

Abstract
Multiple potentially harmful stimuli challenge the liver, the chief metabolic and detoxifying organ of the human body. Due to its central anatomical location, continuous blood flow from the gastrointestinal tract through the hepatic sinusoids allows the metabolically active hepatocytes, the non-parenchymal cells and the various immune cell populations residing and patrolling in the liver to interact with antigens and microbiological components coming from the intestine. Cytokines are key mediators within the complex interplay of intrahepatic immune cells and hepatocytes, because they can activate effector functions of immune cells as well as hepatocytic intracellular signaling pathways controlling cellular homeostasis. Kupffer cells and liver-infiltrating monocyte-derived macrophages are primary sources of cytokines such as tumor necrosis factor (TNF). The liver is also enriched in natural killer (NK) and natural killer T (NKT) cells, which fulfill functions in pathogen defense, T cell recruitment and modulation of fibrogenic responses. TNF can activate specific intracellular pathways in hepatocytes that influence cell fate in different manners, e.g. pro-apoptotic signals via the caspase cascade, but also survival pathways, namely the nuclear factor (NF)-kappaB pathway. NF-kappaB regulates important functions in liver physiology and pathology. The exact dissection of the contribution of recruited and resident immune cells, their soluble cytokine and chemokine mediators and the intracellular hepatocytic response in liver homeostasis and injury could potentially identify novel targets for the treatment of acute and chronic liver disease, liver fibrosis or cirrhosis.
Keywords: inflammation, liver fibrosis, apoptosis, monocytes, cytokines, chemokines, NF-kB, TNF
Introduction
The human liver unites a bandwidth of purposes like metabolic, synthetic and detoxifying functions. Its exceptional role for regulating body’s homeostasis is challenged by liver diseases, whose incidence is increasing globally and which represent an outstanding challenge in health care worldwide. While the main causes of liver disease in the Western world are non-alcoholic steatohepatitis (NASH), alcohol and chronic hepatitis C infections, chronic hepatitis B and C infections are major reasons for liver disease in the Middle East and Asia. Despite different origins of liver injury, virtually all liver diseases trigger specific inflammatory processes, which eventually promote tissue loss, aberrant wound healing resulting in liver fibrosis, cirrhosis as an end-stage and occurrence of hepatocellular carcinoma (Bataller and Brenner, 2005[4]).
The activation of inflammatory pathways is not necessarily harmful in all circumstances. For instance, the liver has also the unique potential to regenerate after tissue loss (a process, which is regulated by inflammatory mediators) and plays an important role in regulating blood glucose or blood lipid levels in response to the current demand of the body (e.g., in conditions of inflammation). The central function of the liver for homeostasis and inflammatory responses is also underscored by its sole anatomical location, allowing continuous blood supply not only from the arterial system (hepatic arteries) but also from the gastrointestinal tract via the portal vein (Figure 1(Fig. 1)). Circulating blood cells, e.g. from the innate or adaptive immune system, are passing a network of sinusoids allowing contact to a variety of intrahepatic cell populations such as parenchymal liver cells (hepatocytes), endothelial cells, liver-resident macrophages (Kupffer cells) or lymphocyte (e.g., NKT cells) populations, hepatic stellate cells and others (Figure 1(Fig. 1)) (Racanelli and Rehermann, 2006[65]).
Figure 1. Intrahepatic cell populations. The healthy liver comprises about 60-80% hepatocytes; the other intrahepatic cell populations include biliary epithelial cells (cholangiocytes), liver sinusoidal endothelial cells (LSECs) lining the liver sinusoids, Kupffer cells (KC), and hepatic stellate cells (HSC) in the Dissé space between hepatocytes and LSECs. In addition, many immune cells are found in the liver, mainly entering from the circulation via hepatic arteries and portal vein branches, including neutrophils (PMN), monocytes (monos), dendritic cells (DCs) and lymphocytes (T, B, NK, NKT cells).
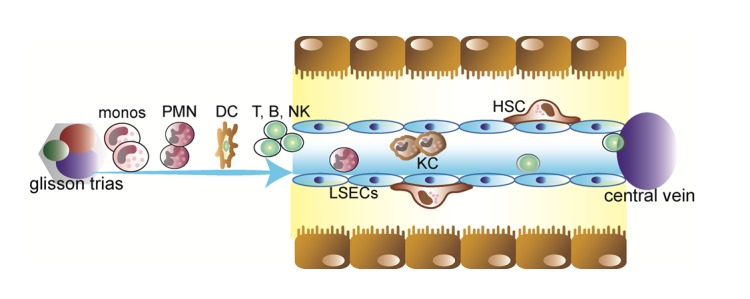
Communication between these cell types and the regulation of hepatic functions are greatly facilitated by cytokines. Cytokines are small molecular weight mesengers secreted by one cell to alter the behavior of the cell itself (autocrine messenger), a closely related cell (paracrine messenger) or cells in different organs (endocrine messenger) (Luedde et al., 2002[53]).
Intrahepatic immune cells as producers and targets of cytokines
Besides its various metabolic functions, the liver may be also viewed as a central immunological organ of the human body. It receives blood coming from the gastrointestinal tract through the portal vain, which is enriched with potential antigens and microorganisms. Immune cells in the liver have the potential to initiate both: a) innate and adaptive immune responses in the case of infections, e.g. in response to lipopolysaccharide (LPS) or bacterial superantigens, or b) immunological tolerance in the vast majority of harmless antigens during homeostasis (Tacke et al., 2009[80]).
