

Abstract
Right ventricular (RV) failure determines outcome in patients with pulmonary hypertension, congenital heart diseases and in left ventricular failure. In 2006, the Working Group on Cellular and Molecular Mechanisms of Right Heart Failure of the NIH advocated the development of preclinical models to study the pathophysiology and pathobiology of RV failure. In this review, we summarize the progress of research into the pathobiology of RV failure and potential therapeutic interventions. The picture emerging from this research is that RV adaptation to increased afterload is characterized by increased contractility, dilatation and hypertrophy. Clinical RV failure is associated with progressive diastolic deterioration and disturbed ventricular–arterial coupling in the presence of increased contractility. The pathobiology of the failing RV shows similarities with that of the LV and is marked by lack of adequate increase in capillary density leading to a hypoxic environment and oxidative stress and a metabolic switch from fatty acids to glucose utilization. However, RV failure also has characteristic features. So far, therapies aiming to specifically improve RV function have had limited success. The use of beta blockers and sildenafil may hold promise, but new therapies have to be developed. The use of recently developed animal models will aid in further understanding of the pathobiology of RV failure and development of new therapeutic strategies.
Keywords: Pressure overload, Congenital heart diseases, Pulmonary hypertension, Pulmonary artery banding, MRI, Pressure–volume analysis
Introduction
Right ventricular (RV) failure is a major determinant of outcome in patients with pulmonary hypertension (PH), corrected congenital heart diseases (CHD) and in left ventricular failure due to ischemic heart disease [1–3]. Improved therapies for these diseases have led to a quickly expanding population of children and young adults at risk of mortality due to RV failure. For example, currently over four million adults in Europe and the USA suffer from late effects after treatment for CHD. Despite the pivotal involvement of the RV in both common and rare cardiovascular diseases, the mechanisms of RV failure have historically gotten little attention. Already in 1988, Reeves pleaded that “One must inquire how increasing pulmonary vascular resistance results in impaired RV function” (Reeves, cited in [4]). However, almost 20 years later in 2006, a Working Group on Cellular and Molecular Mechanisms of Right Heart Failure of the National Heart, Lung and Blood Institute concluded that there was “a paucity of basic knowledge at all levels about the RV’s normal and pathological function” [5]. This observation led to a call to the research community to develop accurate preclinical models, to study the pathophysiology and pathobiology of RV failure and to develop new therapeutic strategies [5]. In this review, we summarize the research in animal models since then into the pathophysiology and pathobiology of RV failure and possible therapeutic interventions.
Modeling and evaluating a unique ventricle
The right ventricle is not a mirror image left ventricle
The RV has specific characteristics that affect the response to abnormal loading conditions, as extensively reviewed previously [6]. In short, firstly, the RV is derived from a distinct set of precursor cells (as compared to the LV), the so-called secondary heart field [7]. It is yet unknown whether this different embryological origin affects the response to abnormal loading conditions [8]. Secondly, the RV is a crescent-shaped structure wrapped around the LV that has a unique contraction pattern, which complicates functional and volumetric analysis. Thirdly, the normal RV is unloaded after birth. During fetal life, the RV and LV work in parallel to support the systemic and pulmonary circulation, but after birth, these circulations are serially connected and the pulmonary vascular resistance, which determines the afterload of the RV, progressively decreases. In patients with CHD, this unloading is often absent [9]. Fourthly, the dominant movement of the RV is longitudinal shortening, pressing the RV-free wall against the septum to create a bellows effect to empty into the low-resistance pulmonary circulation [6]. Finally, in normal adult conditions, the RV has a lower oxygen requirement as compared with the LV and lower coronary flow that mostly occurs during systole [10]. A comprehensive overview of differences between the RV and LV from a clinical and preclinical perspective was published recently [11]. In the current paper, we present a detailed analysis of the experimental literature describing the adaptation of the right ventricle in response to increased afterload.
Evaluation of RV function and failure
To interpret the findings in preclinical models and to translate these to clinical practice, a definition of RV failure is necessary. Right heart failure is not an entity as such but a continuum of disease severities and clinical symptoms and can be defined in congruence with previous definitions of heart failure by Sugawa and Sunagawa [12]. Heart failure is defined as the inability to meet the requirements of the metabolizing tissues of the body. RV failure is defined accordingly, but the clinical signs and symptoms may differ from those in LV failure [13]. The cardinal clinical characteristics of RV failure are low (effective) cardiac output (evident in exercise intolerance, fatigue, dyspnea and poor peripheral circulation) and fluid retention (evident in peripheral edema, effusion and ascites) [6, 13] (Table 1). It is therefore important, in clinical practice but also in preclinical animal experiments, to include these clinical signs in the assessment of RV function or failure, in addition to functional RV parameters.
Table 1.
Evaluation of RV disease in animal models
| Parameter | Examples |
|---|---|
| Type of loading | Proximal pressure load (e.g., pulmonary artery banding) |
| Peripheral pressure load (e.g., pulmonary hypertension) | |
| Volume load (e.g., aorto-caval shunt, pulmonary/tricuspid valve regurgitation) | |
| Combined pressure/volume load (e.g., pulmonary hypertension + aorto-caval shunt | |
| Clinical symptoms | Appearance (decreased grooming or inactivity) |
| Bodyweight changes (cachexia or fluid retention) | |
| Cyanosis or decreased peripheral circulation | |
| Dyspnea/tachypnea (labored breathing) | |
| Effusions (palpable ascites) | |
| Exercise | Voluntary/spontaneous activity |
| Forced exercise testing | |
| Effusion at autopsy | Pleural effusion |
| Ascites | |
| Liver wet/dry weight ratio | |
| Survival | Mortality |
| Human endpoints reached |
In clinical practice, exercise capacity is used as an important guide to grade the severity of heart failure and as a prognostic indicator. Exercise capacity in patients can be determined by maximal cardiopulmonary exercise testing or by voluntary exercise performance, evaluated with a 6-min walk distance. Similarly, in animal models of RV disease, forced exercise capacity can be evaluated by a treadmill test [14, 15] and voluntary exercise capacity by spontaneous activity in a running wheel [8, 16, 17].
Mortality is the ultimate clinical sign of RV failure, and survival analysis may be included in studies. However, in animal models, other factors rather than RV failure that might impede survival (such as pulmonary disease and/or LV dysfunction in the monocrotaline model) should be excluded or accounted for [18]. Once RV disease has been characterized using clinical symptoms, exercise and/or mortality (Table 1), the disease state can be coupled to hemodynamic and cellular adaptation (Table 2).
Table 2.
Overview of hemodynamic changes in models of RV pressure load
| Species | Model | Signs and symptoms | Survival | Exercise | RVP | EDP | Ees | Ees/Ea | Eed | CI or CO | EDV | Ref | Remark |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peripheral pressure load | |||||||||||||
| Rat | mct30 | None | No mortality | n/a | 27 | 56 | 21 | = | −16 | 9 | 13 S | [36] | |
| Rat | mct80 | ↓BW, inactivity | No mortality | n/a | 67 S | 38 | 15 | ↓ | −9 | −26 | 89 S | [36] | |
| Rat | mct80 | Yes (see R1) | No mortality | V ↓ | 96 S | 199 S | 188 S | n/a | 4 | −8 | 30 | [17] | R1, R2 |
| Rat | mct60 | ↓BW, resp distress | ↑ mortality | n/a | 166 S | 200 S | 400 S | ↓ | 700 S | −60 S | n/a | [34] | |
| Rat | mct60 | n/a | n/a | n/a | 325 S | n/a | 766 S | = | 500 S | −64 S | n/a | [102] | |
| Rat | mct40 | None | No mortality | F ↓ | 120 S | 400 S | n/a | n/a | n/a | −45 S | n/a | [56] | |
| Rat | mct60 | Yes (see R3) | n/a | F ↓ | 180 S | 650 S | n/a | n/a | n/a | −10 | n/a | [56] | R3 |
| Rat | mct60 | n/a | n/a | F ↓ | 140 S | 333 S | n/a | n/a | n/a | −30 S | 19 | [32] | |
| Rat | mct60 | n/a | n/a | n/a | 160 S | 200 S | n/a | n/a | n/a | −25 S | n/a | [27] | |
| Rat | mct60 | n/a | n/a | n/a | 110 S | n/a | n/a | n/a | n/a | −60 S | n/a | [69] | |
| Rat | mct40 | n/a | n/a | n/a | 110 S | n/a | n/a | n/a | n/a | −29 S | n/a | [103] | |
| Rat | mct40 | n/a | n/a | n/a | 121 S | n/a | n/a | n/a | n/a | −39 S | n/a | [99] | R4 |
| Rat | SuHx | n/a | n/a | F ↓ | 200 S | n/a | n/a | n/a | n/a | −63 S | n/a | [32] | |
| Rat | SuHx | n/a | ↑ mortality | F ↓ | 222 S | n/a | n/a | n/a | n/a | −42 S | n/a | [88] | |
| Rat | SuHx | n/a | No mortality | n/a | 208 S | n/a | n/a | n/a | n/a | −42 S | n/a | [28] | R5 |
| Rat | SuHx | n/a | n/a | n/a | 283 S | n/a | n/a | n/a | n/a | n/a | n/a | [66] | |
