

Abstract
It is well recognized that cancers develop and grow as a result of disordered function of tumor suppressor genes and oncogenes, which may be exploited for screening purposes. Extensive evidence indicated tumor suppressor protein p53 as candidate marker for mutation identification. We have investigated mutant p53 protein expression in human breast tumors in relation to antioxidant status deficiency. The study included 100 breast cancer patients. p53 protein expression was evaluated by Western blot assay and immunostaining using a CM-1, DO-7 and Pab240 antibodies. Antioxidant parameters and lipid peroxidation were estimated by biochemical analyses. Western blotting with epitopespecific monoclonal antibody Pab240 strongly suggests that nuclear extracts from breast cancer cells express mutant forms of p53. It is of interest that the mutant forms of p53 overexpression in conjunction with the appearance of nuclear bodies are observed in highly aggressive carcinomas. Expression of isoform Δp53 (45 kDa) and isoform of ~ 29 kDa were more common in cases with LN metastasis. These studies point out the molecular consequences of oxidative stress (lipid peroxides, LP, p<0.001) and antioxidant status deficiency (copper, zinc superoxid dismutase, SOD, p<0.001; catalase, CAT, p<0.01; glutathione reductase, GR, p<0.001; glutathione, GSH, p<0.05) and indicate the importance of p53 mutation as the commonest genetic alteration detected in breast cancer cells. The expression of mutant p53 is correlated to increased lipid peroxides (0.346, p<0.05 ) and lowered antioxidant activity of CAT (- 0.437, p<0.01) in the breast cancer patients.
Keywords: antioxidants, breast carcinoma, lipid peroxidation, p53
Introduction
The loss of wild-type p53 gene expression has exceptionally been implicated in the development of a wide variety of human cancers and it is generally accepted that p53 is a component in biochemical pathways central to human carcinogenesis (Braithwaite and Prives, 2006[13]). Although the role of the p53 gene in cancer genesis and development has fueled as many questions (Levine et al., 2006[45]; Meek, 2009[48]), study of p53 has come to the forefront of cancer research and detection of its abnormalities during the development of tumors may have diagnostic, prognostic and therapeutic implications (Royds and Iacopetta, 2006[66]). To be of value in clinical practice, immunohistochemical assessment of p53 protein should provide clinically relevant information. The degree of concordance between p53 gene mutation and the accumulation of p53 protein cannot be perfect, however, the immunohistochemical assay using anti-p53 antibodies is the most widely applicable approach for detection of tumors in routine investigations, particularly with regard to diagnosis or prognosis. The current study has extended the previous analyses in order to characterize potential association of AO status deficiency with breast cancer. As a working hypothesis, we proposed that malignant transformation, antioxidant status deficiency and oxidative stress are closely connected. Detection of p53 mutations may also be of interest of help identifies early lesions that are at a high risk of malignant evolution, particularly in tumors where p53 mutation is considered as an early event. Specifically, in breast cancer detection of a mutation in a dysplastic lesion may be considered as an indicator of high risk of malignant transformation. In addition, the identification of a mutation may help to determine the clonality of a tumor in patients with multiple lesions, and to characterize recurrence as well as distant metastasis. This approach, which has been implemented in the following guidelines, is based on the model proposed by Gannon et al. (1990[22]) that the different activating mutations exert a common conformational effect which results in expression of the Pab240 epitope on mutant p53 molecules.
P53 becomes phosphorylated in response to various cellular stresses, including reactive oxygen species (ROS), which can result in cell cycle arrest, and in some cases leads to apoptosis. While the apoptotic response to p53 is linked to ROS-dependent activation of p53, basal p53 expression drives a number of antioxidant (AO) responses that can limit oxidative stress (Liu et al., 2008[46]; Ladelfa et al., 2011[43]). P53 induces a range of AO targets such as GPx1, MnSOD, ALDH4 and TPP53INP1 (Budanov et al., 2010[15]; Pani and Galeotti, 2011[59]). P53 protein in the breast may function as a sensor of oxidative stress and its up-regulation in response to cellular (oxidative) stress is suggested to be a cytoprotective response (Hofseth et al., 2004[30]; Staib et al., 2005[70]). Oxidative stress is considered to be implicated in the etiology of cancers including breast (Grek and Tew, 2010[26]; Kryston et al., 2011[40]; Ambrosone, 2000[5]), while in the primary cancer site may increase the likelihood of angiogenesis (Xia et al., 2007[83]) and metastasis (Pani et al., 2010[60]). Since oxidants were shown to be mutagenic and may participate in the activation of proto-oncogenes and the inactivation of tumor suppressor genes (Schiff et al., 2000[68]; Hussain et al., 1994[33]) in this work we estimated the association between mutant p53 proteins, oxidative stress and AO protection in breast cancer. Therefore, we explored the concentration of lipid hydroperoxides (LP), as well as AO enzyme activities of copper, zinc superoxid dismutase (CuZnSOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase (GR) and the level of glutathione (GSH) in the blood of patients with primary breast carcinoma. We also explored the relation between activity and protein level of CuZnSOD since previously was shown that increased levels of O2•- and H2O2 are involved in breast cancer (Ray et al., 2000[63]; Yeh et al., 2005[84]). We expect that these analyses may provide more insight into how the knowledge of the cellular p53 protein expression and antioxidant status can help to improve the management of breast cancer.
Materials and Methods
Reagents
All reagents were purchased from Sigma (St. Louis, MO, USA), and Merck (Darmstadt, Germany). Assays for CuZnSOD, GPx, GR, GSH and LP were purchased from Oxis Bioxytech Assays (Oxis International Inc., Portland, OR, USA). Rabbit anti-CuZnSOD polyclonal antibody (SOD-100) and alkaline phosphatase-conjugated goat anti-rabbit IgG (SAB-301) were purchased from Stressgen Biotechnologies (Victoria, Canada). Rabbit anti-human p53 polyclonal antibody (CM-1) was purchased from Dr. Midgley (University of Dundee, United Kingdom). Biotinylated swine anti-rabbit Ig serum (E-353), mouse anti-human p53 monoclonal antibody (clone DO-7, M-7001), biotinylated rabbit anti-mouse antibody (E-354) and ABComplex-HRP (K-377) were purchased from Dako Denmark (Glostrup, Denmark). Monoclonal antibody Pab240 (sc-99), HRP-conjugated rabbit anti-mouse IgG (sc-358917) and rabbit anti-actin antibody (C-11: 1615) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HRP-conjugated goat anti-rabbit IgG (SAB-301) were purchased from Millipore (Billerica, MA 01821, USA). Human breast carcinoma cell line (CL-239M) was purchased from BioGenex (Fremont, CA, USA).
Subjects and breast cancer samples
The study included 100 breast cancer patients (age in years: median=56, min=32, max=86) from the Institute of Oncology and Radiology of Serbia, Belgrade, Serbia. All patients were newly diagnosed and tumors were clinically categorized as stage I (6 cases), stage II (40 cases), stage III (51 cases) and stage IV (3 cases) according to the classification of the International Union Against Cancer (UICC, 1997). Primary breast cancer cases were classified according to TNM classification and tumors were histologically diagnosed as ductal (72), lobular (21), tubular (4) and medullary (3) type. During the study period patients were not submitted to endocrine, chemo- or radiotherapy. Ten normal breast tissues were selected from macroscopically normal areas and 6 from non-tumor patients. Blood samples from sixty healthy women were used as controls for the measurements of AO parameters and LP (age in years: median=54, min=34, max=80). The patients and the controls were nonsmokers, and they used no alcohol consumption, hormones, oral contraceptives, or dietary supplements with antioxidants. None of the subjects had concomitant diseases such as diabetes mellitus, rheumatoid arthritis, liver disorders or any other malignancies. According to the ethical guidelines of the Helsinki Declaration, informed consent was obtained from all participants and the protocol used in this study was approved by the Ethics Committee of Institute of Oncology and Radiology of Serbia, University of Belgrade, Belgrade, Serbia.