Innate immunity within the liver
The liver is especially enriched in macrophages (Kupffer cells), natural killer (NK) and natural killer T (NKT) cells. Although it has been long known that Kupffer cells can principally originate from bone-marrow-derived monocytes (Luedde et al., 2002[53]), Kupffer cells have long been considered a rather sessile, tissue-resident and (fairly) radio-resistant population (Naito et al., 1997[60]; Duffield et al., 2005[21]). Experimental data have challenged this concept, as mouse models revealed that approximately half of the Kupffer cells in a steady state liver may originate directly from bone-marrow precursors, indicating a high turnover rate of macrophages in liver homeostasis (Klein et al., 2007[41]). Nevertheless, using sophisticated cell tracking techniques in mice, recent experiments revealed that the majority of Kupffer cells in homeostasis delineate from local precursors and constantly renew themselves dependent on the growth factors GM-CSF and M-CSF (Yona et al., 2013[93]). In the case of inflammation, blood-derived infiltrating monocytes are attracted to the injured liver, activated by certain cytokines and become dominant for macrophage actions, as suggested by animal models of chronic liver injury and fibrogenesis (Duffield et al., 2005[21]; Imamura et al., 2005[33]). Monocytes consist of at least two major subsets with different migratory and functional properties (Tacke and Randolph, 2006[83]; Tacke et al.; 2007[78] Tacke et al., 2006[79]); their specific roles in liver homeostasis, inflammation and regression are the subject of intensive ongoing research.
Natural killer (NK) cells in liver homeostasis and injury
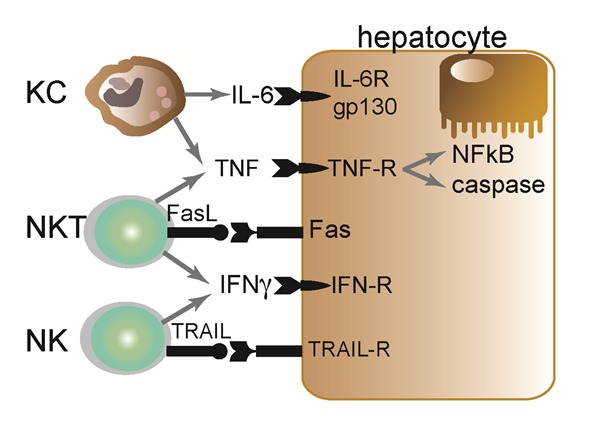
NK cells represent a population of lymphocytes with potent cytolytic activity against virus-infected or tumor cells. Hepatic NK cells are regulated by, for example, Kupffer-cell derived cytokines, like IL-12 and IL-18, as well as IL-4 driven by NKT cells (Wehr et al., 2013[89]). Hepatic NK cells produce large amounts of interferon gamma (IFNγ) upon activation (Figure 2(Fig. 2)), and modulate T cell responses in the liver, promote intracellular changes in endothelial cells as well as hepatocytes (mainly via IFNγ) and can even directly promote necroptosis of hepatocytes or cell lysis (Dong et al., 2007[20]; Bilzer et al., 2006[8]; Fallowfield et al., 2007[23]). In chronic inflammatory conditions, e.g. chronic hepatitis B virus infection, liver injury is closely linked to NK-mediated IL-8 and IFNγ synthesis as well as accumulation and activation of NK cells in the liver expressing the apoptosis-inducing TNF-related apoptosis ligand (TRAIL) (Dunn et al., 2007[22]).
Figure 2. Selected key interactions between innate immune cells and hepatocytes. Kupffer cells (macrophages), NKT and NK cells are abundantly found in the liver. Kupffer cells (KC) can release IL-6 and TNFα, activating hepatocytic gp130-dependent STAT signaling cascades and TNF-R dependent activation of apoptotic caspase and/or anti-apoptotic (pro-inflammatory) NF-kappaB (NFκB) pathways. NKT cells exert their actions via release of TNF, IFNγ or FasL-mediated apoptosis. NK cells can also release IFN and may induce apoptosis via TRAIL in the case of hepatocytic TRAIL-R expression.
In humans an increase of CD16- NK cells in blood and CD16+ NK cell subset in liver are closely associated with liver-disease severity and hepatofibrogenesis (Zimmermann et al., 2013[98]). Furthermore, patients suffering from chronic liver diseases show elevated IL-8 serum levels, according to raised IL-8 producing NK cell populations, which is associated with a hepatic macrophage accumulation via the IL-8 receptor CXCR1 (Zimmermann et al., 2011[99]; Tacke et al., 2011[82]) and may promote profibrogenic responses.
Macrophages in liver homeostasis and injury
While Kupffer cells are important for sensing liver injury and initiating inflammation, infiltrating liver-macrophages (iMϕ) have the capacity to phagocytize and release a broad panel of cytokines, which critically determine the subsequent reactions of other immune cells and hepatocytes as well as the degree of fibrosis by activating myofibroblasts (Bilzer et al., 2006[8]). Depending on their differentiation (classically activated Ly-6Chi vs. alternatively activated Ly-6Clo) iMϕ release a variety of proinflammatory cytokines such as tumor-necrosis factor (TNF), IL-6, IL-1β or leukotrienes (Ly-6Chi iMϕ) (Figure 2(Fig. 2)) (Luedde et al., 2002[53]); or in the context of low “physiological” levels of LPS or during liver damage regression, also anti-inflammatory cytokines like IL-10 and MMP9/12/13 (Ly-6Clo iMϕ) (Knolle, 2006[42]; Baeck et al., 2012[2]). In acute or chronic liver diseases, Kupffer cells and infiltrating monocyte-derived macrophages are known to promote inflammatory cascades by releasing these proinflammatory mediators with consequences for T cell attraction, NKT cell activation, induction of hepatocyte apoptosis (Figure 2(Fig. 2)) or activation of fibrogenic hepatic stellate cells (HSC) (Bilzer et al., 2006[8]; Schumann et al., 2000[72]; Wehr et al., 2013[89]).