| Rat | FHR | n/a | n/a | F ↓ | 36 | n/a | n/a | n/a | n/a | −42 S | n/a | [104] | |
| Rat | mct80 | ↓ BW | No mortality | n/a | n/a | n/a | n/a | n/a | n/a | −50 S | 25 S | [51] | |
| Rat | mct60 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | −83 S | n/a | [105] | |
| Rat | mct60 | n/a | n/a | n/a | 126 S | n/a | n/a | n/a | n/a | n/a | n/a | [106] | |
| Rat | mct60 | n/a | n/a | n/a | 130 S | n/a | n/a | n/a | n/a | n/a | n/a | [107] | |
| Rat | mct60 | Yes (see R6) | ↑ mortality | n/a | 133 S | n/a | n/a | n/a | n/a | n/a | n/a | [108] | R6 |
| Rat | mct60 | Yes (see R7) | ↑ mortality | n/a | 133 S | n/a | n/a | n/a | n/a | n/a | n/a | [109] | R7 |
| Rat | mct60 | n/a | ↑ mortality | n/a | 133 S | n/a | n/a | n/a | n/a | n/a | n/a | [110] | |
| Rat | mct80 | Yes (see R8) | No mortality | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [37] | R8 |
| Rat | mct30 | none | No mortality | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [65] | |
| Rat | mct80 | Yes (see R9) | No mortality | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [65] | R9 |
| Rat | mct60 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [94] | |
| Rat | mct60 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [98] | |
| Rat | mct60 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [83] | |
| Pigs | AVS | n/a | No mortality | n/a | 29 S | n/a | −13 | ↓ | n/a | −44 S | n/a | [81] | R10 |
| Pigs | AVS | n/a | No mortality | n/a | 84 S | n/a | 74 S | = | n/a | 3 | n/a | [39] | R11 |
| Proximal pressure load | |||||||||||||
| Lamb | pab > 8 | None | No mortality | n/a | 433 S | 75 | 281 S | n/a | 62 S | −37 S | −13 | [42] | R12 |
| Rabbit | pab5 | None | No mortality | n/a | 271 S | 30 S | 185 S | n/a | 62 | n/a | n/a | [58] | |
| Dog | pab13 | n/a | No mortality | n/a | 105 S | n/a | 243 S | n/a | 116 S | 0 | n/a | [41] | |
| Rat | pab4 | Mild symptoms | No mortality | V ↓ | 169 S | 500 S | 162 S | n/a | 125 S | −15 S | 60 S | [16] | |
| Rat | pab8 | ABCDE (see R13) | ↑ mortality | n/a | 204 S | 300 S | 338 S | ↓ | 1053 S | −50 S | n/a | [43] | R13 |
| Rat | pab6 | None | n/a | n/a | 117 S | 40 | 100 S | n/a | n/a | −5 | −18 | [26] | |
| Rat | pab12 | None | n/a | n/a | 97 S | 50 | 9 | n/a | n/a | −25 S | n/a | [111] | R14 |
| Rat | pab20 | None | n/a | n/a | 113 S | 17 | −9 | n/a | n/a | −12 | n/a | [111] | R15 |
| Rat | pab3 | n/a | n/a | n/a | 166 S | 200 S | n/a | n/a | n/a | −26 S | n/a | [27] | |
| Mouse | pab4 | None | No mortality | V ↓ | 300 S | n/a | n/a | n/a | n/a | 0 | 20 S | [8] | |
| Rat | pab4 | n/a | n/a | F ↓ | 220 S | n/a | n/a | n/a | n/a | −53 S | n/a | [32] | |
| Rat | pab7 | n/a | n/a | n/a | 152 S | n/a | n/a | n/a | n/a | −37 S | n/a | [69] | |
| Rat | pab6 | Yes (see R16) | No mortality | n/a | 200 S | n/a | n/a | n/a | n/a | 0 | n/a | [86] | R16 |
| Rat | pab6 | n/a | n/a | n/a | 217 S | n/a | n/a | n/a | n/a | n/a | n/a | [66] | |
| Rat | pab22 | n/a | No mortality | n/a | n/a | n/a | n/a | n/a | n/a | 0 | n/a | [28] | |
| Mouse | pab3 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | 0 | 75 S | [38] | |
| Rat | pab4 | n/a | n/a | F ↓ | n/a | n/a | n/a | n/a | n/a | −42 S | n/a | [15] | |
| Rat | pab6 | n/a | No mortality | n/a | n/a | n/a | n/a | n/a | n/a | 0 | n/a | [28] | |
| Rat | pab8 | n/a | n/a | F ↓ | n/a | n/a | n/a | n/a | n/a | −45 S | n/a | [15] | |
| Rat | pab lowcu | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [28] | |
| Rat | pab3 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [50] | |
| Rat | pab6 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [50] | |
| Mouse | pab6 | Yes (see R17) | ↑ mortality | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [29] | R17 |
| Rat | pab9 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | [50] | |
Numbers are percentages increase/decrease versus controls. For some studies and parameters, these are approximations depending on how precise data were reported. S indicates a significant change versus controls. Number behind ‘MCT’ indicates the dosage of monocrotaline in mg/kg. Number behind ‘PAB’ indicates number of weeks after which measurements were performed
MCT monocrotaline, PAB pulmonary artery banding, FHR fawn-hooded rat, SuHx Sugen–Hypoxia, AVS arterio-venous shunt, BW bodyweight, RVP right ventricular peak or systolic pressure, EDP end-diastolic pressure, Ees end-systolic elastance, Ea arterial elastance, Eed end-diastolic elastance, CI cardiac index, CO cardiac output, EDV end-diastolic volume
*for exercise V indicates voluntary exercise testing, F indicates forced exercise testing; ↓ = decreased; ↑ = increased; == unchanged
Remarks: R1 Some weight loss, inactivity and dyspnea. R2 V↓ was trend (p = 0.08). R3 > 5 % loss of body mass a day, lethargy, cyanosis, respiratory distress. R4 CO estimated on ventricular diameters. R5 Pericardial fluid on echo; mortality steeply increased after 6 weeks. R6/R7 BW loss > 10 % for 2 days and arterial oxygen saturation <80 %. R8 ‘signs of heart failure including pleural effusion and ascites’. R9 Weight loss, pleural effusion, ascites. R10/R11 Hemodynamic measurements after shunt clamping. R12 Duration of PAB varied, but exceeded 8 weeks. Eed is stiffness constant. R13 All ABCDE categories (see Table 1). R14/R15 CO is very low in these studies. R16 Failure symptoms are not defined. R17 Failure symptoms were: edema, bodyweight changes (both ↑ and ↓). EDP was 300 S after 10 days in the tightest PAB group
Modeling RV abnormal loading conditions
To study the mechanisms of RV failure, animal models that mimic specific diseases have been developed. The diseases contributing to the spectrum of patients with RV failure can be divided into three main groups: (1) patients with PH, (2) (corrected) CHD and (3) RV failure secondary to left ventricular failure. Although distinct in etiology, these diseases share in common the abnormal loading conditions imposed on the RV, i.e., increased afterload, increased preload or a combination of both [19]. Increased afterload can be peripherally located, as in PH or fixed proximally as in pulmonary stenosis. Either way, the coupling between the RV and the pulmonary arteries is disturbed (Table 2). To represent these chronic abnormal loading conditions models of PH, pulmonary artery banding and/or pre-tricuspid systemic-to-pulmonary shunts have been developed. Knowledge on the RV response to an increase in preload is scarce [16, 20, 21] and will be discussed in a separate paper. In this review, we will focus on the response to increased afterload.
Models of increased afterload
Historically, researchers used the monocrotaline model (MCT) to induce PH (Table 2a). The MCT model has been invaluable for PH research, but it may be questioned whether rats with MCT-induced PH die from progressive PH, from RV failure or from other organ dysfunction [18]. Besides MCT, other models mimicking PH have been developed, e.g., hypoxia (with or without VEGF inhibitor SUGEN) or the induction of increased pulmonary blood flow [22–25]. Although all these models may represent pulmonary vascular disease adequately, the potential direct effects of the triggers used to induce PH on the RV limit their use to study the pathobiology of RVF.
The pulmonary artery banding model (PAB) avoids these limitations of the PH models (Table 2). The banding of the pulmonary artery induces no systemic or toxic effects, and the fixed constriction of the banding ensures a constant afterload, also when pulmonary vasodilators are administered. The time course of the phenotype in this model has shown considerable variability, which may stem from differences in strain, growth rate and size of constriction. Mild constriction will lead to a chronically compensated state with increased RV systolic pressure and RV hypertrophy, but no symptoms of RV failure [16, 26, 27]. Rats have been reported to survive up to 22 weeks in such condition, despite (near) systemic RV pressures [28]. However, a tighter PAB has been demonstrated to lead to clinical symptoms of RV failure, i.e., inactivity, decreased cleaning behavior (raised fur), poor peripheral circulation, dyspnea/tachypnea, ascites and pleural/pericardial effusions and, ultimately, mortality in a high percentage of animals [17, 29–31]. High-intensity exercise capacity [32] and voluntary low-intensity exercise [8, 16, 17] are reduced in this model. These data indicate that a well-sized PAB represents a valuable model of chronic pressure load-induced RV failure.
The physiology of RV adaptation to increased afterload
The RV response to increased afterload shares several features among all in vivo animal models used. The gold standard to assess the hemodynamic properties of the (loaded) RV is pressure–volume analysis, which allows measurement of several parameters that quantify systolic and diastolic function regardless of loading conditions.
The primary response of the RV to match increased afterload is increased contractility, which is defined as end-systolic elastance (Ees) (Table 2). Contractility increases proportionally with increased arterial elastance (Ea, reflecting afterload) to maintain stroke volume (Fig. 1a, b: typical example of PV loops in response to pressure load). When end-systolic elastance increases less than arterial elastance, the Ees/Ea ratio decreases leading to ventricular–arterial uncoupling, which is regarded a physiological sign of RV failure. Indeed, beneficial pharmacological effects in the pressure-loaded RV are not seldom accompanied by a restoration of Ees/Ea ratio [16, 33, 34].
Fig. 1.