Tissue and cellular fractionation
The normal and cancer tissues were surgically dissected and frozen. The nuclear fraction of normal and tumor tissues were obtained by differential centrifugation (Blobel and Potter, 1966[10]). Briefly, the samples of normal and neoplastic tissue were minced by fine dissection and homogenized in 0.25 M sucrose, 5 mM MgCl2, 0.5 % Triton X-100, 1 mM phenylmethylsulfonylfluoride (PMSF) and 50 mM Tris-HCl, pH 7.4, followed by centrifugation of the homogenate at 800 x g for 10 min (Sorvall RC-5B, Sorvall Ltd., Stevenage, England). The supernatants were spun at 11000 x g at 4 °C for 25 min and the resulting supernatant was again centrifuged at 20000 x g, at 4 °C for 45 min (Beckman L7-55 centrifuge, Beckman Instruments Inc., Palo Alto, CA, USA). The cytosolic fraction was obtained by further centrifugation of supernatant at 100000 x g, at 4 °C for 60 min. The pellet obtained after first centrifugation was homogenized in ice-cold buffer (2.2 M sucrose, 5 mM MgCl2, 0.5 % Triton X-100, 1 mM PMSF and 50 mM Tris-HCl, pH 7.4) and further purified by centrifugation. After this step the pellets representing the soluble nuclear proteins were resuspended in 0.25 M sucrose, 5 mM MgCl2, 0.5 % Triton X-100, 1 mM PMSF and 50 mM Tris-HCl, pH 7.4. Protein content in the nuclear fraction was determined by the method of Lowry et al. (1951[47]), using bovine serum albumin as a reference, and samples were aliquoted and stored at 20 ºC.
Western blot analysis of p53
Proteins isolated from normal and breast cancer cells were suspended in sample loading buffer (0.1 M Tris, pH 6.8, 20 % glycerol, 4 % SDS, 0.04 % bromphenolblue, 10 % ß-mercaptoethanol) and separated electrophoretically according to procedure of Laemmli (1970[44]). Equal amounts of nuclear proteins (50 µg) per line were loaded and run on 12 % SDS-polyacrylamide gels using a Mini-Protein® III Electrophoresis Cell (Bio-Rad Laboratories Inc., Hercules, CA, USA). Separated proteins were transferred onto nitrocellulose membrane (Amersham Hybond, GE Healthcare Bio-Sciences AB, Uppsala, Sweden) according to the procedure of Towbin et al. (1979[75]). Transfer of proteins from the acrylamide gel to nitrocellulose was performed in transfer buffer (25 mM Tris, 192 mM glycine, 0.1 % SDS, 20 % methanol, pH 7.5) at 30 V, 40 mA overnight at 4 °C (Trans-Blot® SD Electrophoretic Transfer Cell, Bio-Rad Laboratories Inc., Hercules, CA, USA). Nitrocellulose filters were blocked overnight with blocking buffer (1 % BSA in 50 mM Tris, 0.9 % NaCl, 0.05 % Tween 20, pH 7.5). The filters were then incubated for 1 h 45 min in buffer (50 mM Tris, 0.9 % NaCl, 0.05 % Tween 20, pH 7.5) plus primary antibody CM-1 at the optimal dilution 1:1000. The CM-1 antibody is a rabbit high-titre polyclonal antiserum raised against human recombinant wild-type p53 protein (Midgley et al., 1992[49]). After 1 h of incubation with the biotinylated swine anti-rabbit Ig serum (diluted 1:1000) and strept AB Complex-HRP conjugated (diluted 1:100) the protein bands were visualized in a HRP substrate diaminobenzidine tetrahydrochloride (DAB). The same membranes were stripped and probed for ß-actin (dilution 1:10000) as an internal loading control. To detect actin with rabbit polyclonal antibody we used the HRP-conjugated goat anti-rabbit (1:2000) antibodies as secondary antibodies. Reproducibility was assessed by repeating the protein extraction and the SDS-PAGE three times.
The primary mAb Pab240 was detected with HRP-conjugated rabbit anti-mouse Ig antiserum (diluted 1:50) and diaminobenzidine substrate. It is important to note that the Pab240 is a mAb recognizing an epitope specifically exposed on the p53 protein in an altered-mutated conformation (Gannon et al., 1990[22]). It has been shown that Pab240 antibody recognizes mutant forms of p53 in a wide range of species by cell staining and immunoblotting (Gannon et al., 1990[22]). The epitopes for the Pab240 antibody was mapped between amino acids 213-217 (Stephen and Lane, 1992[71]).
Immunohistochemical analysis
Immunohistochemical (IHC) staining was performed on 3 µm tissue sections by an avidin-biotin-peroxidase complex (ABC) technique (Hsu et al., 1981[32]) using both frozen and formalin-fixed, paraffin-embedded tissues. For formalin-fixed, paraffin-embedded sections, we applied the microwave oven heating technique, shown to be effective for the retrieval of masked epitopes of many antigens. The primary antibodies used were mouse monoclonal antibody (clone DO-7) recognizing a denaturation-resistant epitope between amino acids 35 and 45 and has been shown to recognize both the wild-type and mutant forms of p53 protein (Vojtesek et al., 1992[78]). In addition, IHC with p53 antibodies CM-1 and Pab240 was performed. Briefly, after dewaxing, endogenous peroxidase activity was blocked by incubating the sections with methanol containing 3 % hydrogen peroxide for 20 min. After preincubation with normal rabbit serum (diluted 1:5) for 20 min, a three-step immunoperoxidase procedure was applied: first with the primary antibodies DO-7 at the appropriate working dilution 1:50 in 0.1 % BSA overnight at 4 °C, second with biotinylated rabbit anti-mouse immunoglobulin (diluted 1:250 in 0.1 % BSA) for one hour at room temperature, and the third with strept ABComplex-HRP (diluted 1:100) for 40 min. Finally, the sections were visualized by incubation with diaminobenzidine tetrahydrochloride (DAB) in hydrogen peroxide substrate with 50 mg of imidazole for 10 min. The sections were counterstained with methyl green (0.5 %, 2 min) or Harris haematoxylin to visualize nuclei. We also used the alkaline phosphatase anti-alkaline phosphatase (APAAP) immunohistochemical technique (Cordell et al., 1984[16]). Negative control slides were processed with each slide run and excluded the primary antibody but included all other steps of the procedure. A human breast carcinoma cell line, which expresses p53 suppressor gene product, is used as the positive control. The IHC staining was semiquantitatively scored by 2 of the authors. We studied the tissue samples by assessing the site of staining, the proportion of cell staining (counting at least 1000 cells/ sample) and the intensity of staining: weak (1+), moderate (2+), or strong (3+). Scoring was as follows: 1 % - 10 % tumor cell nuclei staining for p53=1+; 11 % - 30 % tumor cell nuclei staining for p53=2+; 31 % - 50 % tumor cell nuclei staining for p53=3+; > 50 % tumor cell nuclei staining for p53=4+.
Blood samples
Blood samples were obtained after an overnight fast by venous arm puncture in lithium-heparinized tubes. Blood plasma and blood cell preparations used for measurements of LP and AO parameters were prepared from native and in vitro cultured blood according to previously described procedures (Kasapovic et al., 2010[37]).