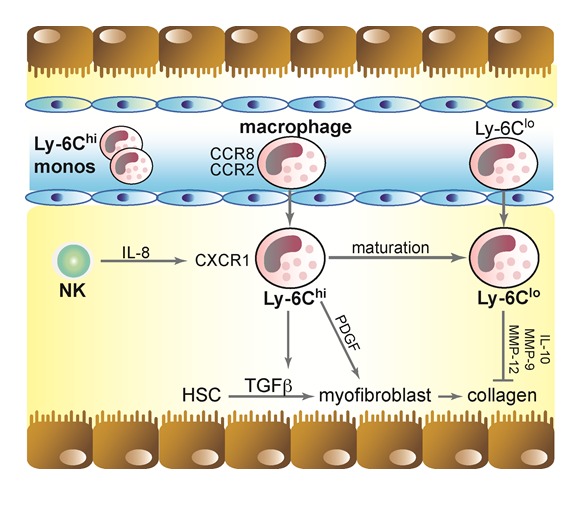
Chemokines, on the other hand, critically control the infiltration of macrophages upon liver injury and thereby promote hepatic inflammation and fibrosis. Chemokine receptors like the C-C motif chemokine receptor 2 (CCR2) and CCR8 can affect trafficking of monocytes/macrophages, DCs and T-helper cells into the liver and are critical for inducing liver-fibrosis as well as regeneration depending on the time point of secretion and the stage of liver disease (Baeck et al., 2012[2]; Heymann et al., 2012[31]; Zimmermann et al., 2012[101]; Zimmermann and Tacke, 2011[100]). Murine models revealed that early inflammatory Ly-6Chi iMϕ infiltrate the acutely injured liver, dependent on chemotaxis through chemokine – receptor interactions CCL2 - CCR2 or CCL1 - CCR8 (Galastri et al., 2012[25]; Heymann et al., 2012[31]; Karlmark et al., 2009[39]; Miura et al., 2012[59]). After infiltration, these Ly-6Chi iMϕ activate HSC and favor their transdifferentiation to myofibroblasts, which leads to enhanced collagen production and scar formation (Baeck et al., 2012[2]; Seki et al., 2007[74]; Fallowfield et al., 2007[23]).
As mentioned above, alternatively activated Ly-6Clo iMϕ appear to constitute key players in regeneration of liver fibrosis and liver injury regression. Recent studies showed that these matured macrophages reduced liver scarring by attracting anti-fibrotic and collagen-degrading immune cells resulting into enhanced levels of anti-fibrotic IL-10 and increased metallopro-teinases MMP-9 and -13 activation (Baeck et al., 2013[3]; Thomas et al., 2011[86]; Imamura et al., 2005[33]). Furthermore, recent murine investigations showed significantly enhanced liver regeneration from fibrosis by pharmacological augmenting the number of Ly-6Clo iMϕ during wound healing (Baeck et al., 2013[3]).
NKT cells in liver homeostasis and injury
NKT cells are a heterogeneous population of unconventional T cells that express markers of NK cells and T cell receptors (TCRs). NKT cells are found at remarkably high numbers in the liver. From studies of experimental liver injury in mice after administration of the plant-derived lectin Concanavalin A (ConA), NKT cells have been identified as critical factors promoting acute liver damage by release of IL-4, IFNγ and direct induction of Fas-mediated hepatocyte apoptosis (Figure 2(Fig. 2)) (Tiegs, 2007[87]). NKT subsets with different functions exist in humans and mice: classical (type I) invariant (iNKT), other CD1d-dependent (type II) NKT cells, as well as CD1d-independent (NKT-like) T cells (Wehr et al., 2013[89]). However, the physiological function of the individual subsets is basis of current investigations. Recent studies revealed an important role of NKT cells in the development of liver fibrosis in mice by accentuating the inflammatory process and activating pro-fibrogenic macrophages through the release of cytokines (Kinjo et al., 2005[40]; Mattner et al., 2005[56]; Wehr et al., 2013[89]). Furthermore, NKT cells are involved in antiviral defense mechanisms by secreting IFNγ, e.g. during chronic hepatitis B virus infection as suggested by studies in transgenic mice (Kakimi et al., 2000[35]).
Antigen presenting cells and adaptive immune cells in the liver
For the initiation of adaptive immune responses, antigens need to be processed and professionally presented to T cells, either in the liver itself or in the draining lymph nodes (Racanelli and Rehermann, 2006[65]). Several hepatic cell populations have antigen-presenting properties. In non-inflammatory conditions, antigen-presentation by liver sinusoidal endothelial cells appears to be a crucial mechanism for the maintenance of immunological tolerance (Limmer et al., 2000[50]). Furthermore, Kupffer cells and also bone-marrow derived dendritic cells, mostly of monocytic origin linking the innate and adaptive part of immune responses, can efficiently prime T cells (Bilzer et al., 2006[8]; Racanelli and Rehermann, 2006[65]; Tacke and Yoneyama, 2013[81]). The nature of the T cell response, e.g. T-cell cytotoxicity in the case of a chronic viral infection or T-cell tolerance in the case of harmless gut-derived antigens or autoantigens, seems to depend on the antigen-presenting cells, the cytokine milieu and the site of primary T cell activation (Bowen et al., 2004[9]; Crispe et al., 2006[12]). HSCs, known for vitamin A storage and collagen synthesis in liver-fibrosis (as activated myofibroblasts), have been suggested as cells with antigen-presenting capacity itself that can activate T cell responses, e.g. after bacterial challenge (Winau et al., 2007[90]; Nevzorova et al., 2012[62]).
The liver is also one of the richest sources of γδ T-cells in the body, which are an unconventional (or innate) T-cell subset that expresses the γδ TCR. These IL-17 and IL-22 expressing γδ T-cells are recruited to the liver via chemokine-chemokine receptor activation (CCL20-CCR6) upon chronic injury and were shown to protect the liver from excessive inflammation and fibrosis by inhibiting HSC (Hammerich et al., 2014[29]; 2011[30]) (Figure 3(Fig. 3)).
Figure 3. Macrophage subsets in chronic liver injury. In the acute phase of hepatic injury increased levels of CCL2 and CCL1 activates ‘classical’ Ly-6Chi monocytes via their receptors that enter the liver, where they develop into infiltrating Ly-6Chi macrophages exhibiting a pro-inflammatory phenotype, which partly resembles M1 macrophages. These macrophages support the progression of chronic liver injury and fibrosis through TGF-ß/PDGF-mediated HSC transdifferentiation, activation and proliferation. This leads to the accumulation of collagen resulting in fibrosis of the organ. After maturation from Ly-6Chi to Ly-6Clo macrophages, these cells can adopt a restorative phenotype characterized by Ly-6Clo expression and capacity to degrade excessive extracellular matrix proteins via metalloproteinases (MMP-9,-12) and to induce HSC apoptosis during fibrosis resolution.