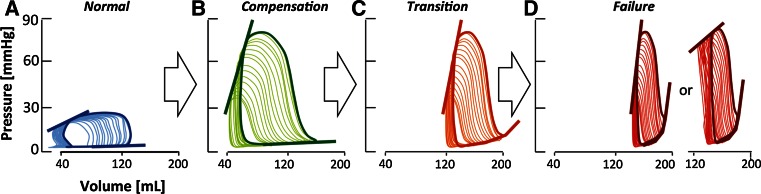
Pathophysiology of the pressure-loaded RV. Conceptual representation of the progression of pathophysiological changes in the pressure-loaded RV. Typical pressure–volume (PV) loops from compensation to failure. Volumetric changes were derived from experimental studies and extrapolated using previously published normal values. Straight lines represent the end-systolic elastance (Ees), dotted lines represent the end-diastolic elastance (Eed). a PV loop in the unloaded RV, showing normal systolic and diastolic function. b PV loop in compensated RV, showing increased systolic function (Ees) and RV dilatation (increased end-diastolic volume) but normal diastolic function (Eed). c PV loop in transition to failure showing increased systolic function (Ees) and impaired diastolic function (Eed). d PV loop in RV failure showing increased or pseudo-normalized systolic function (Ees) and further impaired diastolic function (Eed)
In chronic pressure load induced by pulmonary artery constriction or experimental PH, increased contractility [35] is accompanied by ventricular dilatation (Fig. 1b; [8, 16, 17, 32, 36–38]). This suggests that also the Frank–Starling mechanism contributes to the RV adaptation to chronic pressure load. Indeed, in most animal models of chronic increased pressure load, ventricular dilatation is associated with decreased Ees/Ea ratio, with the exception of mild monocrotaline-induced PH [36] and 3-month duration of flow-induced PH [39]. Unfortunately, ventricular dilatation leads to increased wall stress [8, 40] and is proposed as a hallmark of the failing ventricle [5], rather than an adaptive response. Improvement of the RV–PA coupling ratio does universally lead to reduction in RV dilatation (Table 3) and wall stress.
Table 3.
Treatments for RV failure
| Group | Agent | Clinically available? | Exercise | Symptoms | Mortality | Contractility | Ees/Ea ratio | Diastolic function | RVH | Fibrosis | Tested in fixed afterload? | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta-blockade | Carvedilol | Y | n/a | n/a | Improved | n/a | n/a | n/a | Reduced | Reduced | N | [88] |
| Metoprolol | Y | n/a | n/a | Improved | n/a | n/a | n/a | Unchanged | Reduced | N | [88] | |
| Bisoprolol | Y | n/a | Improved | Improved | Improved | Increased | Improved | Unchanged | Reduced | N | [34] | |
| RAAS inhibition | Losartan | Y | n/a | n/a | n/a | Unchanged | Increased | Improved | Unchanged | n/a | N | [102] |
| Telmisartan | Y | n/a | n/a | n/a | n/a | n/a | n/a | Reduced | Reduced | N | [112] | |
| Losartan/eplerenone | Y | Unchanged | Unchanged | Unchanged | Unchanged | Decreased | Unchanged | Unchanged | Unchanged | Y | [43] | |
| PKG-1-PDE5 pathway | Sildenafil | Y | n/a | n/a | n/a | n/a | n/a | n/a | Unchanged | Unchanged | Y | [50] |
| Sildenafil | Y | n/a | n/a | n/a | n/a | n/a | n/a | Unchanged | n/a | Y | [27] | |
| Sildenafil | Y | Improved | Improved | n/a | Improved | Increased | Unchanged | Unchanged | Increased | Y | [16] | |
| Sildenafil | Y | Unchanged | Unchanged | n/a | Unchanged | Unchanged | Improved | Unchanged | Reduced | Y | [44] | |
| Sildenafil | Y | n/a | n/a | n/a | n/a | n/a | n/a | Unchanged | n/a | N | [103] | |
| Riociguat | N | n/a | n/a | n/a | n/a | n/a | n/a | Reduced | Reduced | N | [95] | |
| BAY 41-2272 | N | n/a | n | n | n/a | n/a | n/a | Unchanged | Unchanged | Y | [113] | |
| Endothelin receptor blockade | Bosentan | Y | n/a | n/a | n/a | n/a | n/a | n/a | Unchanged | n/a | N | [99] |
| Anti-oxidant | Protandim | N | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Reduced | N | [28] |
| Anti-oxidant | EUK-134 | N | n/a | Unchanged | n/a | n/a | n/a | n/a | Unchanged | Reduced | N | [51] |
| Rho-kinase inhibitor | Fasudil | N | n/a | n/a | n/a | n/a | n/a | n/a | Reduced | n/a | N | [103] |
| HDAC inhibitor | Trichostatin A | N | n/a | n/a | n/a | n/a | n/a | n/a | Unchanged | Increased | Y | [86] |
| Fatty acid oxidation blockade | Trimetazidine | Y | Improved | n/a | n/a | n/a | n/a | n/a | Reduced | n/a | Y | [15] |
| Ranolazine | Y | Improved | n/a | n/a | n/a | n/a | n/a | Reduced | n/a | Y | [15] | |
| PDK inhibitor | Dichloroacetate | N | n/a | n/a | n/a | n/a | n/a | n/a | Reduced | n/a | Y | [69] |
| Estrogen receptor-beta agonist | Genistein | N | n/a | n/a | Improved | n/a | n/a | n/a | Reduced | n/a | N | [106] |
Overview of experimental studies reporting on (direct) RV effects of medical treatments in the pressure-loaded RV
RVH right ventricular hypertrophy, HDAC histone deacetylase, PDK pyruvate dehydrogenase kinase, Ees end-systolic elastance, Ea arterial elastance
Evidence is accumulating that RV failure due to chronic pressure load is characterized by both enhanced systolic function and progressive deterioration of diastolic function [16, 31, 34]. Sparse clinical data show that in PH patients, higher right atrial pressure (an indirect measure of RV diastolic function) is associated with worse outcome. This could indicate that diastolic dysfunction contributes to RV failure in patients. From experimental models [41–44] using PAB as afterload (Table 2), it is clear that diastolic dysfunction is an inherent component of increased RV afterload (Fig. 1c, d; [17]). In dogs with a PAB, the diastolic dysfunction of the RV is partly compensated for right atrial adaptation, i.e., right atrial contractility increases, and the right atrium dilates to serve as a reservoir [41]. In a recent study in rats with a PAB separating those with clinical signs of severe RV failure from those without clinical signs but with RV dysfunction, clinical signs of RV failure were associated with a further deterioration of diastolic function despite increased systolic function [31]. These observations are confirmed by studies on isolated myocytes from patients with end-stage RV failure due to PH [45]. Intriguingly, in a recent study in which the RV was subjected to isolated volume load (pulmonary valve regurgitation), diastolic dysfunction was described without changes in Ees [46]. Deterioration of diastolic function might thus play a central role in the transition from compensation to failure (Fig. 1).
Diastolic function has a passive and an active component. The RV (compared to the LV) has been suggested to be particularly vulnerable to disturbed active relaxation, possibly due to insufficient expression of the sodium-calcium exchanger (NCX) [47]. Indeed, in experimental MCT-induced PH, active relaxation is increasingly disturbed with increasing MCT dose [36], while passive stiffness has been reported as normal [17, 36] or increased [48]. Chronic PAB invariably leads to diastolic dysfunction, with disturbances in both active relaxation and passive stiffness [16, 17, 41, 42].
Studies without pressure–volume analysis
Interpretation of the data in the literature is hampered by the lack of pressure–volume analysis in many studies. The alternatives, CMR-derived RV volumes or echocardiographic parameters for systolic function, only indirectly relate to contractility and are dependent on pre- and/or afterload. Systolic displacement of the lateral tricuspid annulus along the base-apex axis (TAPSE) is decreased in chronic pressure load. This reflects reduced longitudinal movement of the RV, but does not relate to RV pumping function, i.e., shifting volume at a certain pressure [49]. Fractional shortening of the RV outflow tract [29] and change in surface area [50] are also reported, but their significance in the setting of abnormal RV loading is unknown. CMR, the gold standard for RV function in clinical practice, has the advantage of being noninvasive, but without simultaneous pressure measurements, only yields load-dependent variables [8, 51, 52]. RV ejection fraction (EF), another outcome parameter, is generally reduced before overt RV failure ensues, although this parameter is of limited value as it is preload dependent [8, 36]. As described above, PV analysis characterizes the initial RV response as increased contractility and ventricular dilatation, two processes that have opposite effects on EF. Depending on the relative size of changes, EF may thus be increased, decreased or remain unchanged. Recently, Vanderpool et al. [53] suggested RV stroke volume divided by the end-systolic volume as an alternative, less preload dependent parameter.
Proximal- versus peripheral-type pressure load
Whether the type of pressure load (PAB vs. PH) determines the RV response is a matter of debate as conflicting data have been reported. Echocardiographic data suggest that at similar levels of pressure overload, the RV is less dilated and has superior function in patients with pulmonary stenosis compared with those with PAH. In contrast, a recent experimental comparison between rats with PAB and PH [17] showed that the PAB rats with moderate RV dysfunction (assessed by exercise and clinical signs and symptoms) had more severe RV dilatation in response to afterload than rats subjected to MCT-induced PH.
Interaction with LV function
The RV and LV functionally interact via shared fibers and the interventricular septum [11]. Interaction of the pressure-loaded RV with LV function has been noted in patients and animal models with PH [54, 55]. In a study in isolated hearts from rats with MCT-PH, pacing improved RV function and reduced interference in the diastolic phase [56]. Also, studies in isolated hearts suggested that the LV contributes to 65 % of the work of the normal RV [57]. In a PAB rabbit model of moderate RV dysfunction, increasing LV afterload via aortic constriction improved systolic RV function [58]. End-diastolic elastance tended to increase, but this effect was not significant.
The increased knowledge on the functional characterization of RV response to increased afterload leads to the question: Can we manipulate this response to support the failing RV? To answer this question, the hemodynamic adaptation has to be coupled to the pathobiology.
Pathobiology of RV failure
In the pressure-loaded RV, a myriad of cellular changes takes place that may initially serve as adaptive remodeling, but are also present in the failing RV (Fig. 2). Rather than caused by a single ‘malignant’ pathway, RV failure is the resultant of many biological changes, both adaptive and maladaptive. The interpretation of these changes is influenced by the state of RV dysfunction assessed by clinical signs and hemodynamic measurements as well as the trigger inducing the afterload (e.g., MCT or PAB). Some of these changes resemble those found in LV remodeling in adaptation to stress [40, 59].
Fig. 2.

