patients with lymphoproliferation and autoimmune cytopenia. Haematologica. 2013;98(12):1948–1955. doi: 10.3324/haematol.2012.081901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goronzy JJ, Li G, Yang Z, Weyand CM. The janus head of T cell aging - autoimmunity and immunodeficiency. Front Immunol. 2013;4:131. doi: 10.3389/fimmu.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudd BD, Venturi V, Li G, et al. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc Natl Acad Sci USA. 2011;108(33):13694–13699. doi: 10.1073/pnas.1107594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li G, Yu M, Lee WW, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. 2012;18(10):1518–1524. doi: 10.1038/nm.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholz JL, Diaz A, Riley RL, Cancro MP, Frasca D. A comparative review of aging and B cell function in mice and humans. Curr Opin Immunol. 2013;25(4):504–510. doi: 10.1016/j.coi.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fülöp T, Larbi A, Pawelec G. Human T cell aging and the impact of persistent viral infections. Front Immunol. 2013;4:271. doi: 10.3389/fimmu.2013.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geier E, Pfeifer G, Wilm M, et al. A giant protease with potential to substitute for some functions of the proteasome. Science. 1999;283(5404):978–981. doi: 10.1126/science.283.5404.978. [DOI] [PubMed] [Google Scholar]
- 11.Reits E, Neijssen J, Herberts C, et al. A major role for TPP2 in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity. 2004;20(4):495–506. doi: 10.1016/s1074-7613(04)00074-3. [DOI] [PubMed] [Google Scholar]
- 12.Rockel B, Peters J, Kühlmorgen B, Glaeser RM, Baumeister W. A giant protease with a twist: the TPP II complex from Drosophila studied by electron microscopy. EMBO J. 2002;21(22):5979–5984. doi: 10.1093/emboj/cdf601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gavioli R, Frisan T, Vertuani S, Bornkamm GW, Masucci MG. c-myc overexpression activates alternative pathways for intracellular proteolysis in lymphoma cells. Nat Cell Biol. 2001;3(3):283–288. doi: 10.1038/35060076. [DOI] [PubMed] [Google Scholar]
- 14.Hong X, Lei L, Glas R. Tumors acquire inhibitor of apoptosis protein (IAP)-mediated apoptosis resistance through altered specificity of cytosolic proteolysis. J Exp Med. 2003;197(12):1731–1743. doi: 10.1084/jem.20020801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huai J, Firat E, Nil A, et al. Activation of cellular death programs associated with immunosenescence-like phenotype in TPP2 knockout mice. Proc Natl Acad Sci USA. 2008;105(13):5177–5182. doi: 10.1073/pnas.0801413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Firat E, Huai J, Saveanu L, et al. Analysis of direct and cross-presentation of antigens in TPP2 knockout mice. J Immunol. 2007;179(12):8137–8145. doi: 10.4049/jimmunol.179.12.8137. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 18.Duensing S, Darr S, Cuevas R, et al. Tripeptidyl peptidase II is required for c-MYC-induced centriole overduplication and a novel therapeutic target in c-MYC-associated neoplasms. Genes Cancer. 2010;1(9):883–892. doi: 10.1177/1947601910389605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcilla M, Villasevil EM, de Castro JA. Tripeptidyl peptidase II is dispensable for the generation of both proteasome-dependent and proteasome-independent ligands of HLA-B27 and other class I molecules. Eur J Immunol. 2008;38(3):631–639. doi: 10.1002/eji.200737444. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 21.Moir S, Ho J, Malaspina A, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. 2008;205(8):1797–1805. doi: 10.1084/jem.20072683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rakhmanov M, Keller B, Gutenberger S, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci USA. 2009;106(32):13451–13456. doi: 10.1073/pnas.0901984106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118(5):1294–1304. doi: 10.1182/blood-2011-01-330530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubtsov AV, Rubtsova K, Fischer A, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isnardi I, Ng YS, Menard L, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115(24):5026–5036. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hambleton S, McDonald DO, Morgan NV, et al. Autosomal recessive combined immunodeficiency due to loss of function mutation in tripeptidyl peptidase II [In: 15th Biennial Meeting European Society for Immunodeficiency (ESID). 2012, Florence, Italy: Springer.]. J Clin Immunol. 2012;32:S385–S386. [Google Scholar]
- 27.Assounga AG, Warner CM. Transcription of major histocompatibility complex class I (Kb) and transporter associated with antigen processing 1 and 2 genes is up-regulated with age. Immunology. 2004;113(3):378–383. doi: 10.1111/j.1365-2567.2004.01967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Appay V, Dunbar PR, Callan M, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8(4):379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 29.Nikolich-Zugich J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat Rev Immunol. 2008;8(7):512–522. doi: 10.1038/nri2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rutishauser RL, Kaech SM. Generating diversity: transcriptional regulation of effector and memory CD8 T-cell differentiation. Immunol Rev. 2010;235(1):219–233. doi: 10.1111/j.0105-2896.2010.00901.x. [DOI] [PubMed] [Google Scholar]
- 31.Orth G. Genetics of epidermodysplasia verruciformis: Insights into host defense against papillomaviruses. Semin Immunol. 2006;18(6):362–374. doi: 10.1016/j.smim.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Crequer A, Troeger A, Patin E, et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J Clin Invest. 2012;122(9):3239–3247. doi: 10.1172/JCI62949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crequer A, Picard C, Patin E, et al. Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS ONE. 2012;7(8):e44010. doi: 10.1371/journal.pone.0044010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moulias R, Proust J, Wang A, et al. Age-related increase in autoantibodies. Lancet. 1984;1(8386):1128–1129. doi: 10.1016/s0140-6736(84)92547-9. [DOI] [PubMed] [Google Scholar]
- 35.Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11(4):289–295. doi: 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- 36.Rockel B, Kopec KO, Lupas AN, Baumeister W. Structure and function of tripeptidyl peptidase II, a giant cytosolic protease. Biochim Biophys Acta. 2012;1824(1):237–245. doi: 10.1016/j.bbapap.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Astle MV, Hannan KM, Ng PY, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2012;31(15):1949–1962. doi: 10.1038/onc.2011.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucas CL, Kuehn HS, Zhao F, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15(1):88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angulo I, Vadas O, Garçon F, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–871. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deau MC, Heurtier L, Frange P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. 2014;124(9):3923–3928. doi: 10.1172/JCI75746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schubert D, Bode C, Kenefeck R, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations [published online ahead of print October 20, 2014]. Nat Med. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuehn HS, Ouyang W, Lo B, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–1627. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Herrera G, Tampella G, Pan-Hammarström Q, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]