TNF and the TNF-receptor in the liver
Besides direct effects of immune cells on hepatocytes and other liver cells, inflammatory processes in the liver are largely regulated by cytokines. Cytokine action is generally mediated by the interaction of cellular receptors with their ligand. These networks have evolved early in the evolution; pathways with strong homology to human cytokine networks are already found in Drosophila and mollusks (Luedde and Trautwein, 2006[54]). E.g. in Drosophila, Nuclear Factor (NF)-kappaB-like transcription factors are activated in order to combat infections; this still represents one major role of cytokine networks in higher organisms like humans. Maintaining the ordered balance between proliferation and controlled cell death (apoptosis) during embryonic development and organogenesis is another important function of cytokines in physiologic conditions. The stability of the cytokine relation to each other is essential for stable conditions of immune cell response within an organ (Luedde et al., 2002[53]). Dysregulated cytokine actions after liver injury, for example, can result in excessive apoptosis, a key finding in various acute and chronic liver diseases, e.g. viral and autoimmune hepatitis, cholestatic disease, alcoholic or drug/toxin-induced liver injury (Neuman, 2001[61]). Among the manifold cytokines relevant for liver homeostasis and injury, we will highlight important findings on TNF as a represent pathway with exceptional significance in the liver. Studies in patients and animal models have strongly implicated that death receptor ligands such as TNF or Fas ligand (FasL) are involved in the induction of apoptosis / necroptosis and in triggering destruction of the liver (Streetz et al., 2000[75]), which leads to final organ failure like liver-fibrosis.
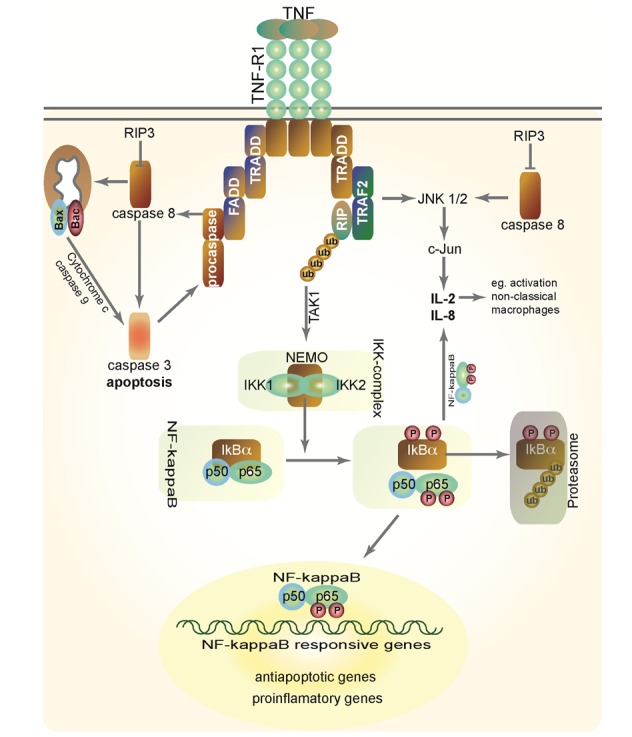
TNF and FasL belongs to a family of nine ligands (TNF, lymphotoxin-α, TNFβ, FasL, OX40L, CD40L, CD27L, CD30L, 4-1BBL and lymphotoxin-β) that activate structurally related receptor proteins known as the TNF receptor superfamily. Currently, twelve different death receptors are well established including TNF receptor 1 (TNF-R1, Figure 4(Fig. 4)), TNF-R2, TNF-RP, Fas, OX-40, 4-1BB, CD40, CD30, CD27, pox virus PV-T2, PV-A53R gene products, and the p75 NGFR. In addition, the apoptosis-signaling receptors death receptor 3 (DR3), DR4 and DR5, their ligand TRAIL, and a nonsignaling decoy receptor TRID/DcR are recently identified members of these superfamilies (Schulze-Osthoff et al., 1998[71]; Tacke et al., 2009[80]). In patients with fulminant hepatic failure, serum levels of TNF, TNF-R1 and TNF-R2 are markedly increased and these changes directly correlated with disease activity. However, in liver physiology the TNF-R1 plays a predominant role (Streetz et al., 2000[76]; 2003[77]).
Figure 4. TNF signaling in the liver. TNF-binds to its receptors, e.g. TNF-R1, and can thereby activate the pro-apoptotic caspase cascades (via TRADD, FADD and cleavage of pro-caspase 8) or the anti-apoptotic NF-kappaB pathway (via activation of the IKK-complex resulting in phosphorylation of IkBalpha, subsequent translocation of NF-kappaB to the nucleus and expression of NF-kappaB re-sponsive genes). For details, please see main text.
The intracellular TNF pathway in liver homeostasis and inflammation
In many instances, hepatic failure might result from an imbalance between damaging and protective signals that are very tightly regulated under physiologic conditions. As mentioned above, TNF and related cytokines are key players in liver homeostasis as they can activate both, pro-apoptotic (mainly caspases) and anti-apoptotic (mainly NF-kappaB) pathways in hepatocytes (Figure 4(Fig. 4)) (Ghosh and Karin, 2002[26]; Liedtke and Trautwein, 2012[49]; Rao et al., 2010[66]; Rothwarf et al., 1998[68]).
