

Abstract
Darier disease (DD) is an autosomal dominant inherited skin disorder, characterized by abnormal keratinization, loss of adhesion between epidermal cells, termed acantholysis, and the development of warty papules and plaques on the central trunk, forehead, scalp and flexures. These symptoms are often exacerbated by heat, sweating, sunburn and stress. Mutations in the ATP2A2 gene, encoding SERCA2, a calcium pump of the sarco/endoplasmic reticulum, are responsible for the disease. The aim of the present study was to investigate two pedigrees of DD and to examine the genetic mutations. DNA was extracted from peripheral blood, which was obtained from four patients with DD, 10 healthy individuals from the two families and 100 ethnicity-matched control individuals, on which subsequent polymerase chain reaction amplification and direct automated DNA sequencing were performed. The results identified two novel missense mutations, p.R603I and p.G749 V. These mutations were not identified in the remaining ten healthy individuals in the same families or in any of the 100 controls. These mutations may contribute to the expanding database of ATP2A2 gene mutations in patients with DD.
Keywords: ATP2A2, Darier disease, mutation
Introduction
Darier disease (DD; OMIM 124200), also termed keratosis follicularis and Darier-White disease, is an autosomal dominant inherited skin disorder, characterized by abnormal keratinization and loss of adhesion between epidermal cells, termed acantholysis, and the development of warty papules and plaques in seborrheic regions, including the central trunk, forehead, scalp and flexures (1). It has a prevalence of 1:30,000–100,000 worldwide. The lesions usually present in the second decade, and penetrance is almost complete, although the phenotypic expression is variable (2). Certain patients exhibit pits or keratotic papules on the palms and distinctive nail abnormalities, whereas patients with severe disease are handicapped by widespread malodour keratotic plaques. Neurological and psychiatric abnormalities have also been reported in patients with DD (1). The symptoms are often exacerbated by heat, sweating, sunburn and stress (3,4).
The ATP2A2 gene encoding SERCA2, a calcium pump of the sarco/endoplasmic reticulum, has been identified as the defective gene in the disease (3). ATP2A2 is positioned on chromosome 12q23–q24.1 and has 21 exons (5). SERCA pumps are important in Ca2+ transport between the cytoplasm and the endoplasmic reticulum (ER), using energy from adenosine triphosphate (ATP) hydrolysis, and is, therefore, important in signal transduction for gene expression and cell differentiation (6,7).
The aim of the present study was to investigate the genetic pedigrees of two families with DD, in order to identify novel mutations.
Materials and methods
Patients
In the present study, two families, containing individuals with DD, were recruited. The proband in pedigree A (Fig. 1A), a 36-year-old male, exhibited widespread follicular papules at the age of 26 years (Fig. 1B), and the proband’s mother exhibited similar clinical manifestations. A 0.4 cm × 0.3 cm biopsy was taken from the skin of all patients. The biopsies were fixed in 10% buffered formalin (Shanghai Shiyi Chemicals Reagent Co.,Ltd., Shanghai, China) and routinely processed and embedded in paraffin. Sections (4 µm) were cut and stained with hematoxylin and eosin (Shanghai Shiyi Chemicals Reagent Co.,Ltd.). Histological examination demonstrated hyperkeratosis with parakeratosis, and suprabasal acantholysis with dyskeratotic cells, including corps ronds and grains. The diagnosis of DD was based on clinical and histopathological examinations (Fig.1C). The proband in pedigree B (Fig. 1D), an 18-year-old man, had DD with a 4-year history of papules and hyperkeratotic plaques over the seborrheic area. Physical examination revealed scattered erythema, and follicular papules on the neck and armpit (Fig. 1E). The proband’s father, a 43-year-old male, had been diagnosed with DD for >10 years. This patient manifested greasy, hyperkeratotic papules on the face, forehead, armpit, upper extremities and inguen. None of the patients presented with any neuropsychiatric symptoms. The diagnosis of DD was based on clinical and histopathological examinations (Fig. 1F).
Figure 1.

(A) Pedigree of family A, the proband is indicated by the black arrow. (B) Clinical characteristics of the proband of family A, demonstrating keratotic papules in a seborrhoeic distribution. (C) Skin biopsy obtained from family A. The cells were stained using hematoxylin and eosin staining (magnification, x100). (D) Pedigree of family B, the proband is indicated by the black arrow. (E) Clinical characteristics of the proband of family B, demonstrating keratotic papules in a seborrhoeic distribution. (F) Skin biopsy obtained from family B. The cells were stained using hematoxylin and eosin staining (magnification, x100).