Overview of the pathobiological changes in the abnormally loaded RV. Pathobiological hallmarks of the abnormally loaded RV. A myriad of genetic and epigenetic changes result in tissue damage-related processes (oxidative stress, fibrosis and apoptosis) and activation of (mal)adaptive processes on the tissue (capillary formation, inflammation) or cellular level (hypertrophy, energy substrate use, mitochondrial function and calcium handling). These processes are regulated by a complex network of signaling pathways related to contactile function, cellular growth, energy metabolism and neurohumoral signaling. ATP adenosine triphosphate, NFAT nuclear factor of activated T cells, MAPK mitogen-activated protein kinase, Mef2 myocyte enhancer factor-2, PKG-1 protein kinase G-1, PDE3/5 phosphodiesterase-type 3/5, PKA protein kinase A, SERCA2 sarcoplasmic reticulum Ca2 + -ATPase, RyR ryanodine receptor, PLB phospholamban, NCX sodium-calcium exchanger, SP3 transcriptional repressor SP3, RAAS renin angiotensin aldosterone system
RV hypertrophy, isoform switch and fibrosis
RV hypertrophy is an adaptive response to reduce wall stress and improve contractility. In the LV, hypertrophy is a strong predictor for outcome [59], but in experimental RV failure, this relation is less clear. In all studies in experimental animals, RV hypertrophy is present but this is not related to functional adaptation [16, 17, 26, 28]. In one study, RV hypertrophy was more severe in a PH model than in a PAB model [28], in other studies it was not, possibly due to differences in the degree of loading [14, 17, 26]. This is important to note, as some authors compare different models using the degree of RV hypertrophy to classify the state of adaptation, which may yield contradictory results [15].
Increased afterload of the RV induces a switch in myosin heavy chain (MHC) isoform composition from the fast alpha-MHC to the slower but energetically favorable beta-MHC in the RV myocardium [8, 16, 17, 27]. This response is not related to the degree of RV dysfunction. The hypertrophic response is induced by similar pathways as in the LV, e.g., the calcineurin−NFAT pathway [8], which may render the RV a putative target for calcineurin interference or other therapeutic strategies that have been shown to target LV hypertrophy [20].
RV fibrosis in response to increased afterload has been reported in some PH models [28, 34, 60] as well as in PAB models [17, 28], although there is a wide variation in the amount of collagen measured across models and the timeframe in which it develops. Given the putative relation between fibrosis and diastolic dysfunction, fibrosis may be a therapeutic target. However, a recent study shows there was no relation between the amount of (interstitial myocardial) fibrosis and the degree of RV dysfunction [17]. One may hypothesize that RV fibrosis is a by-product of the (mal)adaptive RV response to pressure load and has limited pathophysiological significance. Either way, fibrosis can be reduced with several interventions, i.e., beta-blockade [34], ROS scavenger [51], prostacyclin [60], but whether this is a direct effect or secondary to afterload reduction is unclear. Interestingly, an effective strategy to target specifically fibrosis (in LV failure) did not reduce fibrosis nor improve RV function in experimental RV failure [43]. Fibrosis may thus have ventricular-specific characteristics that require further exploration of its pathophysiological significance and therapeutic amenability in RV failure.
RV capillary formation/RV oxygen supply
Capillary rarefaction can play an important role in the development of RV failure. In the unstressed RV, coronary perfusion is present throughout the cardiac cycle [10], but in patients with PH, the coronary perfusion occurs primarily during diastole [61]. Because RV oxygen consumption also increases; a chronic oxygen demand–supply mismatch may be an underlying mechanism in the transition to failure. Coronary flow has not been measured yet in experimental models, but capillary density was reduced in several PH models [28, 34, 60]. Recently, two studies in various pressure load-induced disease states, i.e., MCT [14] and PAB [31], reported increased capillary density in compensated RV hypertrophy and pseudo-normalized density in decompensated RV hypertrophy. Further support for the significance of insufficient myocardial perfusion comes from the observation that prostacyclin therapy in rats with PH improves both capillary density and survival in the absence of beneficial effects on pulmonary hemodynamics or vascular remodeling [60]. Given its putative importance in the pathophysiology of RV failure, it is questioned which cellular processes are most affected by reduced myocardial perfusion. Candidate pathways are substrate metabolism, mitochondrial function and calcium handling.
Cardiac metabolism
In the LV, significant shifts in myocardial metabolism have been reported during the transition from adaptation to failure. Fatty acids are the main substrate for the adult heart under normal conditions, but under stress, a switch in substrate use occurs toward glucose and lactate [62]. In addition, glucose metabolism shifts from complete oxidation via the Krebs cycle to glycolysis only, which yields less ATP but also uses less oxygen per ATP molecule. Metabolic shifts are also observed in the pressure-loaded RV. Fatty acid oxidation (FAO) is reduced in PH patients, but only in those with severe hypertrophy [63]. Also, in rats with MCT-induced PH, the expression of CPT-1b (a rate limiting enzyme in the uptake of long-chain fatty acids [64]) was reduced [65, 66]. A reduction in the use of fatty acids seems to be an adaptive mechanism, since preventive inhibition via trimetazidine (which inhibits an essential step in oxidation of FA), increased cardiac output measured by echocardiography in a model of mild RV dysfunction due to a PAB [15].
In addition to the reduction in FAO, patients with PH have an increased uptake of the glucose analog 18FDG in the RV [67]. Also, in rats with MCT-induced PH, the expression of glycolysis-related genes is increased [68] as well as the enzymatic rates of glycolysis [69]. Increased expression of glycolysis-related genes has been shown in rats with MCT-PH [69], hypoxic-PH [70], Fawn-Hooded rats [71] and rats with a PAB [15, 69, 72], but none of the rats in these experimental studies had clinical symptoms of RV failure. Similarly, upregulation of pyruvate dehydrogenase kinase (PDK), which uncouples glycolysis from the Krebs cycle, has been shown in both PH and PAB models of adaptive RV remodeling [71]. Inhibition of PDK has been shown to increase RV O2 consumption and improve RV function, although a concomitant reduction in RV systolic pressure in the setting of fixed afterload (PAB model) in this study complicated the interpretation of these results [71]. Further evaluation of these effects in models of more severe RV dysfunction is needed since this therapy has also been shown to reverse pulmonary vascular remodeling in experimental PH [73], which could indicate a therapeutic strategy that may benefit both the RV and pulmonary vasculature.
Mitochondrial function
Mitochondria are the “powerhouses” of the cardiomyocyte, but also regulate many processes involved in the response to stress: formation of oxygen radicals, oxygen sensing, induction of apoptosis and inflammation [74]. The first notion that the RV in PH may be subjected to increased “oxidative stress” came from observations in rats with MCT-induced PH. These rats had increased myocardial activity of complex II and oxygen radicals, and also increased production of radical scavengers [37]. Treatment with a radical scavenger (EUK-134) improved RV systolic function but did not affect diastolic dysfunction [51]; however, in this study, pulmonary vascular resistance was also decreased after EUK-134 treatment, which may have contributed to the beneficial RV effects [75]. In another PH model (SUGEN + hypoxia), treatment with protandim (a plant extract inducing nrf2 expression) increased cardiac output, suggesting that at least in PH rats, increasing defense mechanisms against oxidative stress may be beneficial [28]. These pathways have not been tested in PAB models, i.e., independent from the pulmonary circulation.
Apoptosis and inflammation
Apoptosis and inflammation, both important mechanisms in the vascular pathology of PH [76–78], have not been explored in detail in the myocardium of models of RV pressure load. However, cardiomyocyte apoptosis and inflammation have been reported in both PH and PAB models [58, 79–81]. Pressure load is linked to apoptosis in multiple ways (mechanic damage, oxidative stress and neurohumoral signaling) and even mildly increased rates of apoptosis can contribute to heart failure [40]. Inflammation, marked by the presence and activation of immune cells and increased activity of inflammatory cytokines also connects pressure load and apoptosis. In addition, in LV pressure, mediators such as TNF-alpha and interleukins interact with neurohumoral signaling, induce fibrosis and affect contractile function and myocardial gene expression [40]. Data on the functional importance of inflammation in the development of RV failure are lacking.
PKG−PDE5 pathway
The importance of the protein kinase G (PKG) and phosphodiesterase 5 (PDE5) pathway in LV remodeling was shown in mice with transverse aortic constriction [82]. PDE5 catabolizes cGMP, which activates PKG and generally suppresses proliferative pathways. Hence, PDE5 inhibitors (e.g., sildenafil) may enhance the protective effects of PKG. Patients with increased RV afterload showed increased PDE5 expression [83], but only few studies in rat report data on PDE5 expression or PKG activity. PKG activity is not uniformly lowered in all models of experimental RV afterload [16]. Therefore, currently little is known about the importance of this pathway in RV failure. Positive results of intervention studies targeting this pathway (see Treatment of RV failure below) underline the importance of more detailed exploration of the PKG−PDE5 pathway in the RV as it may provide new treatment options for RV failure.
Other pathways
What are the consequences of the unique embryologic origin of the RV for the response to stress [8, 84]? In a rat model of PAB, the expression of dHand, an RV-specific precursor, as well as GATA-4, MEF2 and NKX2.5 were increased [85]. On the other hand, also signaling pathways involved in LV remodeling, such as the calcineurin pathway, have been shown to be activated in a murine model of PAB [8]. However, suppression of calcineurin activation in transgenic mice induced RV dilatation even without pressure load [20]. Similarly, inhibitors of histone deacytelases, which reduce adverse remodeling and improve function in experimental LV pressure load, worsen RV function in rats with a PAB [86]. Such incidental reports suggest that the regulation of adaptation to stress is indeed (partly) chamber specific.
The use of microarrays yields divergent results on genes and pathways possibly involved in the development of RV failure [31, 38, 65, 72, 87]. This may be due to the differences in models used (e.g., MCT vs. PAB), species differences (rat, mouse and rabbit) and different degrees of RV adaptation (compensated vs. decompensated). Additionally, array studies have provided evidence for chamber specificity of gene expression. Table 4 provides a summary of differences in signals between RV and LV in response to increased afterload.
Table 4.