Activation of pro-apoptotic signaling cascades
FasL and TNF facilitate programmed cell death in a similar manner by activation of caspases. The most important target of both pathways orchestrating cellular death is the aspartate-specific cysteine protease- or caspase-cascade, consisting of initiator caspases such as caspases 8 and 9 and executioner caspases, e.g. caspase 3, 6 and 7. Since proteolytic cleavage generates the mature caspases, one way by which these enzymes are activated is via the action of proteases, including other caspases (Martin and Green, 1995[55]). TNF signals through two distinct cell surface receptors, TNF-R1 and TNF-R2, of which TNF-R1 initiates the majority of TNF’s biological activities. Binding of TNF to its receptor leads to the recruitment of the inhibitory protein silencer of death domains (SODD) from TNF-R1’s intracellular domain. This results in the activation of the adaptor protein TNF receptor associated death domain (TRADD), the central molecule of the TNF signaling, which in turn recruits Fas-associated death domain (FADD). FADD recruits caspase-8 to the TNF-R1 complex, where it becomes activated and initiates the protease cascade leading to activation of executioner caspases and apoptosis (Figure 4(Fig. 4)) (Chen and Goeddel, 2002[11]; Cubero et al., 2013[13]; Das et al., 2009[14]).
In hepatocytes, caspase-8-mediated signals are also influenced by a mitochondrial pathway: The cytosolic protein Bid (BH3-interacting domain death agonist) is proteolytically activated by caspase-8 and thereby converted into its active form tBID which translocates into the mitochondrial membrane and contributes to increased mitochondrial permeability and subsequent to release cytochrome C from the mitochondria into the cytosol via the membrane proteins Bax and Bac. The release of cytochrome C triggers the activation of caspase-3 via caspase-9. Via positive feedback loops, caspase-3 may then activate further procaspase-8 molecules, but also directly triggers necroptosis (Liedtke et al., 2011[48]; Ashkenazi and Dixit, 1998[1]; Streetz et al., 2000[75]; Gupta et al., 2005[28]). Interestingly, caspase-8 has two faces within liver homeostasis: mice with hepatocellular deletion of caspase-8 were protected against liver injury and hepatocarcinogenesis, while caspase-8-deficient immune cells were responsible for increased liver injury in models of cholestatic hepatitis (Chaudhary et al., 2013[10]; Liedtke et al., 2011[48]).
In contrast to TNF-dependent signaling, FasL can interact directly with the death domain of FADD without recruiting TRADD (Ashkenazi and Dixit, 1998[1]; Chen and Goeddel, 2002[11]).
Activation of the NF-kappaB pathway
In addition the activation of caspases, binding of TNF to its receptor also leads to the activation of the NF-kappaB pathway (Figure 4(Fig. 4)). NF-kappaB is a heterodimer or a homodimer of members of the Rel family of DNA-binding proteins, and regulates the transcription of genes that contain kappaB binding sites. The mammalian NF-kappaB family includes five cellular DNA-binding subunits: p50 (NF-ϰB1), p52 (NF-ϰB2), c-Rel (Rel), p65 (RelA) and RelB (Ghosh et al., 1998[27]). The N-terminal Rel homology domain (RHD), which is shared by NF-kappaB DNA-binding subunits, is responsible for DNA-binding, dimerization, nuclear translocation and interaction with the inhibitory I-kappaB proteins. In addition, p65, RelB and cRel contain a C-terminal transactivation domain, which activates transcription (Ghosh and Karin, 2002[26]). NF-kappaB leads to different gene transcription with kappaB binding sites within the liver, which are involved in the regulation of inflammation, cell survival and immune response (Pahl, 1999[63]).
In the liver, NF-kappaB mediates protective and anti-apoptotic effects. Thus, hepatocytes with a specific deletion for the adapter molecule NEMO that abrogates NF-kappaB activation, are hypersensitive for massive TNF-induced apoptosis in vivo and in vitro (Beraza et al., 2007[7]).
TNF-induced activation of NF-kappaB relies on the phosphorylation of two conserved serines (S32 and S36 in human I-kappaBα) in the N-terminal regulatory domain of I-kappaBs. After phosphorylation, the I-kappaBs undergo a second post-translational modification: polyubiquitination by a cascade of enzymatic reactions, followed by the degradation of IκB proteins by the proteasome, thus releasing NF-kappaB from its inhibitory I-kappaB-binding partner, so it can translocate to the nucleus and activate transcription of NF-kappaB dependent target genes (Figure 4(Fig. 4)) (Beraza et al., 2007[7]; Karin, 1999[36]; Yamamoto and Gaynor, 2004[92]). Since the enzymes that catalyze the ubiquitination of I-kappaB are constitutively active, the only regulated step in NF-kappaB activation appears to be in most cases the phosphorylation of I-kappaB molecules (Liedtke and Trautwein, 2012[49]).
The high-molecular-weight “IKK-complex” mediates the phosphorylation of I-κB (Figure 4(Fig. 4)). This complex consists of three tightly associated I-κB kinase (IKK) polypeptides: IKK1 (also called IKKα) and IKK2 (IKKβ), the catalytic subunits of the kinase complex (DiDonato et al., 1997[18]; Mercurio et al., 1999[57]; Regnier et al., 1997)[67], and a regulatory subunit called NEMO (NF-kappaB Essential Modulator, IKKγ) (Mercurio et al., 1999[57]; Rothwarf et al., 1998[68]). In vitro, IKK1 and IKK2 can form homo- and heterodimers (Zandi et al., 1998[94]). Both IKK1 and IKK2 are able to phosphorylate I-κB in vitro, but IKK2 has a higher kinase activity in vitro compared with IKK1 (Delhase et al., 1999[15]; Mercurio et al., 1999[57]; 1997[58]; Woronicz et al., 1997[91]; Zandi et al., 1997[95]).
Activation of the IKK complex upon TNF stimulation involves IKK recruitment to the TNF-R1 (Devin et al., 2000[16]; 2001[17]; Zhang et al., 2000[96]). Besides TNF-R1, this process involves TNF-receptor-associated-factor 2 (TRAF2) and the death-domain kinase receptor-interacting protein (RIP). After TNF treatment, TRAF2 recruits the IKK-complex to TNF-R1 via the interaction of the RING-finger motifs of TRAF2 with the leucin-zipper motif of both IKK1 and IKK2 (Devin et al., 2000[16], 2001[17]).