Genetic analysis
Ethical approval for the present study was obtained from the Ethical Committee of Fuzhou Dermatology Hospital for human studies (Fuzhou, China). Informed consent was obtained from the two family members. A total of 5 ml median cubital vein, basilic vein or cephalic vein blood was collected into a tube with 2% EDTA (0.5 ml), then genomic DNA was extracted from EDTA blood samples, using DNA extraction regents (TIANamp Blood DNA Midi Kit DP332-01, Tiangen, Shanghai, China). Polymerase chain reaction (PCR) was performed in a 25 µl reaction volume, containing 12.5 µl PCR mix, 11 µl double distilled H2O, 0.5 µl forward primer, 0.5 µl reverse primer and 0.5 µl (200 ng) genomic DNA. The PCR amplification of ATP2A2 was performed from the genomic DNA using 19 pairs of primers, spanning all 21 exons and flanking splice sites of the gene, as presented in Table I (5). The amplification was performed at 94°C for 5 min, followed by 32 cycles of 30 sec denaturation at 94°C, 30 sec annealing at 50–62°C, 45 sec extension at 72°C, and termination for 10 min at 72°C (Mastercycler ep gradient S; Eppendorf, Hamburg, Germany).
Table I.
Polymerase chain reaction primers for amplification of ATP2A2 from genomic DNA.
| Exon | Forward primer sequence | Reverse primer sequence | Annealing temperature (°C) |
|---|---|---|---|
| 1 | CGAGGCGGAGGCGAGGAG | GGAGCCGAAGCCCACGCG | 62.0 |
| 2 + 3 | ACCTCCCTCTTGACACATTG | GACAACTCCTAACCACACTG | 55.1 |
| 4 | CGTGCCATTTCTCTTCTAGG | CTCAACACATCAGGAAAAACAG | 55.1 |
| 5 | AGTGTCAGGCAGGTCTTTAC | AGGAAGGGAGGTGCTAAAAC | 55.1 |
| 6 | AGCCTCATTCTCTTCCTTCC | ATGGAGCGAGACTAAAGCAC | 55.1 |
| 7 | CTTGGTGTGGGTCGCAGAG | CCTTTAGAATGATAGCCAGTG | 50.3 |
| 8 | GTTGTATGGCTGGTTGCTTG | GAACAAAGAACCACGACACG | 52.0 |
| 9 | GGTTGTTTGCCTTTGTCCTAA | ATAACAAACACAAATCCCTCTT | 50.3 |
| 10 | GGCGACCATACCCTGCTC | CCCACCCCACCCTTGAAC | 55.0 |
| 11 | TCAGAGGAGGATAAAAATGGC | CTGTAAGTTTGAGGAGATAAGG | 52.0 |
| 12 + 13 | ATTGCCACCCAGTAGTATCC | GAACTGTTTGACCTTTTGCTTG | 55.1 |
| 14 | CTAGAACTTGCCACTTTTATTTA | GAGGCTACTATGTGCTTGTG | 50.0 |
| 15 | TTTCCTCCTGCTTCCCATTC | GCAATCTGGAGAGCAAACTG | 55.0 |
| 16 | TCATTTATTTTTCTGGAGGAGG | CATCTCTGTCTTTTGCTACCC | 53.8 |
| 17 | TGATCTTCGTCCTTGTGGGG | TGATAGATACCGAAACCACAG | 53.8 |
| 18 | GGGTTGGAGCCTGGACTTG | TTTTGGGAAGGGAAGAACTGT | 55.0 |
| 19 + 20 | TCCCCACCTCTCCTTGCTC | CCTCCATCACCAGCCAGTAT | 57.5 |
| 21 | GTTCCTTTTCATCTGTCGCTG | TCTTTTTCCCCAACATCAGTC | 53.8 |
The purified PCR products were detected by bidirectional direct sequencing using an ABI3130 automated sequencing system (Applied Biosystems, Foster City, CA, USA). DNA, extracted from blood samples obtained from 100 ethnicity-matched healthy individuals, were used as controls. The ATP2A2 sequence, which was published in the NCBI database (http://www.ncbi.nlm.nih.gov), was used as a reference sequence, which was compared with the experimental results to identify mutations. The nucleotide numbering followed the NM_001681.3 sequence and the amino acid numbering followed the NP_001672.1 ATP2A2 protein sequence. To determine whether the mutations were pathogenic, in silico analysis was performed using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) (8). The categories of pathogenicity included benign, possibly damaging and probably damaging. The mutations were assigned a score between 0 and 1, with the highest score reflecting the closest adherence to be damaging. The mutation (R603I) was predicted to be ‘probably damaging’ with a score of 1 and the mutation (G749V) was predicted to be ‘probably damaging’ with a score of 0.998. Scale-invariant feature transform (SIFT) (http://sift.jcvi.org/) (9), was also used which predicts the effect of a specific amino acid change at the protein level. Amino acids with probabilities <0.05 are predicted to be deleterious. Substitutions at position 603 from R to I and position 749 from G to V are predicted to ‘affect protein function’ with a score of 0.00.