Assay of SOD activity
SOD-525™ assay is based on SOD-mediated enhance of autoxidation of 5,6,6a,llb-tetrahydro-3,9,10-tryhydroxybenzo[c]fluorene in aqueous alkaline solution to produce a chromophore with the absorbance maximum at 525 nm (Perkin Elmer spectrophotometer, λ25, Perkin Elmer Instruments, Norwalk, CT, USA). The activity of SOD is calculated from the proportion of the autoxidation rates in the conditions of SOD presence (Vs) and absence (Vc). One SOD-525 activity unit is defined as the activity that doubles the rate of autoxidation in the control blank (Vs/Vc=2).
Assay of CAT activity
CAT activity was measured by the method of Beutler (Beutler, 1984[9]), as the rate of H2O2 decomposition by CAT contained in the examined samples. The reaction was carried out in an incubation mixture containing 1 M Tris–HCl, 5 mM EDTA, pH 8.0 and monitored spectrophotometrically at 230 nm. One CAT activity unit is defined as 1 µmol of H2O2 decomposed per minute under the assay conditions.
Assay of GPX activity
The principle of GPx-340™ assay is that oxidized glutathione (GSSG) formed during reduction of an organic peroxide by GPx, is immediately regenerated to its reduced form (GSH) with associated oxidation of NADPH to NADP+. The oxidation of NADPH was monitored spectrophotometrically as a decline in absorbance at 340 nm. One GPx-340 activity unit is defined as 1 µmol of NADH oxidized per minute under the assay conditions.
Assay of GR activity
The principle of GR-340™ assay is that oxidation of NADPH to NADP+ during the reduction of oxidized glutathione (GSSG) is catalyzed by a limiting concentration of GR. The oxidation of NADPH was spectrophotometrically measured as a decline in absorbance at 340 nm. One GR-340 activity unit is defined as 1 µmol of NADH oxidized per minute under the assay conditions.
GSH concentration
GSH-420™ assay is based on the reaction of 4-chloro-1-methyl-7-trifluoromethylquinolinium and all thiols to produce thioethers. Following the increase of pH above 13 by addition of base, a ß-elimination specific to the GSH-thioether produces the chromophoric thione, measured spectrophotometrically at 420 nm.
Lipid hydroperoxide concentration
The principle of LPO-560™ assay is that under acidic conditions, hydroperoxides promote oxidation of Fe2+ to Fe3+ ions which binds the indicator xylenol orange and produces a colored complex measured spectrophotometrically at 560 nm. Since biological samples have much higher content of H2O2 than that of other hydroperoxides, pretreatment with CAT decomposed the existing H2O2 and eliminated the interference.
Protein concentration
Determination of protein concentration was performed by the method of Lowry et al. (1951[47]).
Western blot analysis of CuZnSOD
Equal amounts of protein were dissolved in SDS-PAGE sample loading buffer and subjected to SDS-PAGE electrophoresis in 10 % polyacrylamide gel (Mini-protean®3 Cell, Bio-Rad Laboratories Inc.) (Laemmli, 1970[44]). Separated proteins were transferred to nitrocellulose membranes for Western blot analysis of CuZnSOD (Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell, Bio-Rad Laboratories Inc.). Non-specific binding sites on membranes were blocked with TBST (10 mM Tris, 150 mM NaCl, 0.1 % Tween 20) containing 1 % BSA and then exposed to rabbit anti-CuZnSOD polyclonal antibody and rabbit anti-actin antibody. For detection purposes we used alkaline phosphatase-conjugated goat anti-rabbit IgG. Reproducibility was assessed by repeating the SDS-PAGE and blotting three times. The density of bands was determined by ImageJ processing program, normalized to the level of actin and expressed as percent of value found in the control samples considered as 100 %.
Statistical analysis
The significance of differences between different experimental values was assessed by means of ANOVA, Student’s t-test and Pearson’s product moment correlation. Data were tested at a statistical significance level of p<0.05 and expressed as mean±SEM. All statistical manipulations were performed using the SPSS for Windows Software System and OriginPro 8.0.
Results
Expression of p53 protein in breast carcinoma: immunohistochemistry and Western blot analysis
Forty-seven cases of breast cancer were screened for the presence of the p53 protein by immunohistochemical and Western blot assay using a polyclonal antiserum CM-1 or monoclonal antibodies DO-7 and Pab240 with multiple anti-p53 specificities. The majority of the immunohistochemical studies used either DO-7 or Pab240 as the antibody for identifying abnormal accumulation of p53 and so results for these antibodies are shown separately. A positive staining was observed in 30/47 (64 %) specimens with variations in the staining percentage and intensity. Three groups were detected: 25 cases (53 %) had high levels of p53 in the nucleus of the cancer cells (Figure 1a(Fig. 1); 2a,b(Fig. 2); 3a(Fig. 3)), 17 cases (36 %) had a complete lack of detectable staining (Figure 2c(Fig. 2)) and 5 cases (11 %) showed a pattern of cytoplasmic and nuclear staining. In 2 cases, a strong nuclear and weaker cytoplasmic localization of p53 was observed (Figure 1a(Fig. 1)). The same breast cancer (Figure 2a and b(Fig. 2)) gave strong nuclear staining with Pab 240 (Figure 3a(Fig. 3)). Surprisingly, 17 specimens (36 %) showed complete absence ofdetectable p53 protein in multiple sections despite the presence of abundant malignant tissue. Normal levels of p53 protein present in stromal cells, or in normal breast epithelium were undetectable and thus served as internal control (Figure 4a(Fig. 4)). Control experiments that substituted normal mouse IgG or phosphate-buffered saline for the p53-specific monoclonal antibody showed no detectable staining. IHC data verified the difference in the localization patterns of the p53 wt forms and/or their mutants. Moreover, the localization of the p53 protein and each of the mutants differs significantly in various breast cancer cells, suggesting a multipotent role of the protein when it comes to activation pathways and localization patterns within the cell. In fact, immunohistochemical detection of mutant forms of p53 overexpression results in extensive mammary epithelial cell hyperplasia and a disorganized basement membrane, all features associated with malignancy. The higher expression levels of mutant forms of p53 protein in most breast cancer analyzed was associated with more frequent lymph node metastasis. We observed a significant difference for the progression of the disease between p53-positive tumors and p53-negative tumors (Student’s t-test, p<0.01). In general, tumor p53 overexpression correlates with a more aggressive tumor phenotype and negative survival prognosis. As expected, the MAbs DO-7 and Pab240 appears as a good marker for recognition of p53 mutated cases including those with a worse prognosis (Figure 4b(Fig. 4); Figure 1a(Fig. 1)).
Figure 1. Figure 2: (a) Carcinoma of the breast with immunohistochemical evidence of p53 overexpression. Intense nuclear staining is evident. P53 associates with nucleolar structures.
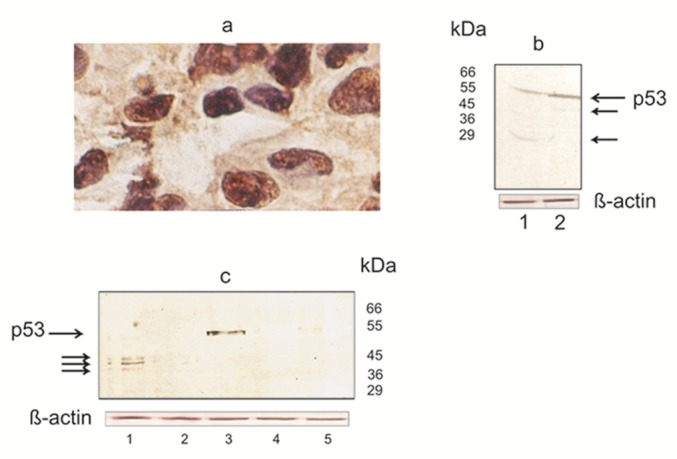
Frozen section was stained with anti-p53 antibodies DO-7 by the immunoperoxidase technique, with hematoxylin counterstain. Magnification, x 250.