In addition, association of TRADD with TRAF2 can activate the Jun-(N)-terminal kinases (JNK1/2) via the N-terminal zinc-finger domain of TRAF2. It has been demonstrated that activation of the upstream kinases ASK1 (apoptosis signal-related kinase) and the mitogen-activated kinases MKK4 and MKK7 are essential for TNF-mediated JNK1/2 activation (Liedtke et al., 2011[48]), which phosphorylates and activates transcriptional factor c-Jun and so regulates the expression of interleukins IL-2 and IL-8 (Baud et al., 1999[5]). As a possible result IL-2 and its soluble IL-2 recepor is an immune cell activator in chronic liver diseases in humans by attracting non-classical CD14+ and CD16+ monocytes (Seidler et al., 2012)[73].
Recent studies highlighted new potential molecules for drug targeting related to NF-kappaB signaling. The molecule receptor interacting protein 3 (RIP3) interacts with inflammatory pathways in hepatocytes, because it limits pro-inflammatory immune response by inhibiting caspase-8-dependent activation of JNK1/2 (Vucur et al., 2013[88]). RIPs can also directly interact with NEMO and mediate IKK activation, although the enzymatic activity of RIP is not required for this process (Zhang et al., 2000[96]; Zhu et al., 2007[97], Israel, 2006[34]). The mechanism by which recruitment of the IKK-complex to the TNF receptor leads to IKK activation is not clear, but might involve NEMO induced autophosphorylation of the IKK-complex. Moreover, ubiquitination of multiple factors that regulate the IKK-complex, like TRAF6/TAK1 or c-IAP1, an inhibitor of apoptosis that is also part of the TNF receptor complex, modulate the activity of the NF-kappaB pathway (Yamamoto and Gaynor, 2004[92]).
Nemo and IKKs themselves have also NF-kappaB-independent functions, which seem to play a role in the regulation of cell death, carcinogenesis and inflammation (Karin et al., 2004[38]).
Consequences of NF-kappaB activation in the liver
After cytokine-consolidation with a death receptor additional to the resulting apoptosis of the cell (e.g. liver cell), survival signals are simultaneously activated. In case of the TNF-R, this is provided by NF-kappaB. This process is assumed to involve the transcriptional induction of various apoptotic suppressors (Liu et al., 1996[51]). Evidence that NF-kappaB governs critical anti-apoptotic proteins comes from well-described animal models. Injection of TNF into mice and addition of TNF to hepatic cells in culture resulted in activation of nuclear translocation and of DNA binding of NF-kappaB (Fitzgerald et al., 1995)[24]. Moreover, hepatocytes are resistant to apoptosis induced by TNF or LPS, a potent inductor for endogenous TNF in the liver, unless they are treated with inhibitors of transcription of (anti-apoptotic) proteins like cycloheximide or actinomycin D (Lehmann et al., 1987[43]; Leist et al., 1994[44], 1997[45]). Knockout mice lacking the p65 subunit of NF-kappaB die between days E15 and E16 post-coitum as a result of fetal hepatocyte apoptosis (Beg and Baltimore, 1996[6]). This is caused by increased sensitivity towards TNF, since TNF/p65 double-deficient mice are rescued from embryonic lethality (Doi et al., 1999[19]).
Interestingly, IKK subunits in TNF-mediated liver apoptosis have also differential functions. Genetic experiments have highlighted that mice lacking IKK1 die shortly after birth and display a phenotype marked by thickening of skin and limb as well as skeletal defects (Hu et al., 1999[32]; Takeda et al., 1999[84]). IKK2-deficient mice die in utero approximately at embryonic day 12.5 as a result of massive apoptosis in the liver, and fibroblasts from these mice show no activation of NF-kappaB in response to TNF (Li et al., 1999[46][47]; Tanaka et al., 1999[85]). However, deletion of IKK/NEMO causes spontaneous progression of TNF-mediated chronic hepatitis to HCC, while this deletion of IKK/NEMO in TNFR1 knockout mice decreased apoptotic cell death, liver-fibrosis and infiltration of immune cells (Cubero et al., 2013[13]; Schneider et al., 2012[70]; Rudolph et al., 2000[69]). Therefore, at least during embryogenesis, IKK2 and NEMO appear to be the critical subunits for NF-kappaB activation and protection of liver cells from proinflammatory cytokines like TNF.
IKK subunits in the adult animal and its role during TNFR1 activation are less well understood. Conditional knockout mice based on the cre/loxP system have emerged as new powerful tools to study gene functions in the adult animal in vivo (Luedde and Trautwein, 2006[54]; Pasparakis et al., 2006[64]). It was shown that hepatocyte-specific ablation of IKK2 does not lead to a strongly impaired activation of NF-kappaB or increased apoptosis after TNF stimulation, probably because IKK1 homodimers can take over this function in the absence of IKK2 in the adult mouse (Luedde et al., 2005[52]). In contrast, conditional hepatocyte-specific knockout of NEMO resulted in complete block of NF-kappaB activation and massive hepatocyte apoptosis, underlining that NEMO is the only irreplaceable IKK subunit for prevention of TNF mediated liver apoptosis (Beraza et al., 2007[7]).
Furthermore, TNF mediated activation of NF-kappaB seems to have, besides its anti-apoptotic function, also essential functions in various models of experimental liver injury. For instance, NF-kappaB DNA-binding occurs quickly upon hepatic ischemia-reperfusion (I/R) injury (Zwacka et al., 1998[102]), but it has also long been unclear whether NF-kappaB dependent signaling withholds a protective or damaging role in I/R injury. It was shown that hepatocyte-specific conditional knockout mice for IKK2 show a defect in NF-kappaB activation after I/R (Luedde et al., 2005[52]). Inhibition of NF-kappaB activation in conditional IKK2-deficient mice protected from liver injury due to I/R (Luedde et al., 2005[52]), thus underlining that, depending on the experimental model, the NF-kappaB pathway does not serve as a survival pathway, but instead can aggravate hepatocyte death and liver damage. However, complete abolishment of NF-kappaB activation in conditional NEMO-knockout mice resulted in massive hepatic inflammation and apoptosis after I/R injury (Beraza et al., 2007[7]).