Differences in signals between RV and LV in response to increased afterload
| Model | Mechanism | Difference with LV | Ref |
|---|---|---|---|
| Mouse PAB | Extracellular matrix proteins | ↑ expressed in RV | [38] |
| Proteases and inhibitors | ↑ expressed in RV | [38] | |
| Developmentally regulated proteins | only expressed in RV | [38] | |
| Rat PAB | PINK1 | ↓ in RVF, ↑ in LVF | [31, 114] |
| Mouse PAB vs TAC | miRNA 28,148a,93 | ↑ in RVF (in non-myocyt fraction) | [87] |
| Mouse PAB | Wnt signalling | ↑↑ in RVF > LVF | [29] |
| Rat MCT | Mef2c | ↑ compensated RV, ↓ RVF, no change LVF | [115] |
Overview of studies reporting differences in myocardial signaling between the RV and LV, in response to increased afterload
PAB pulmonary artery banding, TAC transverse aorta constriction, RVF RV failure, LVF LV failure
A study in mice, comparing pressure load of the RV via PAB with pressure load of the LV via aortic constriction, showed differences between the two ventricles in expression of genes involved in (1) extracellular matrix proteins, (2) proteases and inhibitors and (3) developmentally regulated proteins [38]. In a similar study, four microRNAs were upregulated in the pressure-loaded RV, but these were all located in the non-myocyte fraction of the RV [80]. Gain/loss of function studies is the next step to determine the functional relevance of these findings.
Treatment of RV failure
Currently, no RV-specific medical treatment strategies exist. Reports of experimental RV drugs (Table 3) fall into one of three categories: (1) treatment strategies for LV failure (e.g., beta-adrenergic blockade, RAAS inhibition), (2) drugs that target the pulmonary vasculature in PH (e.g., PDE5 inhibitors, endothelin antagonists) and (3) proof-of-concept studies with experimental treatments.
Beta-adrenergic blockade and RAAS inhibition
The cornerstones of treatment of LV failure are inhibition of the beta-adrenergic receptors and the renin−angiotensin aldosterone system (RAAS). Clinicians are reluctant to prescribe beta blockers to patients with failing RVs in the setting of PH because of their negative inotropic effects, but preclinical studies have shown beneficial effects on RV remodeling. In the hypoxia + SUGEN model of PH, carvedilol, a non-selective beta blocker with also alpha-1-receptor blocking effects, reduced the development of hypertrophy, fibrosis, capillary rarefaction and attenuated reduction in cardiac output and TAPSE [88]. After treatment with Metoprolol, a selective beta blocker, similar effects were observed, although these can be attributed to reduction in the pulmonary vascular remodeling [88]. In the MCT model, bisoprolol (another selective beta blocker) does not prevent hypertrophy or capillary rarefaction, but prevents fibrosis along with delaying the decline in cardiac output and TAPSE [34]. Interestingly, bisoprolol increased phosphorylation of myocardial titin, suggesting a direct RV effect. In conclusion, beta blockers have variable effects in PH models. Unfortunately, no data are published on beta-blockade in the failing RV due to stenosis-type pressure overload, which might elucidate the direct protective effects of beta-blockade on the RV.
Clinical data in PH suggest that the RAAS is activated in at least a subgroup of patients. There are also suggestions that the RAAS system is active in congenital heart disease, and that combinations of RAAS-inhibiting drugs that are used in the standard care for LV failure patients (such as angiotensin receptor blockers + eplerenone) might potentiate the effect and target oxidative stress, fibrosis and improve diastolic dysfunction like in the LV. However, recently no effect of RAAS inhibition was shown in a rat model of PAB [43], in accordance with the results from a study in patients with a systemic RV in CHD [89].
PDE5 inhibitors
PDE5 inhibition would be an excellent therapeutic approach for RV failure in PH, as it also reduces pulmonary vasculature resistance [90]. However, the first two studies reporting on PDE5 inhibition in the PAB model showed no prevention or reduction in hypertrophy and fibrosis [27, 50]. In these studies, the functional analysis was limited to thermodilution-measured cardiac output and echocardiography. Using pressure–volume analysis, it was shown that preventive sildenafil increases contractility, reduces dilatation and attenuates the decline of spontaneous exercise, but leaves diastolic function unchanged [16], whereas in established RV pressure load, sildenafil treatment predominantly improved diastolic dysfunction, with a small effect on contractility [44]. The differences between the first and the latter studies may be due to differences in the severity of the loading condition, as in the LV it was shown that the effects of PDE5 inhibition are dependent upon the severity of the loading condition [91].
The mechanisms by which sildenafil exerts its effects are pleiotropic, incompletely known and not limited to the PDE5−PKG system. Mechanistically, sildenafil inhibits PDE5, increases compartmentalized cGMP and thereby activates PKG-1, which in turn has inhibitory effects on pathways of pathological remodeling [92]. PKG-1 also phosphorylates titin, thereby reducing stiffness [93]. In the pressure-loaded RV, sildenafil treatment as a preventive strategy increased fibrosis [16, 27, 50]. However, when given in a later stage of established pressure load, sildenafil reduced fibrosis and reduced ventricular stiffness [44], supporting the concept that timing and loading severity determines cardiac response (Fig. 3). Sildenafil also stimulates the PDE3−PKA pathway and targets mitochondrial (k)ATP channels, mitochondria and inflammation [94]. Other pharmacological approaches to manipulate the PDE5−PKG-1 axis include stimulation of soluble guanylate cyclase by riociguat, but results so far indicate effects on the pulmonary vasculature, rather than direct beneficial RV effects [95].
Fig. 3.

Effects of sildenafil depend on the stage of pressure load-induced RV dysfunction sildenafil effects in the early (0–4 weeks) and late (4–8 weeks) stage of pressure load-induced RV dysfunction are compared. These data illustrate the concept that the effects of pharmacological intervention in the pressure-loaded RV (in these studies with sildenafil) depend on the stage of RV failure when treatment is started. Data of rats with a PAB at week 4 are derived from [16], in which sildenafil treatment was started at the day of surgery (preventive strategy); data of rats with PAB at week 4–8 are derived from [44], in which sildenafil treatment was started 4 weeks after the PAB surgery, when RV dysfunction was already present (therapeutic strategy). a End-systolic elastance (Ees). b End-diastolic elastance (Eed). To allow comparison of Eed between the studies, the Eed of the initial 2 mmHg end-diastolic pressure drop during occlusion was used here (see methods on characterization of diastolic function). c Permillage RV fibrosis per unit surface area. d Ejection fraction. Mean ± SEM, * p < 0.05. PAB pulmonary artery banding, VEH vehicle treated, SIL sildenafil treated. Figure adapted from [44]; used without permission
Endothelin receptor antagonists
Endothelin receptor antagonists (ERA) have an anti-hypertrophic and anti-fibrotic effect on the RV in PH [96]. It is unclear whether this is a direct effect on the RV or a consequence of the vasodilatation and anti-proliferative effect on the pulmonary vasculature. Unfortunately, no studies have addressed this issue. Endothelin-1 increases contractility in mouse RV cardiomyocytes via an increase in intracellular Ca2+ transients through activation of the Endothelin Receptor-A and the Na+–Ca2+ exchanger [97]. Upregulation of endothelin-1 in RV myocardium of patients and models of compensated hypertrophy [98] suggests a potential rationale for ERAs. However, isolated heart studies showed that ERAs suppress both contractility and relaxation in the hypertrophic RV [98] which might explain the negative outcomes of preclinical studies on ERAs in RV pressure load without pulmonary vascular remodeling [99]. Intriguingly, the negative inotropic effect of ERAs is absent in LV failure, which may be due to differences in endothelin-1 activation. It may even be possible that ERAs have contrasting therapeutic effects in compensated and failing RVs, but so far, this has not been studied.
Apart from medical intervention, exercise training may be beneficial for the pressure-loaded RV. In patients with a pressure-loaded RV, exercise training induces a modest increase in exercise capacity [100]. In rats with MCT-induced PH; however, exercise training was beneficial in mild PH and deleterious in severe PH [101]. Exercise training has not been tested yet in other models of RV load.
Conclusion
Since the Working Group Statement, progress has been made in understanding the pathophysiological and pathobiological mechanisms of RV failure due to chronic abnormal loading conditions, specifically increased afterload. The RV adaptation to increased afterload is characterized by increased contractility, dilatation and hypertrophy, whereas clinical RV failure is associated with progressive diastolic deterioration and despite increased contractility, disturbed ventricular−arterial coupling. The pathobiology of the failing RV has several characteristic features. An important factor seems to be the lack of adequate increase in capillary density leading to a hypoxic environment and oxidative stress. Additionally, there is a metabolic switch from FA to glucose utilization in the process of RV adaptation, but the role of this switch in RV failure is yet unclear. So far, therapies aiming to specifically improve RV function have had limited success. The use of beta blockers and sildenafil may hold some promise, but new therapies have to be developed. Finally, a lot of insight has been gained with regard to the specific values and limitations of the different models of RV pressure load and the methods used to define and characterize RV function and failure. In the future, this may aid in the understanding of the pathobiology of RV failure and development of new therapeutic strategies.
Acknowledgments
This study was supported by the Sebald foundation and the Netherlands Heart Foundation [Grant #:2006T038; 2007T068].
Conflict of interest
Drs. Borgdorff, Dickinson, Berger and Bartelds have no conflicts of interest or financial ties to disclose.