(b) Western blotting analyses of p53 expressed in nuclear extracts of breast carcinoma cells. As indicated, the anti-p53 antibody detected one band with the classical molecular mass of 53 kDa and another at 45 and 29 kDa. Truncated protein representing the p53 can be seen by smaller size.
Proteins in nuclear extracts were resolved on a 12 % SDS-PAGE and then transferred to a nitrocellulose membrane. p53 present in the nuclear extract were detected with CM-1 antibody (see Materials and methods). This experiment was repeated in triplicate with reproducible results. Lane 1, 2- breast cancer cases
(c) Western blot analysis shows that p53 in nuclear extracts of breast cancer cells also migrated as three closely spaced bands at 38-45 kDa (Lane 1). The arrow indicates a single immunoreactive band representing p53 (Lane 3). Molecular mass markers are presented on the right.
Procedures for SDS-PAGE and Western blot analyses were the same as those used for the experiment described in Figure 2b, and detailed in Materials and Methods. Each analysis was repeated three times with similar results. Lane 1, 2 and 3- breast cancer cases; Lane 4, 5- normal breast tissue
Figure 2. Figure 3: Immunohistochemical analysis of p53 protein expression in carcinoma of the breast.

(a) A breast tumor displaying a more generalized and intense p53 nuclear staining. A diffuse pattern of staining was identified in the cytoplasm of tumor cells. The labelled cells for the p53 show a brown precipitate, a green staining underlines nuclei of unlabelled cells.
(b) The positive cases demonstrated characteristic staining patterns that were either predominantly nuclear or both nuclear and cytoplasmic. P53 is located at the cytoplasmic surfaces of the nuclear envelope. Different structural systems of the nucleus are targets for p53. Nuclear p53 is found in the nucleoplasm, chromatin fraction and also attached to the nuclear matrix. Insert: focal staining showing the fine nucleolar distribution characteristic of p53. DAB staining is prevalent throughout the nuclear bodies.
(c) Breast carcinoma cells show negative staining for p53. Unlabelled nuclei show only counterstain, indicated by the blue color.
Avidin-biotin immunoperoxidase technique on frozen sections counterstained with methyl green (a, b) or haematoxylin (b insert, c). All panels were photographed at the same magnification.
Figure 3. Figure 4: (a) Immunohistochemical detection of mutant forms of p53 in breast cancer cells.

High-power view showing granular staining pattern confined to nuclei. The epithelial levels contain cells with enlarged, darkly stained, irregularly shaped, dysplastic nuclei. Even among the positive cells the strength of staining was quite variable. Different structural systems of the nucleus are targets for p53. Nuclear bodies display punctuate patterns.
P53 was immunostained with Pab240 antibody (diluted 1:50) using the avidin-biotin-peroxidase complex (ABC) method. The appropriate secondary antibodies were diluted 1:200, and histochemical detection was done by developing with aminoethylcarbazole. Nuclei were counterstained with hematoxylin. Frozen tumor sample (magnification, x 320).
(b) Western blot analysis of p53 expressed in the nuclear extract of breast cancer cells.
A single immunoreactive band representing the mutant forms of p53 were visualized in nuclear extracts of breast carcinoma cells (Lane 1, 2). The p53 protein of relative molecular mass 53kDa, was absent in normal breast (Lane 3).
Equal amounts of nuclear proteins (50µg/lane) from extracts of normal and breast cancer tissues were separated on 12 % SDS-PAGE and transferred to nitrocellulose membrane. The primary mAb Pab240 was detected with HRP-conjugated rabbit anti-mouse Ig antiserum (diluted 1:50). The reactive proteins were visualized with diaminobenzidine (DAB) as chromogen. Bottom panel shows actin loading control. Lane 1, 2- invasive ductal breast carcinomas. Lane 3- normal breast tissue
Molecular mass markers are presented on the right. These results are representative of three experiments.
Figure 4. Figure 1: p53 immunostaining in normal breast tissue and breast carcinoma (a) Normal breast tissue is negative for p53 (b) Positive p53 immunostaining in the epithelial cancer cells using the anti-p53 mAb DO-7. A mutant case with intense nuclear staining. Immunoperoxidase staining with nuclear counterstain with hematoxylin. Magnification, x 40. Microwave antigen retrieval was for 15 min.
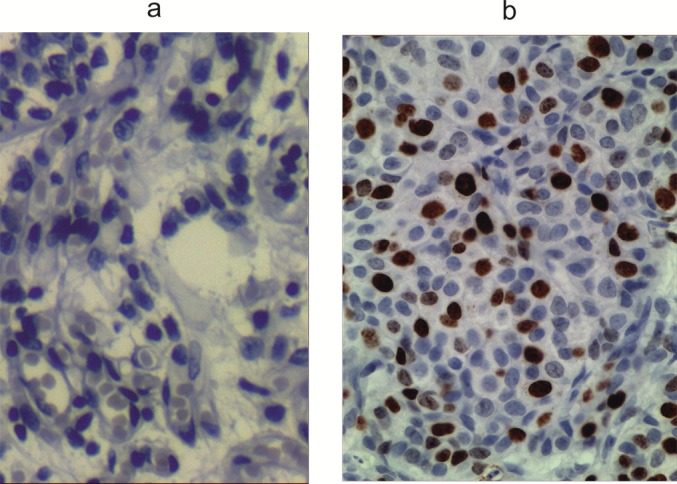
P53 proteins overexpressed ectopically form a subnuclear structure. It is noteworthy that positive immunostaining of p53 is in conjunction with the appearance of nuclear bodies (Figure 1a(Fig. 1); 2a, b(Fig. 2); 3a(Fig. 3)). Nuclear bodies display punctate patterns suggesting p53 and additional proteins co-localized in the nuclear bodies which were not seen in control cells. The nuclear body structure was detected in a high percentage of p53 positive cells but not in a small population of cells which stained negatively for p53 (Figure 1a(Fig. 1); 2a, b(Fig. 2); 3a(Fig. 3)), and is a general phenomenon of the proteins co-expression. Additionally, it is possible that anti-p53 antibodies cross-react with structurally similar epitopes on unrelated molecules.
The expression of p53 detected by Western blotting was compatible with the results of immunohistochemistry in most cases examined. Immunoreactive proteins were visualized in nuclear extracts of breast carcinoma cells (Figure 1b, c(Fig. 1); 3b(Fig. 3)). As indicated, the anti-p53 antibody detected one band with the classical molecular mass of 53 kDa and another at 45 and 29 kDa (Figure 1b(Fig. 1)). Truncated protein representing the p53 can be seen by smaller size of the 45- and 29-kDa (Figure 1b(Fig. 1), Lane 1, 2). Notably, Western blot analysis with a p53 specific antibody CM-1 revealed three closely spaced bands migrated at 36-45 kDa (Figure 1c(Fig. 1), Lane 1). Expression of p53 and its isoforms Δp53 (45 kDa) and isoform of ~ 29 kDa were more common in cases with LN metastasis (Figure 1b(Fig. 1), Lane 1, 2). Taken together, these results indicate that in nuclear extracts of breast carcinoma cells p53 exists in various assemblies, with the smallest oligomer being a threemer. Western blotting with epitopespecific monoclonal antibody Pab240 strongly suggests that nuclear extracts from breast cancer cells express mutant forms of p53 (Figure 3b(Fig. 3)).
Antioxidant parameters and lipid hydroperoxides
In the blood cells of native blood, AO parameters of breast cancer patients were found to decrease when compared to age-matched controls (Figure 5(Fig. 5)). The decline of enzyme activities were recorded for SOD (p<0.001), CAT (p<0.01) and GR (p<0.001), whereas GPx activity was found to be unchanged (p>0.05). This was followed with the decline of GSH (p<0.05) and the increase of LP concentrations (p<0.001).