Conclusions
A growing number of studies have implicated immune cells, cytokines and cytokine-dependent pathways in the development of liver failure, chronic liver disease, hepatic inflammation and liver fibrogenesis. Resident and infiltrating immune cells depending on their differentiation, detectable by their cell-surface markers, have been linked to progression (especially Ly-6Chi expressing iMϕ) as well as regression of liver injury (e.g., Ly-6Clo iMϕ) (Baeck et al., 2012[2], 2013[3]). On the other hand, parenchymal and non-parenchymal survival pathways like NF-kappaB withhold a protective function in many experimental liver disease models and thus are an attractive target for a pharmacological intervention (Karin et al., 2002[37]). However, many experimental models highlighted dual functions of these key players, namely beneficial or adverse effects. For instance, blockage of monocyte infiltration into the liver limits disease progression in different murine models, but also negatively affect the resolution of liver fibrosis (Duffield et al., 2005[21]). Thus, an inflammatory immune cell is not necessarily detrimental in any context. Similarly, an inhibition of NF-κB in the liver can have different outcomes depending on the experimental model applied: protecting from apoptosis in a model of TNF-dependent cell death vs. aggravating cellular necrosis in a model of ischemia/reperfusion injury (Luedde and Trautwein, 2006[54]). Thus, a ‘survival’ pathway is not necessarily protective in any context. Dissecting the cellular and molecular inflammatory pathways during liver homeostasis and during liver injury will hopefully provide the basis for novel specific therapeutic approaches in the near future.
References
- 1.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 2.Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–426. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- 3.Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) accelerates liver fibrosis regression by suppressing Ly-6C+ macrophage infiltration. Hepatology. 2013;doi:10.1002/hep.26783. doi: 10.1002/hep.26783.. Available from: http://dx.doi.org/10.1002/hep.26783. [DOI] [PubMed] [Google Scholar]
- 4.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 7.Beraza N, Ludde T, Assmus U, Roskams T, Vander Borght S, Trautwein C. Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132:2504–2517. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 8.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 9.Bowen DG, Zen M, Holz L, Davis T, Mccaughan GW, Bertolino P. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J Clin Invest. 2004;114:701–712. doi: 10.1172/JCI21593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhary K, Liedtke C, Wertenbruch S, Trautwein C, Streetz KL. Caspase 8 differentially controls hepatocytes and non-parenchymal liver cells during chronic cholestatic liver injury in mice. J Hepatol. 2013;59:1292–1298. doi: 10.1016/j.jhep.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 11.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 12.Crispe IN, Giannandrea M, Klein I, John B, Sampson B, Wuensch S. Cellular and molecular mechanisms of liver tolerance. Immunol Rev. 2006;213:101–118. doi: 10.1111/j.1600-065X.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- 13.Cubero FJ, Singh A, Borkham-Kamphorst E, Nevzorova YA, Al Masaoudi M, Haas U, et al. TNFR1 determines progression of chronic liver injury in the IKKgamma/Nemo genetic model. Cell Death Differ. 2013;20:1580–1592. doi: 10.1038/cdd.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das M, Sabio G, Jiang F, Rincon M, Flavell RA, Davis RJ. Induction of hepatitis by JNK-mediated expression of TNF-alpha. Cell. 2009;136:249–260. doi: 10.1016/j.cell.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 16.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 17.Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Z. The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol Cell Biol. 2001;21:3986–3994. doi: 10.1128/MCB.21.12.3986-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Didonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 19.Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, et al. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci U S A. 1999;96:2994–2999. doi: 10.1073/pnas.96.6.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Z, Wei H, Sun R, Tian Z. The roles of innate immune cells in liver injury and regeneration. Cell Mol Immunol. 2007;4:241–252. [PubMed] [Google Scholar]
- 21.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204:667–680. doi: 10.1084/jem.20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 24.Fitzgerald MJ, Webber EM, Donovan JR, Fausto N. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ. 1995;6:417–427. [PubMed] [Google Scholar]
- 25.Galastri S, Zamara E, Milani S, Novo E, Provenzano A, Delogu W, et al. Lack of CC chemokine ligand 2 differentially affects inflammation and fibrosis according to the genetic background in a murine model of steatohepatitis. Clin Sci (Lond) 2012;123:459–471. doi: 10.1042/CS20110515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 28.Gupta S, Su H, Bi R, Agrawal S, Gollapudi S. Life and death of lymphocytes: a role in immunesenescence. Immun Ageing. 2005;2:12. doi: 10.1186/1742-4933-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammerich L, Bangen JM, Govaere O, Zimmermann HW, Gassler N, Huss S, et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T-cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. 2014;59:630–642. doi: 10.1002/hep.26697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol. 2011;2011:345803. doi: 10.1155/2011/345803.. Available from: http://dx.doi.org/10.1155/2011/345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology. 2012;55:898–909. doi: 10.1002/hep.24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 33.Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology. 2005;128:138–146. doi: 10.1053/j.gastro.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Israel A. NF-kappaB activation: Nondegradative ubiquitination implicates NEMO. Trends Immunol. 2006;27:395–397. doi: 10.1016/j.it.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Kakimi K, Guidotti LG, Koezuka Y, Chisari FV. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med. 2000;192:921–930. doi: 10.1084/jem.192.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 37.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 38.Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 39.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 40.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingo-lipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 41.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knolle PA. Involvement of the liver in the induction of CD8 T cell tolerance towards oral antigen. Z Gastroenterol. 2006;44:51–56. doi: 10.1055/s-2005-858988. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann V, Freudenberg MA, Galanos C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 45.Leist M, Gantner F, Naumann H, Bluethmann H, Vogt, K, Brigelius-Flohe RA. Tumor necrosis factor-induced apoptosis during the poisoning of mice with hepatotoxins. Gastroenterology. 1997;112:923–934. doi: 10.1053/gast.1997.v112.pm9041255. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 47.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ, et al. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology. 2011;141:2176–2187. doi: 10.1053/j.gastro.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 49.Liedtke C, Trautwein C. The role of TNF and Fas dependent signaling in animal models of inflammatory liver injury and liver cancer. Eur J Cell Biol. 2012;91:582–589. doi: 10.1016/j.ejcb.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. 2000;6:1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 51.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 52.Luedde T, Assmus U, Wustefeld T, Meyer Zu Vilsendorf A, Roskams T, Schmidt-Supprian M, et al. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J Clin Invest. 2005;115:849–859. doi: 10.1172/JCI23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luedde T, Liedtke C, Manns MP, Trautwein C. Losing balance: cytokine signaling and cell death in the context of hepatocyte injury and hepatic failure. Eur Cytokine Netw. 2002;13:377–383. [PubMed] [Google Scholar]