Abbreviations
- CHD
Congenital heart diseases
- Ees
End-systolic elastance
- Eed
End-diastolic elastance
- ERA
Endothelin receptor antagonists
- FAO
Fatty acid oxidation
- LV
Left ventricle
- MCT
Monocrotaline
- PAB
Pulmonary artery banding
- PDE5
Phosphodiesterase 5
- PDK
Pyruvate dehydrogenase kinase
- PKG
Protein kinase G
- PH
Pulmonary hypertension
- RAAS
Renin−angiotensin aldosterone system
- RV
Right ventricle
References
- 1.Norozi K, Wessel A, Alpers V, Arnhold JO, Geyer S, Zoege M, Buchhorn R. Incidence and risk distribution of heart failure in adolescents and adults with congenital heart disease after cardiac surgery. Am J Cardiol. 2006;97:1238–1243. doi: 10.1016/j.amjcard.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 2.van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE, Vonk-Noordegraaf A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28:1250–1257. doi: 10.1093/eurheartj/ehl477. [DOI] [PubMed] [Google Scholar]
- 3.Meyer P, Filippatos GS, Ahmed MI, Iskandrian AE, Bittner V, Perry GJ, White M, Aban IB, Mujib M, Dell’Italia LJ, Ahmed A. Effects of right ventricular ejection fraction on outcomes in chronic systolic heart failure. Circulation. 2010;121:252–258. doi: 10.1161/CIRCULATIONAHA.109.887570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naeije R, Brimioulle S, Dewachter L. Biomechanics of the right ventricle in health and disease (2013 Grover Conference series) Pulm Circ. 2014;4:395–406. doi: 10.1086/677354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114:1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 6.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117:1717–1731. doi: 10.1161/CIRCULATIONAHA.107.653584. [DOI] [PubMed] [Google Scholar]
- 7.Zaffran S, Kelly RG, Meilhac SM, Buckingham ME, Brown NA. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004;95:261–268. doi: 10.1161/01.RES.0000136815.73623.BE. [DOI] [PubMed] [Google Scholar]
- 8.Bartelds B, Borgdorff MA, Smit-van Oosten A, Takens J, Boersma B, Nederhoff MG, Elzenga NJ, van Gilst WH, De Windt LJ, Berger RM. Differential responses of the right ventricle to abnormal loading conditions in mice: pressure vs. volume load. Eur J Heart Fail. 2011;13:1275–1282. doi: 10.1093/eurjhf/hfr134. [DOI] [PubMed] [Google Scholar]
- 9.Bartelds B, Berger RMF. The right ventricle in congenital heart diseases. In: Gaine SP, Naeije R, Paecock AJ, editors. The right heart. Berlin: Springer; 2014. p. 131. [Google Scholar]
- 10.Zong P, Tune JD, Downey HF. Mechanisms of oxygen demand/supply balance in the right ventricle. Exp Biol Med (Maywood) 2005;230:507–519. doi: 10.1177/153537020523000801. [DOI] [PubMed] [Google Scholar]
- 11.Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation. 2014;129:1033–1044. doi: 10.1161/CIRCULATIONAHA.113.001375. [DOI] [PubMed] [Google Scholar]
- 12.Sagawa K, Maughan L, Suga H, Sunagawa K. Cardiac contraction and the pressure–volume relationship. New York: Oxford University Press; 1988. [Google Scholar]
- 13.Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013;62:D22–D33. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 14.Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, Michelakis ED. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med. 2013;91:1315–1327. doi: 10.1007/s00109-013-1059-4. [DOI] [PubMed] [Google Scholar]
- 15.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med. 2012;90:31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borgdorff MA, Bartelds B, Dickinson MG, Boersma B, Weij M, Zandvoort A, Sillje HH, Steendijk P, de Vroomen M, Berger RM. Sildenafil enhances systolic adaptation, but does not prevent diastolic dysfunction, in the pressure-loaded right ventricle. Eur J Heart Fail. 2012;14:1067–1074. doi: 10.1093/eurjhf/hfs094. [DOI] [PubMed] [Google Scholar]
- 17.Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, de Vroomen M, Berger RM. Distinct loading conditions reveal various patterns of right ventricular adaptation. Am J Physiol Heart Circ Physiol. 2013;305:H354–H364. doi: 10.1152/ajpheart.00180.2013. [DOI] [PubMed] [Google Scholar]
- 18.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012;302:L363–L369. doi: 10.1152/ajplung.00212.2011. [DOI] [PubMed] [Google Scholar]
- 19.Bartelds B, Borgdorff MAJ, Berger RMF. Right ventricular adaptation in congenital heart diseases. J Cardiovasc Dev Dis. 2014;1:83. doi: 10.3390/jcdd1010083. [DOI] [Google Scholar]
- 20.Bartelds B, Borgdorff MA, Boersma B, Takens J, Smit-van Oosten A, De Windt LJ, Berger RMF. Right ventricular adaptation to pressure load in mice is improved after blockade of calcineurin activation. Eur Heart J. 2011;31(suppl):305. [Google Scholar]
- 21.Reddy S, Zhao M, Hu DQ, Fajardo G, Katznelson E, Punn R, Spin JM, Chan FP, Bernstein D. Physiologic and molecular characterization of a murine model of right ventricular volume overload. Am J Physiol Heart Circ Physiol. 2013;304:H1314–H1327. doi: 10.1152/ajpheart.00776.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickinson MG, Bartelds B, Borgdorff MA, Berger RM. The role of disturbed blood flow in the development of pulmonary arterial hypertension: lessons from preclinical animal models. Am J Physiol Lung Cell Mol Physiol. 2013;305:L1–L14. doi: 10.1152/ajplung.00031.2013. [DOI] [PubMed] [Google Scholar]
- 23.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 24.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 25.Ghobadi G, Bartelds B, van der Veen SJ, Dickinson MG, Brandenburg S, Berger RM, Langendijk JA, Coppes RP, van Luijk P. Lung irradiation induces pulmonary vascular remodelling resembling pulmonary arterial hypertension. Thorax. 2012;67:334–341. doi: 10.1136/thoraxjnl-2011-200346. [DOI] [PubMed] [Google Scholar]
- 26.Faber MJ, Dalinghaus M, Lankhuizen IM, Steendijk P, Hop WC, Schoemaker RG, Duncker DJ, Lamers JM, Helbing WA. Right and left ventricular function after chronic pulmonary artery banding in rats assessed with biventricular pressure–volume loops. Am J Physiol Heart Circ Physiol. 2006;291:H1580–H1586. doi: 10.1152/ajpheart.00286.2006. [DOI] [PubMed] [Google Scholar]
- 27.Schafer S, Ellinghaus P, Janssen W, Kramer F, Lustig K, Milting H, Kast R, Klein M. Chronic inhibition of phosphodiesterase 5 does not prevent pressure-overload-induced right-ventricular remodelling. Cardiovasc Res. 2009;82:30–39. doi: 10.1093/cvr/cvp002. [DOI] [PubMed] [Google Scholar]
- 28.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 29.Urashima T, Zhao M, Wagner R, Fajardo G, Farahani S, Quertermous T, Bernstein D. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol. 2008;295:H1351–H1368. doi: 10.1152/ajpheart.91526.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LekanneDeprez RH, van den Hoff MJ, de Boer PA, Ruijter PM, Maas AA, Chamuleau RA, Lamers WH, Moorman AF. Changing patterns of gene expression in the pulmonary trunk-banded rat heart. J Mol Cell Cardiol. 1998;30:1877–1888. doi: 10.1006/jmcc.1998.0753. [DOI] [PubMed] [Google Scholar]
- 31.Borgdorff MA, Koop AM, Bloks VW, Dickinson MG, Steendijk P, Sillje HH, van Wiechen MP, Berger RM, Bartelds B. Clinical symptoms of right ventricular failure in experimental chronic pressure load are associated with progressive diastolic dysfunction. J Mol Cell Cardiol. 2015;79:244–253. doi: 10.1016/j.yjmcc.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 32.Piao L, Fang YH, Parikh KS, Ryan JJ, D’Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012;126:2859–2869. doi: 10.1161/CIRCULATIONAHA.112.109868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brimioulle S, Wauthy P, Naeije R. Single-beat evaluation of right ventricular contractility. Crit Care Med. 2005;33:917–918. doi: 10.1097/01.CCM.0000156236.49988.31. [DOI] [PubMed] [Google Scholar]
- 34.de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5:97–105. doi: 10.1161/CIRCHEARTFAILURE.111.964494. [DOI] [PubMed] [Google Scholar]
- 35.de Vroomen M, Cardozo RH, Steendijk P, van Bel F, Baan J. Improved contractile performance of right ventricle in response to increased RV afterload in newborn lamb. Am J Physiol Heart Circ Physiol. 2000;278:H100–H105. doi: 10.1152/ajpheart.2000.278.1.H100. [DOI] [PubMed] [Google Scholar]
- 36.Hessel MH, Steendijk P, den Adel B, Schutte CI, van der Laarse A. Characterization of right ventricular function after monocrotaline-induced pulmonary hypertension in the intact rat. Am J Physiol Heart Circ Physiol. 2006;291:H2424–H2430. doi: 10.1152/ajpheart.00369.2006. [DOI] [PubMed] [Google Scholar]
- 37.Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res. 2007;75:770–781. doi: 10.1016/j.cardiores.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 38.Kreymborg K, Uchida S, Gellert P, Schneider A, Boettger T, Voswinckel R, Wietelmann A, Szibor M, Weissmann N, Ghofrani AH, Schermuly R, Schranz D, Seeger W, Braun T. Identification of right heart-enriched genes in a murine model of chronic outflow tract obstruction. J Mol Cell Cardiol. 2010;49:598–605. doi: 10.1016/j.yjmcc.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 39.Rondelet B, Dewachter L, Kerbaul F, Dewachter C, Hubloue I, Fesler P, Franck S, Remmelink M, Brimioulle S, Naeije R. Sildenafil added to sitaxsentan in overcirculation-induced pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2010;299:H1118–H1123. doi: 10.1152/ajpheart.00418.2010. [DOI] [PubMed] [Google Scholar]
- 40.Bogaard HJ, Abe K, Noordegraaf AV, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. doi: 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 41.Gaynor SL, Maniar HS, Bloch JB, Steendijk P, Moon MR. Right atrial and ventricular adaptation to chronic right ventricular pressure overload. Circulation. 2005;112:I212–I218. doi: 10.1161/CIRCULATIONAHA.104.517789. [DOI] [PubMed] [Google Scholar]