Figure 5. Antioxidant enzyme activities and concentration of GSH and lipid peroxides (LP) in native blood of control subjects (A) and breast cancer patients (B). The data are given in mean values ± SEM, *p<0.05, **p<0.01, ***p<0.001 compared to A (Student’s independent t-test).

The decline of CuZnSOD activity (p<0.001) and concentration of GSH (p<0.01) in breast cancer patients were the only effects which were maintained after 48 h of in vitro blood incubation, while the effects on CAT (p>0.05) and GR (p>0.05) activities, detected in native blood, disappeared (Figure 6(Fig. 6)). Prolonged decline of CuZnSOD activity was indication for the change in protein synthesis of this enzyme. The disappearance of effects on other AO enzymes in incubated blood is likely the consequence of different modulatory milieu of blood cells presented in native blood and incubation medium.
Figure 6. Antioxidant enzyme activities and concentration of GSH in in vitro incubated blood of control subjects (A) and breast cancer patients (B). The data are given in mean values ± SEM, **p<0.01, ***p<0.001 compared to A (Student’s independent t-test).

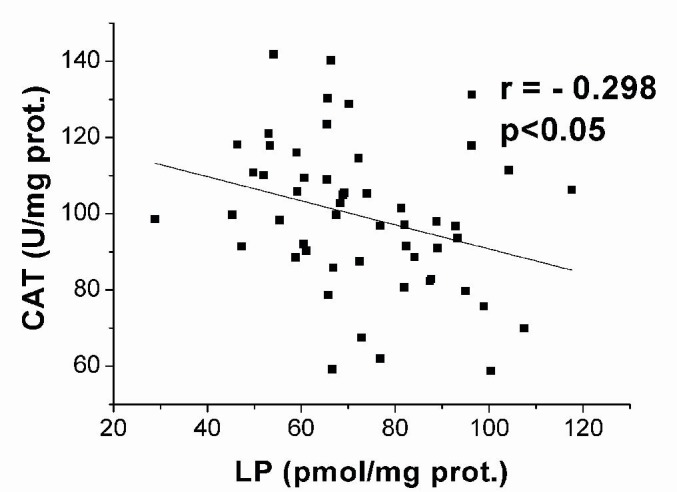
Analysis of correlations between the activities of AO enzymes or GSH concentration and the concentration of LP revealed negative correlation between the activity of CAT and concentration of LP (r = - 0.298, p<0.05) (Figure 7(Fig. 7)).
Figure 7. Data plot and coefficient of Pearson’s correlation r between the activity of CAT and the concentration of lipid peroxides (LP) in native blood of breast cancer patients.
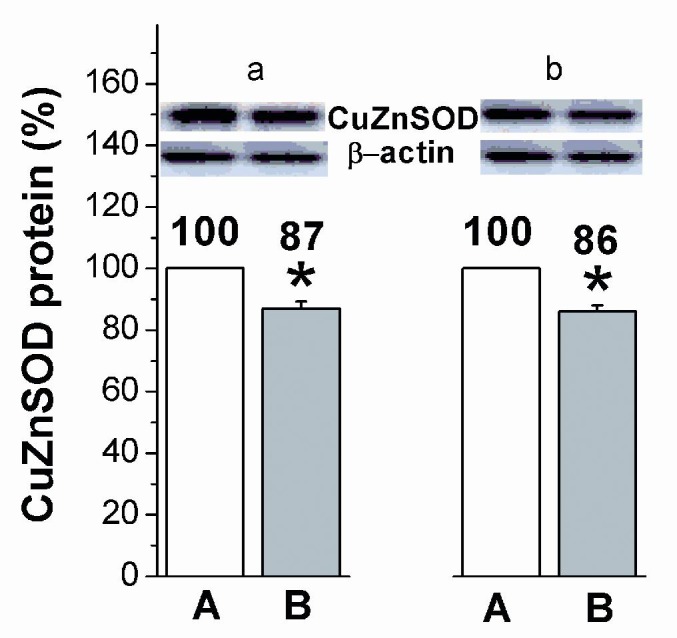
CuZnSOD protein level was reduced in blood cells of breast cancer patients in native blood (p<0.05), as well as in incubated blood (p<0.05) when compared to healthy controls considered as 100 % (Figure 8(Fig. 8)).
Figure 8. Relative levels of CuZnSOD protein in native (a) and in vitro incubated (b) blood of control subjects (A) and breast cancer patients (B). Relative values are expressed as percent of value found in the control subjects, which is considered as 100 %. * p<0.05 compared to A (Student’s independent t-test).
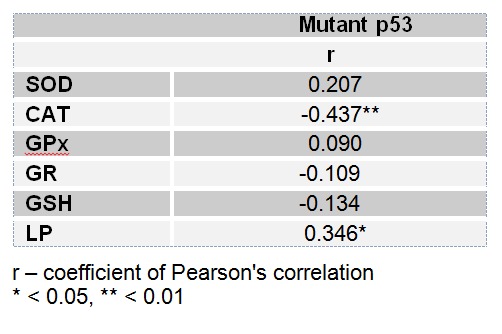
Analysis of correlations between the AO parameters or concentration of LP and the level of mutant p53 protein expression showed negative correlation for the activity of CAT (- 0.437, p<0.01) and positive correlation for the concentration of LP (0.346, p<0.05) (Table 1(Tab. 1)).
Table 1. Correlation between AO parameters or concentration of LP and expression of mutant p53 proteins.
Discussion
The p53 tumor suppressor gene has a critical role in the protection of the genome from the consequences of DNA damage and its inactivation is a critical step in cellular transformation. p53 serves as transcriptional regulator, genomic stabilizer, inhibitor of cell cycle progression, facilitator of apoptosis, and inhibitor of angiogenesis (Hollstein and Hainaut, 2010[31]). Major variants of the p53 protein may be formed through alternative splicing of mRNA or protein cleavage. To date, the human p53 gene has been reported to encode for nine different p53 proteins (isoforms) through variously spliced mRNA (Bourdon et al., 2005[12]; Rohaly et al., 2005[65]). These are the full-length proteins, three NH2-terminally truncated isoforms translated from an alternative point of initiation at codon 40 or by alternative splicing of intron 2 (Bourdon et al., 2005[12]; Courtois et al., 2002[17]; Ghosh et al., 2004[24]), two COOH-terminally truncated isoforms produced by alternative splicing of intron 9 (Bourdon et al., 2005[12]; Flaman et al., 1996[21]), three isoforms produced from an internal promoter in intron 4 (Bourdon et al., 2005), and one isoform produced by alternative splicing within exons 7 to 9 (Rohaly et al., 2005[12]). In addition, truncated p53 protein products are known produced as a result of protease action most likely through an autoproteolysis mechanism (Okorokov et al., 1997[56]). However, it is also possible that some new p53 isoforms arise due to mutations (Varley et al., 2001[77]). Mutations in p53 gene and elevated levels of mutated forms of p53 protein in breast tumors suggesting that the deregulated p53 expression and reduced apoptosis may be important in breast tumor development (Zhou et al., 2009[86]; Abdel-Fatah et al., 2010[1]). P53 gene mutations are seen in all stages of breast cancers, from in situ lesions to invasive carcinomas, suggesting that the p53 gene alteration can occur in the earliest recognized phase of breast cancer and that this alteration is maintained during tumor progression (Weigelt et al., 2003[81]; Torres et al., 2007[74]; Suzuki and Tarin, 2007[72]). Furthermore, mutations of the p53 gene seem to be associated with clonal evolution and metastatic progression of the disease (Weigelt et al., 2005[82]; Nicolini et al., 2006[54], 2007[55]; Riethdorf et al., 2008[64]). Abrogation of p53 function should therefore lead to a more aggressive breast cancer phenotype and a worse clinical outcome (Abdel-Fatah et al., 2008[2]). Molecular analysis of the p53 gene is complex and poorly suited to routine diagnostic use: p53 mutations have been found in 90 of the 393 codons required for synthesis of the protein. This considerable heterogeneity makes diagnosis more difficult, because the region to be analyzed extends over virtually the entire gene. Many groups have focused their invesinvestigations on exons 5 to 8, which are the target of 80-90 % of all mutations. Mutations in exons 4 to 8 of the p53 gene were evaluated using PCR-SSCP sequencing analysis as described Olivier et al. (2006[57]) and Shah et al. (2012[69]). The vast majority of the TP53 mutations detected in the earlier study by Angèle et al. (2000[6]), are among those frequently found and are in accordance with the published TP53 mutation spectra in breast cancer (IARC TP53 mutations database in breast tumors). The difficulty to link p53 mutation status to clinical outcome and cancer treatment can be explained by the fact that mutations of the p53 gene do not necessarily result in inactivation of p53 transcriptional activity. Hence, 60 % of the mutations that can occur in the p53 gene do not alter p53 transcriptional activity. Only 15 % of the mutations lead to mutant p53 completely inactive in transactivation. In the remaining 25 %, mutant forms of p53 present differential transcriptional activity. They can transactivate some promoters, but are completely inactive on others (Petitjean et al., 2007[61], [62]).