Results
The entire 21 exons and exon-intron boundaries of the ATP2A2 gene were sequenced in the two families, in which two novel heterozygous mutations were identified (Fig. 2). In family A, the mutation analysis revealed a nucleotide alteration between G and T (2246G>T) in exon 15 in the two affected individuals, which caused an amino acid substitution from glycine to valine and was designated as p.G749V. In family B, a heterozygous nucleotide change in G1808T in exon 14 was identified in the two affected individuals. This mutation caused an amino acid substitution from arginine to isoleucine acid at position 603 (p.R603I). No ATP2A2 variants were detected in the unaffected individuals, in either the healthy relatives in the two families or in the 100 ethnicity-matched healthy controls. Furthermore, PolyPhen-2 and SIFT were used to predict the pathogenicity of the mutations. The two novel missense mutations were scored as ‘probably damaging’ using PolyPhen. The prediction scores of the SIFT for each mutation were 0, therefore, p.R603I and p.G749V were considered deleterious mutations, affecting the protein structure and/or function of SCRCA2.
Figure 2.
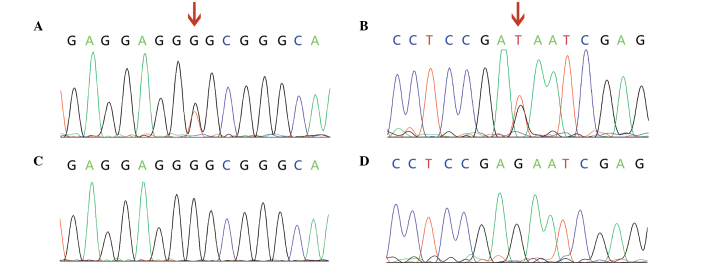
Mutation analysis of the ATP2A2 gene with Chromas, version 2. 31. (A) Proband of family A exhibited a missense mutation (p.G749V) in exon 15; whereas the (B) proband of family B exhibited a missense mutation (p.R603I) in exon 14. Wild-type alleles of (C) exon 15 and (D) exon 14 of the ATP2A2 gene are presented.
Discussion
In the present study, two novel mutations in the ATP2A2 gene were identified following genetic analyses of two families containing patients with DD. The G1808T mutation was detected in codon 603, located within the ATP-binding domain. This missense mutation resulted in the substitution of hydrophilic polar amino acid, arganine, to the hydrophobic amino acid, isoleucine acid, which may have affected the affinity of ATP, leading to alterations in the steric structural and biochemical properties of SERCA2.
The present study also identified G2246T as a second mutation in DD, which was positioned in codon 749. An amino acid substitution from glycine to arginine (G749R) has been previously reported (3), however, the present study demonstrated that a different amino acid substitution at the same amino acid position in the ATP2A2 gene may have also lead to a steric clash and disrupt the function of SERCA2, leading to DD. These amino acid changes were not observed in the remaining unaffected family members or in the 100 healthy control individuals, therefore, these changes were unlikely to represent silent polymorphisms.
SERCA2, encoded by the ATP2A2 gene, transports Ca2+ between the cytoplasm and the ER and is important in maintaining calcium homeostasis (3,5,10,11). It has been recognized that extracellular calcium ([Ca2+]e) is important in epidermal differentiation and intra-epidermal cohesion (12). The differentiation of keratinocytes is triggered by increasing the [Ca2+]e >0.1 mM (13). Another previous study demonstrated that the calcium concentration in the basal layer in the skin of patients with DD skin is lower than in normal skin in vivo (11, 14).
In addition, the keratinocytes of patients with DD exhibit depleted ER Ca2+ stores due to the loss of SERCA2 Ca2+ transport (15,16). This depletion of Ca2+ ER stores potentially triggers the ER stress response, and persistent ER stress, which cannot be overcome due to the defective upregulation of SERCA2, may result in apoptotic keratinocytes, as observed in DD (12). A previous study demonstrated that SERCA2 mutant proteins, which are not degraded by the proteasome, form insoluble aggregates and cause ER stress and apoptosis (17).
Previous studies have also provided evidence to support the role of SERCA2 in the assembly of the desmosomal complex. In normal human keratinocytes, the inhibition of SERCA by thapsigargin, an inhibitor of SERCA2, impairs the trafficking of desmoplakin to the cell surface, and the trafficking of desmoplakin is inhibited in Darier keratinocytes (12). Defective addressing to the desmosomal plaque of desmoplakin, which links the cytoskeleton to the desmosomal complexes, may affect the stability and cell surface expression of desmosomal proteins, impairing cell-to-cell adhesion in the DD epidermis and leading to acantholysis (12,17).