- 42.Leeuwenburgh BP, Steendijk P, Helbing WA, Baan J. Indexes of diastolic RV function: load dependence and changes after chronic RV pressure overload in lambs. Am J Physiol Heart Circ Physiol. 2002;282:H1350–H1358. doi: 10.1152/ajpheart.00782.2001. [DOI] [PubMed] [Google Scholar]
- 43.Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, Berger RM. A cornerstone of heart failure treatment is not effective in experimental right ventricular failure. Int J Cardiol. 2013;169:183–189. doi: 10.1016/j.ijcard.2013.08.102. [DOI] [PubMed] [Google Scholar]
- 44.Borgdorff MA, Bartelds B, Dickinson MG, van Wiechen MP, Steendijk P, de Vroomen M, Berger RM. Sildenafil treatment in established right ventricular dysfunction improves diastolic function and attenuates interstitial fibrosis independent from afterload. Am J Physiol Heart Circ Physiol. 2014;307:H361–H369. doi: 10.1152/ajpheart.00843.2013. [DOI] [PubMed] [Google Scholar]
- 45.Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmuller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013;128(2016–25):1–10. doi: 10.1161/CIRCULATIONAHA.113.001873. [DOI] [PubMed] [Google Scholar]
- 46.Bove T, Vandekerckhove K, Bouchez S, Wouters P, Somers P, Van Nooten G. Role of myocardial hypertrophy on acute and chronic right ventricular performance in relation to chronic volume overload in a porcine model: relevance for the surgical management of tetralogy of Fallot. J Thorac Cardiovasc Surg. 2014;147:1956–1965. doi: 10.1016/j.jtcvs.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 47.Chugh SS, Whitesel S, Turner M, Roberts CT, Jr, Nagalla SR. Genetic basis for chamber-specific ventricular phenotypes in the rat infarct model. Cardiovasc Res. 2003;57:477–485. doi: 10.1016/S0008-6363(02)00703-4. [DOI] [PubMed] [Google Scholar]
- 48.Lamberts RR, Caldenhoven E, Lansink M, Witte G, Vaessen RJ, St Cyr JA, Stienen GJ. Preservation of diastolic function in monocrotaline-induced right ventricular hypertrophy in rats. Am J Physiol Heart Circ Physiol. 2007;293:H1869–H1876. doi: 10.1152/ajpheart.00294.2007. [DOI] [PubMed] [Google Scholar]
- 49.Buechel ERV, Mertens LL. Imaging the right heart: the use of integrated multimodality imaging. Eur Heart J. 2012;33:949–960. doi: 10.1093/eurheartj/ehr490. [DOI] [PubMed] [Google Scholar]
- 50.Andersen A, Nielsen JM, Peters CD, Schou UK, Sloth E, Nielsen-Kudsk JE. Effects of phosphodiesterase-5 inhibition by sildenafil in the pressure overloaded right heart. Eur J Heart Fail. 2008;10:1158–1165. doi: 10.1016/j.ejheart.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 51.Redout EM, van der Toorn A, Zuidwijk MJ, van de Kolk CW, van Echteld CJ, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H1038–H1047. doi: 10.1152/ajpheart.00097.2009. [DOI] [PubMed] [Google Scholar]
- 52.Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, Weber O, Higgins CB, Ewert P, Fleck E, Nagel E, Schulze-Neick I, Lange P. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation. 2004;110:2010–2016. doi: 10.1161/01.CIR.0000143138.02493.DD. [DOI] [PubMed] [Google Scholar]
- 53.Vanderpool RR, Pinsky MR, Naeije R, Deible C, Kosaraju V, Bunner C, Mathier MA, Lacomis J, Champion HC, Simon MA. RV-pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart. 2015;101:37–43. doi: 10.1136/heartjnl-2014-306142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gan C, Lankhaar JW, Marcus JT, Westerhof N, Marques KM, Bronzwaer JG, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Impaired left ventricular filling due to right-to-left ventricular interaction in patients with pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H1528–H1533. doi: 10.1152/ajpheart.01031.2005. [DOI] [PubMed] [Google Scholar]
- 55.Ghobadi G, van der Veen S, Bartelds B, de Boer RA, Dickinson MG, de Jong JR, Faber H, Niemantsverdriet M, Brandenburg S, Berger RM, Langendijk JA, Coppes RP, van Luijk P. Physiological interaction of heart and lung in thoracic irradiation. Int J Radiat Oncol Biol Phys. 2012;84:e639–e646. doi: 10.1016/j.ijrobp.2012.07.2362. [DOI] [PubMed] [Google Scholar]
- 56.Handoko ML, Lamberts RR, Redout EM, de Man FS, Boer C, Simonides WS, Paulus WJ, Westerhof N, Allaart CP, Vonk-Noordegraaf A. Right ventricular pacing improves right heart function in experimental pulmonary arterial hypertension: a study in the isolated heart. Am J Physiol Heart Circ Physiol. 2009;297:H1752–H1759. doi: 10.1152/ajpheart.00555.2009. [DOI] [PubMed] [Google Scholar]
- 57.Damiano RJ, Jr, La Follette P, Jr Cox JL, Lowe JE, Santamore WP. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261:H1514–H1524. doi: 10.1152/ajpheart.1991.261.5.H1514. [DOI] [PubMed] [Google Scholar]
- 58.Apitz C, Honjo O, Humpl T, Li J, Assad RS, Cho MY, Hong J, Friedberg MK, Redington AN. Biventricular structural and functional responses to aortic constriction in a rabbit model of chronic right ventricular pressure overload. J Thorac Cardiovasc Surg. 2012;144:1494–1501. doi: 10.1016/j.jtcvs.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 59.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Albada ME, Berger RM, Niggebrugge M, van Veghel R, Cromme-Dijkhuis AH, Schoemaker RG. Prostacyclin therapy increases right ventricular capillarisation in a model for flow-associated pulmonary hypertension. Eur J Pharmacol. 2006;549:107–116. doi: 10.1016/j.ejphar.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 61.Wong YY, Ruiter G, Lubberink M, Raijmakers PG, Knaapen P, Marcus JT, Boonstra A, Lammertsma AA, Westerhof N, van der Laarse WJ, Vonk-Noordegraaf A. Right ventricular failure in idiopathic pulmonary arterial hypertension is associated with inefficient myocardial oxygen utilization. Circ Heart Fail. 2011;4:700–706. doi: 10.1161/CIRCHEARTFAILURE.111.962381. [DOI] [PubMed] [Google Scholar]
- 62.Bartelds B, Knoester H, Smid GB, Takens J, Visser GH, Penninga L, van der Leij FR, Beaufort-Krol GC, Zijlstra WG, Heymans HS, Kuipers JR. Perinatal changes in myocardial metabolism in lambs. Circulation. 2000;102:926–931. doi: 10.1161/01.CIR.102.8.926. [DOI] [PubMed] [Google Scholar]
- 63.Kim Y, Goto H, Kobayashi K, Sawada Y, Miyake Y, Fujiwara G, Chiba H, Okada T, Nishimura T. Detection of impaired fatty acid metabolism in right ventricular hypertrophy: assessment by I-123 beta-methyl iodophenyl pentadecanoic acid (BMIPP) myocardial single-photon emission computed tomography. Ann Nucl Med. 1997;11:207–212. doi: 10.1007/BF03164765. [DOI] [PubMed] [Google Scholar]
- 64.Bartelds B, Takens J, Smid GB, Zammit VA, Prip-Buus C, Kuipers JR, van der Leij FR. Myocardial carnitine palmitoyltransferase I expression and long-chain fatty acid oxidation in fetal and newborn lambs. Am J Physiol Heart Circ Physiol. 2004;286:H2243–H2248. doi: 10.1152/ajpheart.00864.2003. [DOI] [PubMed] [Google Scholar]
- 65.Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, Eijk PP, van Hardeveld C, Kasanmoentalib S, Visser FC, Ylstra B, Simonides WS. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol Genomics. 2005;21:314–323. doi: 10.1152/physiolgenomics.00185.2004. [DOI] [PubMed] [Google Scholar]
- 66.Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG, Drake JI, Farkas L, Kraskauskas D, Wijesinghe DS, Chalfant CE, Bigbee J, Abbate A, Lesnefsky EJ, Bogaard HJ, Voelkel NF. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail. 2013;6:136–144. doi: 10.1161/CIRCHEARTFAILURE.111.966127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, Takahashi T, Nawata J, Ido T, Watanabe J, Shirato K. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–1855. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 68.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF, Natarajan R. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45:1239–1247. doi: 10.1165/rcmb.2010-0412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med. 2010;88:47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 71.Piao L, Marsboom G, Archer SL. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J Mol Med. 2010;88:1011–1020. doi: 10.1007/s00109-010-0679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Dekkers DH, Duncker DJ, Helbing WA, Lamers JM. Proteomic changes in the pressure overloaded right ventricle after 6 weeks in young rats: correlations with the degree of hypertrophy. Proteomics. 2005;5:2519–2530. doi: 10.1002/pmic.200401313. [DOI] [PubMed] [Google Scholar]
- 73.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 74.Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. 2013;75:95–126. doi: 10.1146/annurev-physiol-030212-183804. [DOI] [PubMed] [Google Scholar]
- 75.Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2013;127:115–125. doi: 10.1161/CIRCULATIONAHA.112.133413. [DOI] [PubMed] [Google Scholar]
- 76.Bartelds B, van Loon RL, Mohaupt S, Wijnberg H, Dickinson MG, Boersma B, Takens J, van Albada M, Berger RM. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest. 2012;141:651–660. doi: 10.1378/chest.11-0663. [DOI] [PubMed] [Google Scholar]
- 77.van Albada ME, Bartelds B, Wijnberg H, Mohaupt S, Dickinson MG, Schoemaker RG, Kooi K, Gerbens F, Berger RM. Gene expression profile in flow-associated pulmonary arterial hypertension with neointimal lesions. Am J Physiol Lung Cell Mol Physiol. 2010;298:L483–L491. doi: 10.1152/ajplung.00106.2009. [DOI] [PubMed] [Google Scholar]