In this study we used a panel of anti-p53 antibodies CM-1, DO-7 and Pab240 for cell staining and found abnormalities in at least 30 % of the cases. Based on the literature, DO-7 is one of the most sensitive for the detection of p53 overexpression (Vojtesek et al., 1992[78]). Other published data also support that rabbit p53 antibody CM-1 is highly sensitive for the p53 staining (Midgley et al., 1992[49]). It is widely accepted that in normal tissues, p53 protein is present in such low quantities that it is not readily detectable by immunochemical techniques. Thus, the accumulation of p53 protein in normal breast epithelial cells is a highly unusual finding because the abnormally stabilized p53 protein has only been associated with malignant disease and DNA damage (Hollstein and Hainaut, 2010[31]). It is thought that the monoclonal PAb240 antibody reliably detects a wide variety of p53 mutations and these mutations have a common effect on the structure of p53. An antibody PAb240 does not immunoprecipitate wild-type p53 (Gannon et al., 1990[22]). Based on the analysis of breast tumor samples, it became clear that DO-7 and CM-1 antibodies are potentially useful in the detection of p53 overexpression. In accordance with the before mentioned data, the Pab240 antibody is extremely useful for the detection of p53 mutations of the type that occur in breast cancer.
Western blotting analyses showed that the faster migrating forms of p53 are truncated version of the p53 protein, a Δp53 (~ 45 kDa) and the isoform of ~ 29 kDa (Figure 1b(Fig. 1)). Understandably, as tetramerization domain (residues 325-356) is preserved in Δp53 (Murray-Zmijewski et al., 2006[52]), the p53 isoform has the ability to oligomerize. The oligomeric state of Δp53 in nuclear extracts of breast carcinoma cells (Figure 1c(Fig. 1)), suggesting that Δp53 may exert a functions, differential transcriptional activity per se independent from multi-functional transcription factor p53, and a gene activation pattern different from that of p53 (Rohaly et al., 2005[65]). Additionally, that Δp53 does not interact with p53 was confirmed by Western blot (Figure 1b, c(Fig. 1)). From the present results, we can conclude that out of the detected p53 isoforms, (Bourdon et al., 2005[12]; Bourdon, 2007[11]), Δp53 is the most easily identified isoform in nuclear extracts of breast carcinoma cells and the other isoforms are more difficult to identify. However, this should be interpreted with cautions, as Bourdon et al. (2005[12]) showed that p53ß, p53γ and Δ40p53 migrate all at 45 kDa. In this respect, the 45 kDa band identified by Rohaly et al. (2005)[65] is probably composed of a mix of Δp53, p53ß, p53γ and Δ40p53. Δp53 is an alternative splice form that differs from the full-length p53 form by lacking parts of exon 7, complete elimination of exon 8 and partial removal of exon 9 (Rohaly et al., 2005[65]). The study of Baumbusch et al. (2006[7]) is one of the first confirming that mRNA expression of both the full-length p53 and the Δp53 form are elevated in tumors with missense and in frame mutations. The mRNA expression of p53 and Δp53 level showed a wide range in p53 wild-type tumors with significant association to molecular breast cancer subtype distribution. Although our data suggests that p53 variants an ~ 45 and 29 kDa represent a minority of total cellular p53, a role for these isoforms cannot be discounted in view of their potential for interaction with p53-family member to alter the balance of p53, p63 and p73 transcriptional activities during normal and neoplastic cell growth. We should note that in the most tumor analyzed p53 levels were extremely low for detection, as in the case of the tumor type shown (Figure 1c, Lane 2(Fig. 1)), suggesting deletion, complex mutations generating stop codons, overexpression of the p53 ubiquitin ligase MDM2, or wild-type p53 status.
The three staining pattern of p53 protein (nuclear, nuclear and cytoplasmic and absent) have commonly been explained by the fact that mutant p53 proteins are more stable (have a longer half-life) than wild-type p53 proteins. The non-detectable levels of p53 in the p53 negative tumors are either the very low wild-type levels or due to loss of p53 (deletion mutants). In contrast, the elevated levels of nuclear and cytoplasmic p53 proteins were previously thought to be mutant p53 proteins (Gannon et al., 1990). The high level of p53 protein seen in mutated tumors has previously been explained by accumulation of the protein due to lack of degradation of the mutated protein and not by overexpression at the mRNA level (Royds and Iacopetta, 2006[66]). Mutant forms of p53 in breast cancer cells may accelerate tumor progression under these circumstances via default of p53-mediated cell cycle arrest, DNA repair and/or apoptosis. It is now generally believed that different forms of mutant p53 proteins may have different functional and biological effects (Petitjean et al., 2007[61], [62]; Vousden and Prives, 2005[79]). All available information suggests that mutant forms of p53 are at a crossroad for multiple signaling pathways (Oren and Rotter, 2010[58]). Further analyses of point and truncated mutant expression products of the p53 gene has shown that mutated forms of p53 not only lose their tumor suppressive functions but may also gain new abilities that enhance tumorigenesis (Oren and Rotter, 2010[58]; Heinlein et al., 2008[29]; Adhikari and Iwakuma, 2009[3]; Muller et al., 2009[51]; Brosh and Rotter, 2009[14]).
Tumor derived p53 mutants act as transactivators of various cancer-associated genes in tumor progression. Indeed, the p53 mutation is linked with chemo-resistance and transformation to a more aggressive disease in many tumor types (Al-Joudi et al., 2008[4]). In the present study, we found that the higher expression levels of mutant p53 protein in most breast cancer analyzed was associated with more frequent lymph node metastasis, advanced TNM stage and poor survival which is consistent with other reports (Olivier et al., 2006[57]; Miller et al., 2005[50]). A detailed overview of mutant form p53’s role in tumor formation is beyond the scope of this article and the reader is referred to several excellent reviews on this topic (Oren and Rotter, 2010[58]; Adhikari and Iwakuma, 2009[3]; Muller et al., 2009[51]). Clearly, p53 mutations impact on clinical outcome (Olivier et al., 2006[57]), but it would be over-simplistic to assume that p53 missense mutations are the sole contributors from the p53 network to the outcome of breast cancer. To date, attempts to group breast cancers according to p53 immuno-histological phenotype have lagged behind transcriptional array approaches of tumor p53 transcriptional profiling (Miller et al., 2005[50]) and stroma profiling (Finak et al., 2008[19]), demonstrating significant association with outcome in breast cancer. The comparison of p53 mutation to immunohistochemistry in solid tumors initially identified patterns of protein expression using p53, mdm2, mdm4 and p21, which in the 44 breast cancers examined closely, reflected the presence and class of p53 mutation (Nenutil et al., 2005[53]). Abdel-Fatah et al. (2010[1]) have taken this further and hypothesize that the functional status of p53 pathways, rather then p53 protein expression alone, could accurately discriminate between patients with good or poor outcome. Novel p53 isoforms suggest alternative splicing as a regulatory feature of p53 activity. Importantly, it has also been reported that interactions between p53 and its isoforms Delta40p53 or Delta133p53 play critical roles in intracellular signaling by ROS (Hafsi and Hainaut, 2011[28]).