The p.R603I and p.G749V mutations identified in the present study were located within the ATP-binding and stalk domain regions of SERCA2, respectively. These functional domains have been demonstrated to be highly conserved in different species and likely to be critical for the normal function of SERCA2 (Fig. 3) (5). Therefore, it is possible that these missense mutations, which altered polarity and hydrophobicity, may have interfered with the function of SERCA2.
Figure 3.

Evolutionary conservation analysis for the two identified ATP2A2 gene mutations using Clustalx 2.1.
In conclusion, the present study identified two novel missense mutations, p.R603I and p.G749V, in the ATP2A2 gene in two families containing individuals diagnosed with DD. These results contribute to the expanding database of ATP2A2 gene mutations. Supplemental functional trials are imperative to confirm the relevance between the mutations and the disease.
Acknowledgments
The authors would like to thank the patients with DD and the healthy individuals for their participation in the study. The current study was supported by the Jiangsu Technology Project of Chinese Medicine Pharmacy (grant no. LZ11082); the Fifth High-level Talent Project of “Six Talent Peaks” of Jiangsu Province (grant no. 2008.101); and the Fujian Science and Technology Project (grant no. 2011-5-81-1).
References
- 1.Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40–50. doi: 10.1016/0190-9622(92)70154-8. [DOI] [PubMed] [Google Scholar]
- 2.Munro CS. The phenotype of Darier’s disease: penetrance and expressivity in adults and children. Br J Dermatol. 1992;127:126–130. doi: 10.1111/j.1365-2133.1992.tb08044.x. [DOI] [PubMed] [Google Scholar]
- 3.Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271–277. doi: 10.1038/6784. [DOI] [PubMed] [Google Scholar]
- 4.Green EK, Gordon-Smith K, Burge SM, et al. Novel ATP2A2 mutations in a large sample of individuals with Darier disease. J Dermatol. 2013;40:259–266. doi: 10.1111/1346-8138.12082. [DOI] [PubMed] [Google Scholar]
- 5.Sakuntabhai A, Burge S, Monk S, Hovnanian A. Spectrum of novel ATP2A2 mutations in patients with Darier’s disease. Hum Mol Genet. 1999;8:1611–1619. doi: 10.1093/hmg/8.9.1611. [DOI] [PubMed] [Google Scholar]
- 6.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 7.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 8.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 10.Dhitavat J, Fairclough RJ, Hovnanian A, Burge SM. Calcium pumps and keratinocytes: lessons from Darier’s disease and Hailey-Hailey disease. Br J Dermatol. 2004;150:821–828. doi: 10.1111/j.1365-2133.2004.05904.x. [DOI] [PubMed] [Google Scholar]
- 11.Leinonen PT, Hägg PM, Peltonen S, et al. Reevaluation of the normal epidermal calcium gradient and analysis of calcium levels and ATP receptors in Hailey-Hailey and Darier epidermis. J Invest Dermatol. 2009;129:1379–1387. doi: 10.1038/jid.2008.381. [DOI] [PubMed] [Google Scholar]
- 12.Savignac M, Edir A, Simon M, Hovnanian A. Darier disease: a disease model of impaired calcium homeostasis in the skin. Biochim Biophys Acta. 2011;1813:1111–1117. doi: 10.1016/j.bbamcr.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Elias PM, Ahn SK, Denda M, et al. Modulations in epidermal calcium regulate the expression of differentiation-specific markers. J Invest Dermatol. 2002;119:1128–1136. doi: 10.1046/j.1523-1747.2002.19512.x. [DOI] [PubMed] [Google Scholar]
- 14.Dhitavat J, Cobbold C, Leslie N, Burge S, Hovnanian A. Impaired trafficking of the desmoplakins in cultured Darier’s disease keratinocytes. J Invest Dermatol. 2003;121:1349–1355. doi: 10.1046/j.1523-1747.2003.12557.x. [DOI] [PubMed] [Google Scholar]
- 15.Foggia L, Aronchik I, Aberg K, Brown B, Hovnanian A, Mauro TM. Activity of the hSPCA1 Golgi Ca2+ pump is essential for Ca2+-mediated Ca2+ response and cell viability in Darier disease. J Cell Sci. 2006;119:671–679. doi: 10.1242/jcs.02781. [DOI] [PubMed] [Google Scholar]
- 16.Leinonen PT, Myllylä RM, Hägg PM, et al. Keratinocytes cultured from patients with Hailey-Hailey disease and Darier disease display distinct patterns of calcium regulation. Br J Dermatol. 2005;153:113–117. doi: 10.1111/j.1365-2133.2005.06623.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Bruce AT, Tu C, et al. Protein aggregation of SERCA2 mutants associated with Darier disease elicits ER stress and apoptosis in keratinocytes. J Cell Sci. 2011;124:3568–3580. doi: 10.1242/jcs.084053. [DOI] [PMC free article] [PubMed] [Google Scholar]