- 54.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 55.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 56.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;34:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 57.Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 59.Miura K, Yang L, Van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310–G1321. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Naito M, Hasegawa G, Takahashi K. Development, differentiation, and maturation of Kupffer cells. Microsc Res Tech. 1997;39:350–364. doi: 10.1002/(SICI)1097-0029(19971115)39:4<350::AID-JEMT5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 61.Neuman MG. Apoptosis in diseases of the liver. Crit Rev Clin Lab Sci. 2001;38:109–166. doi: 10.1080/20014091084182. [DOI] [PubMed] [Google Scholar]
- 62.Nevzorova YA, Bangen JM, Hu W, Haas U, Weiskirchen R, Gassler N, et al. Cyclin E1 controls proliferation of hepatic stellate cells and is essential for liver fibrogenesis in mice. Hepatology. 2012;56:1140–1149. doi: 10.1002/hep.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 64.Pasparakis M, Luedde T, Schmidt-Supprian M. Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ. 2006;13:861–872. doi: 10.1038/sj.cdd.4401870. [DOI] [PubMed] [Google Scholar]
- 65.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 66.Rao P, Hayden MS, Long M, Scott ML, West AP, Zhang D, et al. IkappaBbeta acts to inhibit and activate gene expression during the inflammatory response. Nature. 2010;466:1115–1119. doi: 10.1038/nature09283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 68.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 69.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/ IKKgamma-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61:1733–1743. doi: 10.1136/gutjnl-2011-301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 72.Schumann J, Wolf D, Pahl A, Brune K, Papadopoulos T, Van Rooijen N, et al. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol. 2000;157:1671–1683. doi: 10.1016/S0002-9440(10)64804-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seidler S, Zimmermann HW, Weiskirchen R, Trautwein C, Tacke F. Elevated circulating soluble interleukin-2 receptor in patients with chronic liver diseases is associated with non-classical monocytes. BMC Gastroenterology. 2012;12:38. doi: 10.1186/1471-230X-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 75.Streetz K, Leifeld L, Grundmann D, Ramakers J, Eckert K, Spengler U, et al. Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology. 2000a;119:446–460. doi: 10.1053/gast.2000.9364. [DOI] [PubMed] [Google Scholar]
- 76.Streetz KL, Luedde T, Manns MP, Trautwein C. Interleukin 6 and liver regeneration. Gut. 2000b;47:309–312. doi: 10.1136/gut.47.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Streetz KL, Tacke F, Leifeld L, Wustefeld T, Graw A, Klein C, et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology. 2003;38:218–229. doi: 10.1053/jhep.2003.50268. [DOI] [PubMed] [Google Scholar]
- 78.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tacke F, Ginhoux F, Jakubzick C, Van Rooijen N, Merad M, Randolph GJ. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J Exp Med. 2006;203:583–597. doi: 10.1084/jem.20052119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 81.Tacke F, Yoneyama H. From NAFLD to NASH to fibrosis to HCC: role of dendritic cell populations in the liver. Hepatology. 2013;58:494–496. doi: 10.1002/hep.26405. [DOI] [PubMed] [Google Scholar]
- 82.Tacke F, Zimmermann HW, Trautwein C, Schnabl B. CXCL5 plasma levels decrease in patients with chronic liver disease. J Gastroenterol Hepatol. 2011;26:523–529. doi: 10.1111/j.1440-1746.2010.06436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tacke, F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–618. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 84.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai, et al. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 85.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, et al. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 86.Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53:2003–2015. doi: 10.1002/hep.24315. [DOI] [PubMed] [Google Scholar]
- 87.Tiegs G. Cellular and cytokine-mediated mechanisms of inflammation and its modulation in immune-mediated liver injury. Z Gastroenterol. 2007;45:63–70. doi: 10.1055/s-2006-927397. [DOI] [PubMed] [Google Scholar]
- 88.Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 2013;4:776–790. doi: 10.1016/j.celrep.2013.07.035. [DOI] [PubMed] [Google Scholar]
- 89.Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol. 2013;190:5226–5236. doi: 10.4049/jimmunol.1202909. [DOI] [PubMed] [Google Scholar]
- 90.Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito cells are liver-resi-dent antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 91.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 92.Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 93.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 95.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 96.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 97.Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17:1438–1443. doi: 10.1016/j.cub.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 98.Zimmermann HW, Mueller JR, Seidler S, Luedde T, Trautwein C, Tacke F. Frequency and phenotype of human circulating and intrahepatic natural killer cell subsets is differentially regulated according to stage of chronic liver disease. Digestion. 2013;88:1–16. doi: 10.1159/000350821. [DOI] [PubMed] [Google Scholar]
- 99.Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C, et al. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One. 2011;6,e21381. doi: 10.1371/journal.pone.0021381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets. 2011;10:509–536. doi: 10.2174/187152811798104890. [DOI] [PubMed] [Google Scholar]
- 101.Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol. 2012;3:56. doi: 10.3389/fphys.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zwacka RM, Zhang Y, Zhou W, Halldorson J, Engelhardt JF. Ischemia/reperfusion injury in the liver of BALB/c mice activates AP-1 and nuclear factor kappaB independently of IkappaB degradation. Hepatology. 1998;28:1022–1030. doi: 10.1002/hep.510280417. [DOI] [PubMed] [Google Scholar]