Here we also demonstrated that the nuclear bodies formed by p53 although variable in size are clearly visible by light microscopy, which suggests that they likely contain a large number of additional proteins and are potentially associated with chromatin. As we exclude the possibilities that these nuclear bodies are nucleoli or nucleolus break-down products, one interpretation of these results is that the nuclear bodies are formed to regulate the sequence-specific transactivity of p53. Additionally, it is possible that they may function in a DNA synthesis or chromatin structure-related events. Experimental evidence suggests that p53 may be directly involved in the regulation of DNA synthesis during either DNA replication or repair (Kim and Deppert, 2006[38]; Gatz and Wiesmüller, 2006[23]; Vousden and Prives, 2009[79]). Given their discrete and higher order structure, it is likely that p53 facilitates the formation of and associates with nuclear bodies. The interaction between p53 and additional proteins co-localized in the nuclear bodies is likely critical in the development of the neoplastic phenotype. In this regard, it is of note that mutations associated with the different forms of breast tumors are not only associated with alterations in oxidative stress but also cell cycle alterations. Based on the findings that in a more aggressive tumor phenotype mutant forms of p53 are localized into the nucleus in a form of nuclear bodies, this subnuclear structure may be a possible immunocytochemical biomolecular marker for the identification of this cancer type. Many forms of genotoxic stress can increase nuclear bodies number and alter their biochemical composition suggesting that these characteristic changes in nuclear bodies are likely a universal response to DNA damage (Dellaire and Bazett-Jones, 2004[18]; Bernardi and Pandolfi, 2007[8]).
Oxy-radicals were found to induce mutations of hotspot codons 248 and 249 of human fibroblast p53 gene (Hussain et al., 1994[33]). The alteration of TP53 gene or post-translational modification of p53 protein can alter its response to cellular stress. Based on this, hypothesis was made that the generation of oxygen and nitrogen species and aldehydes induce a high frequency of p53 mutations in oxy-radical overload disease that may contribute to the increased risk of cancer (Hussain and Harris, 2006[35]). Our results support this view, showing that expression of mutant p53 proteins associates with increase of oxidative damages and decrease of AO detoxifying capacity in breast cancer. Elevated level of plasma LP (Figure 5(Fig. 5)) is in agreement with previous observations of elevated different lipid peroxidation products in the blood in those patients, such as TBARS (Kumaraguruparan et al., 2002[41]) and MDA (Gonenc et al. 2001[25]). Diminished activities of SOD and CAT enzymes in blood cells (Figure 5(Fig. 5)) are also in accordance with previous records in erythrocytes and blood of breast cancer patients (Ray et al., 2000[63]; Kumaraguruparan et al., 2002[41]). Under oxidative stress, elevated level of ROS can damage AO enzyme molecules, leading to their reduced activities (Kono and Fridovich, 1982[39]; Salo et al., 1988[67]) and autocatalysis of oxidative damage process.
Increased amounts of H2O2 produced by human tumor cells keeps them under persistent oxidative stress (Szatrowski and Nathan, 1991[73]). Enlarged production of O2•- and H2O2 was also recorded in the blood of breast cancer patients independent of clinical stage of disease and menopausal status (Ray et al., 2000[63]; Yeh et al., 2005[84]). This may be due to decline of CuZnSOD and CAT activities (Figure 5(Fig. 5)) showing decreased capacity for O2•- and H2O2 detoxification in native blood of these patients. Lowered CAT activity correlating with an increase of LP concentration (Figure 7(Fig. 7)), indicates elevated lipid peroxidation mediated by increased production of H2O2. Since GPx activity in blood cells was unchanged in patients (Figure 5(Fig. 5)), it seems that oxidative damage is related mostly to suppressed activity of CAT. The lower GSH level in patients (Figure 5(Fig. 5)), supports the hypothesis that GSH status is inversely correlated to malignant transformation (Kumaraguruparan et al., 2002[42]). This may be due to decreased GR capacity for GSH restoration (Figure 5(Fig. 5)).
Suppression of CuZnSOD activity has been the only prolonged effect of breast cancer on AO enzymes recorded after 48 h of blood incubation (Figure 6(Fig. 6)), indicating drop of SOD protein, which was confirmed by Western blot analyses in native, as well as in incubated blood (Figure 8(Fig. 8)). It seems that disturbed expression of AO enzymes for O2•- and peroxides detoxification in human cancerogenesis, besides previously observed MnSOD (glioma) (Zhong and Oberly, 1997[85]), and GPx (breast cancer) (Gromadzinska et al., 1997[27]), includes CuZnSOD enzyme as well. Association between diminished CAT activity, increased oxidative damages and the expression level of mutant p53 forms (Figure 7(Fig. 7), Table 1(Tab. 1)), indicates H2O2 as most possible mediator of mutagenic impact on p53 in breast cancer, regarding its non-restricted diffusion mobility (Hussain et al., 2004[34]) and involvement in signal transduction pathways (Finkel, 2011[20]) and carcinogenesis (Ambrosone, 2000[5]). This is supported with recent findings of crucial function of CAT in p53-mediated ROS regulation. p53 and its downstream targets physically and functionally cooperate with CAT causing p53 to express either antioxidant or prooxidant functions depending on stress intensity (Kang et al., 2013[36]). After genotoxic stress, high levels of p53 and p53-inducible gene 3 (IG3) cooperate to reduce CAT activity and shift the intracellular environment toward prooxidative conditions to induce apoptosis. Following this, it can be assumed that chronic oxidative stress in breast cancer, by inhibiting the detoxifying capacity for H2O2 and elevating the expression of mutant p53 proteins, disturb apoptotic interplay of p53, AO system and ROS contributing to breast carcinogenesis.
Acknowledgements
We wish to thank Professor David P. Lane and Dr. Carol A. Midgley (University of Dundee, United Kingdom) for the gift of CM-1 antibodies and helpful discussions. This work is supported by the Ministry of Education and Science of the Republic of Serbia (Grant 173034).