- 78.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 79.Rajagopalan V, Zhao M, Reddy S, Fajardo GA, Wang X, Dewey S, Gomes AV, Bernstein D. Altered ubiquitin-proteasome signaling in right ventricular hypertrophy and failure. Am J Physiol Heart Circ Physiol. 2013;305:H551–H562. doi: 10.1152/ajpheart.00771.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dewachter C, Dewachter L, Rondelet B, Fesler P, Brimioulle S, Kerbaul F, Naeije R. Activation of apoptotic pathways in experimental acute afterload-induced right ventricular failure. Crit Care Med. 2010;38:1405–1413. doi: 10.1097/CCM.0b013e3181de8bd3. [DOI] [PubMed] [Google Scholar]
- 81.Rondelet B, Dewachter C, Kerbaul F, Kang X, Fesler P, Brimioulle S, Naeije R, Dewachter L. Prolonged overcirculation-induced pulmonary arterial hypertension as a cause of right ventricular failure. Eur Heart J. 2012;33:1017–1026. doi: 10.1093/eurheartj/ehr111. [DOI] [PubMed] [Google Scholar]
- 82.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 83.Nagendran J, Gurtu V, Fu DZ, Dyck JR, Haromy A, Ross DB, Rebeyka IM, Michelakis ED. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg. 2008;136:168–178. doi: 10.1016/j.jtcvs.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 84.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bar H, Kreuzer J, Cojoc A, Jahn L. Upregulation of embryonic transcription factors in right ventricular hypertrophy. Basic Res Cardiol. 2003;98:285–294. doi: 10.1007/s00395-003-0410-2. [DOI] [PubMed] [Google Scholar]
- 86.Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med. 2011;183:1402–1410. doi: 10.1164/rccm.201007-1106OC. [DOI] [PubMed] [Google Scholar]
- 87.Reddy S, Zhao M, Hu DQ, Fajardo G, Hu S, Ghosh Z, Rajagopalan V, Wu JC, Bernstein D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics. 2012;44:562–575. doi: 10.1152/physiolgenomics.00163.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. doi: 10.1164/rccm.201003-0335OC. [DOI] [PubMed] [Google Scholar]
- 89.van der Bom T, Winter MM, Bouma BJ, Groenink M, Vliegen HW, Pieper PG, van Dijk AP, Sieswerda GT, Roos-Hesselink JW, Zwinderman AH, Mulder BJ. Effect of valsartan on systemic right ventricular function: a double-blind, randomized, placebo-controlled pilot trial. Circulation. 2013;127:322–330. doi: 10.1161/CIRCULATIONAHA.112.135392. [DOI] [PubMed] [Google Scholar]
- 90.Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361:1864–1871. doi: 10.1056/NEJMct0904473. [DOI] [PubMed] [Google Scholar]
- 91.Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Pressure-overload magnitude-dependence of the anti-hypertrophic efficacy of PDE5A inhibition. J Mol Cell Cardiol. 2009;46:560–567. doi: 10.1016/j.yjmcc.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hsu S, Nagayama T, Koitabashi N, Zhang M, Zhou L, Bedja D, Gabrielson KL, Molkentin JD, Kass DA, Takimoto E. Phosphodiesterase 5 inhibition blocks pressure overload-induced cardiac hypertrophy independent of the calcineurin pathway. Cardiovasc Res. 2009;81:301–309. doi: 10.1093/cvr/cvn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, Brozovich FV, Burnett JC, Jr, Linke WA, Redfield MM. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. 2011;124:2882–2891. doi: 10.1161/CIRCULATIONAHA.111.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 95.Lang M, Kojonazarov B, Tian X, Kalymbetov A, Weissmann N, Grimminger F, Kretschmer A, Stasch JP, Seeger W, Ghofrani HA, Schermuly RT. The soluble guanylate cyclase stimulator riociguat ameliorates pulmonary hypertension induced by hypoxia and SU5416 in rats. PLoS one. 2012;7:e43433. doi: 10.1371/journal.pone.0043433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choudhary G, Troncales F, Martin D, Harrington EO, Klinger JR. Bosentan attenuates right ventricular hypertrophy and fibrosis in normobaric hypoxia model of pulmonary hypertension. J Heart Lung Transplant. 2011;30:827–833. doi: 10.1016/j.healun.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagasaka T, Izumi M, Hori M, Ozaki H, Karaki H. Positive inotropic effect of endothelin-1 in the neonatal mouse right ventricle. Eur J Pharmacol. 2003;472:197–204. doi: 10.1016/S0014-2999(03)01936-8. [DOI] [PubMed] [Google Scholar]
- 98.Nagendran J, Sutendra G, Paterson I, Champion HC, Webster L, Chiu B, Haromy A, Rebeyka IM, Ross DB, Michelakis ED. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ Res. 2013;112:347–354. doi: 10.1161/CIRCRESAHA.111.300448. [DOI] [PubMed] [Google Scholar]
- 99.Mouchaers KT, Schalij I, Versteilen AM, Hadi AM, van Nieuw Amerongen GP, van Hinsbergh VW, Postmus PE, van der Laarse WJ, Vonk-Noordegraaf A. Endothelin receptor blockade combined with phosphodiesterase-5 inhibition increases right ventricular mitochondrial capacity in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2009;297:H200–H207. doi: 10.1152/ajpheart.00893.2008. [DOI] [PubMed] [Google Scholar]
- 100.Winter MM, van der Bom T, de Vries LC, Balducci A, Bouma BJ, Pieper PG, van Dijk AP, van der Plas MN, Picchio FM, Mulder BJ. Exercise training improves exercise capacity in adult patients with a systemic right ventricle: a randomized clinical trial. Eur Heart J. 2012;33:1378–1385. doi: 10.1093/eurheartj/ehr396. [DOI] [PubMed] [Google Scholar]
- 101.Handoko ML, de Man FS, Happe CM, Schalij I, Musters RJ, Westerhof N, Postmus PE, Paulus WJ, van der Laarse WJ, Vonk-Noordegraaf A. Opposite effects of training in rats with stable and progressive pulmonary hypertension. Circulation. 2009;120:42–49. doi: 10.1161/CIRCULATIONAHA.108.829713. [DOI] [PubMed] [Google Scholar]
- 102.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, Francois C, Schalij I, Dorfmuller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, van der Velden J, Vonk-Noordegraaf A, Humbert M, Eddahibi S, Guignabert C. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:780–789. doi: 10.1164/rccm.201203-0411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mouchaers KT, Schalij I, de Boer MA, Postmus PE, van Hinsbergh VW, van Nieuw Amerongen GP, Vonk Noordegraaf A, van der Laarse WJ. Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur Respir J. 2010;36:800–807. doi: 10.1183/09031936.00130209. [DOI] [PubMed] [Google Scholar]
- 104.Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, Archer SL. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med . 2013;91:333–346. doi: 10.1007/s00109-012-0982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, Wang LC, Weiss RM, Grumbach IM, Anderson ME, Song LS. Sildenafil prevents and reverses transverse-tubule remodeling and Ca(2+) handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension. 2012;59:355–362. doi: 10.1161/HYPERTENSIONAHA.111.180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matori H, Umar S, Nadadur RD, Sharma S, Partow-Navid R, Afkhami M, Amjedi M, Eghbali M. Genistein, a soy phytoestrogen, reverses severe pulmonary hypertension and prevents right heart failure in rats. Hypertension. 2012;60:425–430. doi: 10.1161/HYPERTENSIONAHA.112.191445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nadadur RD, Umar S, Wong G, Eghbali M, Iorga A, Matori H, Partow-Navid R, Eghbali M. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J Appl Physiol. 2012;113:149–158. doi: 10.1152/japplphysiol.01349.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, van der Laarse A, Eghbali M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184:715–723. doi: 10.1164/rccm.201101-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Umar S, Lee JH, de Lange E, Iorga A, Partow-Navid R, Bapat A, van der Laarse A, Saggar R, Saggar R, Ypey DL, Karagueuzian HS, Eghbali M. Spontaneous ventricular fibrillation in right ventricular failure secondary to chronic pulmonary hypertension. Circ Arrhythm Electrophysiol. 2012;5:181–190. doi: 10.1161/CIRCEP.111.967265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Umar S, Nadadur RD, Li J, Maltese F, Partownavid P, van der Laarse A, Eghbali M. Intralipid prevents and rescues fatal pulmonary arterial hypertension and right ventricular failure in rats. Hypertension. 2011;58:512–518. doi: 10.1161/HYPERTENSIONAHA.110.168781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Verhoeven AJ, Duncker DJ, Helbing WA, Lamers JM. Time dependent changes in cytoplasmic proteins of the right ventricle during prolonged pressure overload. J Mol Cell Cardiol. 2007;43:197–209. doi: 10.1016/j.yjmcc.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 112.Okada M, Harada T, Kikuzuki R, Yamawaki H, Hara Y. Effects of telmisartan on right ventricular remodeling induced by monocrotaline in rats. J Pharmacol Sci. 2009;111:193–200. doi: 10.1254/jphs.09112FP. [DOI] [PubMed] [Google Scholar]
- 113.Andersen A, Nielsen JM, Holmboe S, Vildbrad MD, Nielsen-Kudsk JE. The effects of cyclic guanylate cyclase stimulation on right ventricular hypertrophy and failure alone and in combination with phosphodiesterase-5 inhibition. J Cardiovasc Pharmacol. 2013;62:167–173. doi: 10.1097/FJC.0b013e318294a1cf. [DOI] [PubMed] [Google Scholar]
- 114.Lu B, Yu H, Zwartbol M, Ruifrok WP, van Gilst WH, de Boer RA, Silljé HH. Identification of hypertrophy- and heart failure-associated genes by combining in vitro and in vivo models. Physiol Genomics. 2012;44:443–454. doi: 10.1152/physiolgenomics.00148.2011. [DOI] [PubMed] [Google Scholar]
- 115.Paulin R, Sutendra G, Gurtu V, Dromparis P, Haromy A, Provencher S, Bonnet S, Michelakis ED. A miR-208-Mef2 axis drives the decompensation of right ventricular function in pulmonary hypertension. Circ Res. 2015;116:56–69. doi: 10.1161/CIRCRESAHA.115.303910. [DOI] [PubMed] [Google Scholar]