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Abdel-Fatah TM, Powe DG, Agboola J, Adamowicz-Brice M, Blamey RW, Lopez-Garcia MA, et al. The biological, clinical and prognostic implications of p53 transcriptional pathways in breast cancers. J Pathol. 2010;220:419–34. doi: 10.1002/path.2663. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Fatah TM, Powe DG, Hodi Z, Reis-Filho JS, Lee AH, Ellis IO. Morphologic and molecular evolutionary pathways of low nuclear grade invasive breast cancers and their putative precursor lesions: further evidence to support the concept of low nuclear grade breast neoplasia family. Am J Surg Pathol. 2008;132:513–23. doi: 10.1097/PAS.0b013e318161d1a5. [DOI] [PubMed] [Google Scholar]
- 3.Adhikari AS, Iwakuma T. Mutant p53 gain of oncogenic function: in vivo evidence, mechanism of action and its clinical implications. Fukuoka Igaku Zasshi. 2009;100:217–28. [PubMed] [Google Scholar]
- 4.Al-Joudi FS, Iskandar ZA, Rusli J. The expression of p53 in invasive ductal carcinoma of the breast: a study in the North-East States of Malaysia. Med J Malaysia. 2008;63:96–9. [PubMed] [Google Scholar]
- 5.Ambrosone CB. Oxidants and antioxidants in breast cancer. Antioxid Redox Signal. 2000;2:903–17. doi: 10.1089/ars.2000.2.4-903. [DOI] [PubMed] [Google Scholar]
- 6.Angèle S, Treilleux I, Tanière P, Martel-Planche G, Vuillaume M, Bailly C, et al. Abnormal expression of the ATM and TP53 genes in sporadic breast carcinomas. Clin Cancer Res. 2000;6:3536–44. [PubMed] [Google Scholar]
- 7.Baumbusch LO, Myhre S, Langerød A, Bergamaschi A, Geisler SB, Lønning PE, et al. Expression of full-length p53 and its isoform Δp53 in breast carcinomas in relation to mutation status and clinical parameters. Mol Cancer. 2006;5:47. doi: 10.1186/1476-4598-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 9.Beutler E. Red cell metabolism. 3rd ed. Orlando, FL: Grune and Stratton Inc.; 1984. pp. 105–106. [Google Scholar]
- 10.Blobel G, Potter VR. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154:1662–5. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 11.Bourdon JC. p53 and its isoforms in cancer. Br J Cancer. 2007;97:277–82. doi: 10.1038/sj.bjc.6603886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–37. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braithwaite AW, Prives CL. p53: more research and more questions. Cell Death Differ. 2006;13:877–80. doi: 10.1038/sj.cdd.4401938. [DOI] [PubMed] [Google Scholar]
- 14.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field: lessons from the distribution of TP53 mutations. Nat Rev Cancer. 2009;9:701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 15.Budanov AV, Lee JH, Karin M. Stressin' sestrins take an aging fight. EMBO Mol Med. 2010;2:388–400. doi: 10.1002/emmm.201000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordell JL, Falini B, Erber WN, Ghosh AK, Abdulaziz Z, MacDonald S, et al. Immunoenzymatic labeling of monoclonal antibodies using immune complexes of alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes) J Histochem Cytochem. 1984;32:219–29. doi: 10.1177/32.2.6198355. [DOI] [PubMed] [Google Scholar]
- 17.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, et al. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–8. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
- 18.Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 2004;26:963–77. doi: 10.1002/bies.20089. [DOI] [PubMed] [Google Scholar]
- 19.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 20.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaman JM, Waridel F, Estreicher A, Vannier A, Limacher JM, Gilbert D, et al. The human tumour suppressor gene p53 is alternatively spliced in normal cells. Oncogene. 1996;12:813–8. [PubMed] [Google Scholar]
- 22.Gannon JV, Greaves R, Iggo R, Lane DP. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J. 1990;9:1595–602. doi: 10.1002/j.1460-2075.1990.tb08279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–16. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol. 2004;24:7987–97. doi: 10.1128/MCB.24.18.7987-7997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonenc A, Ozkan Y, Torun M, Simsek B. Plasma malondialdehyde (MDA) levels in breast and lung cancer patients. J Clin Pharm Ther. 2001;26:141–4. doi: 10.1046/j.1365-2710.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- 26.Grek CL, Tew KD. Redox metabolism and malignancy. Curr Opin Pharmacol. 2010;10:362–8. doi: 10.1016/j.coph.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gromadziňska J, Wasowicz W, Andrijewski M, Sklodowska M, Zambrano Quispe O, Wolkanin P, et al. Glutathione and glutathione metabolizing enzymes in tissues and blood of breast cancer patients. Neoplasma. 1997;44:45–51. [PubMed] [Google Scholar]
- 28.Hafsi H, Hainaut P. Redox control and interplay between p53 isoforms: roles in the regulation of basal p53 levels, cell fate, and senescence. Antioxid Redox Signal. 2011;15:1655–67. doi: 10.1089/ars.2010.3771. [DOI] [PubMed] [Google Scholar]
- 29.Heinlein C, Krepulat F, Löhler J, Speidel D, Deppert W, Tolstonog GV. Mutant p53(R270H) gain of function phenotype in a mouse model for oncogene-induced mammary carcinogenesis. Int J Cancer. 2008;122:1701–9. doi: 10.1002/ijc.23317. [DOI] [PubMed] [Google Scholar]
- 30.Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25:177–81. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Hollstein M, Hainaut P. Massively regulated genes: the example of TP53. J Pathol. 2010;220:164–73. doi: 10.1002/path.2637. [DOI] [PubMed] [Google Scholar]
- 32.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–80. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- 33.Hussain SP, Aguilar F, Amstad P, Cerutti PA. Oxy-radical induced mutagenesis of hotspot codons 248 and 249 of the human p53 gene. Oncogene. 1994;9:2277–81. [PubMed] [Google Scholar]
- 34.Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–6. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 35.Hussain SP, Harris CC. p53 biological network: at the crossroads of the cellular-stress response pathway and molecular carcinogenesis. J Nippon Med Sch. 2006;73:54–64. doi: 10.1272/jnms.73.54. [DOI] [PubMed] [Google Scholar]
- 36.Kang MY, Kim H-B, Piao C, Lee KH, Hyun JW, Chang I-Y, et al. The critical role of catalase in prooxidant and antioxidant function of p53. Cell Death Differ. 2013;20:117–29. doi: 10.1038/cdd.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasapović J, Pejić S, Stojiljković V, Todorović A, Radošević-Jelić Lj, Saičić ZS, et al. Antioxidant status and lipid peroxidation in the blood of breast cancer patients of different ages after chemotherapy with 5-fluorouracil, doxorubicin and cyclophosphamide. Clin Biochem. 2010;43:1287–1293. doi: 10.1016/j.clinbiochem.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Kim E, Deppert W. The versatile interactions of p53 with DNA: when flexibility serves specificity. Cell Death Differ. 2006;13:885–9. doi: 10.1038/sj.cdd.4401909. [DOI] [PubMed] [Google Scholar]
- 39.Kono Y, Fridovich I. Superoxide radical inhibits catalase. J Biol Chem. 1982;57:1571–8. [PubMed] [Google Scholar]
- 40.Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. 2011;711:193–201. doi: 10.1016/j.mrfmmm.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 41.Kumaraguruparan R, Subapriya R, Kabalimoorthy J, Nagini S. Antioxidant profile in the circulation of patients with fibroadenoma and adenocarcinoma of the breast. Clin Biochem. 2002;35:275–9. doi: 10.1016/s0009-9120(02)00310-7. [DOI] [PubMed] [Google Scholar]
- 42.Kumaraguruparan R, Subapriya R, Viswanathan P, Nagini S. Tissue lipid peroxidation and antioxidant status in patients with adenocarcinoma of the breast. Clin Chim Acta. 2002;325:165–70. doi: 10.1016/s0009-8981(02)00292-9. [DOI] [PubMed] [Google Scholar]
- 43.Ladelfa MF, Toledo MF, Laiseca JE, Monte M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid Redox Signal. 2011;15:1749–61. doi: 10.1089/ars.2010.3652. [DOI] [PubMed] [Google Scholar]
- 44.Leammli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 45.Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–36. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 46.Liu B, Chen Y, StClair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–35. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowry OH, Rosebrough NJ, Farr AL, Randal RJJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 48.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714–23. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 49.Midgley CA, Fisher CJ, Bártek J, Vojtĕsek B, Lane D, Barnes DM. Analysis of p53 expression in human tumours: an antibody raised against human p53 expressed in Escherichia coli. J Cell Sci. 1992;101:183–9. doi: 10.1242/jcs.101.1.183. [DOI] [PubMed] [Google Scholar]
- 50.Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–5. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–41. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 52.Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–72. doi: 10.1038/sj.cdd.4401914. [DOI] [PubMed] [Google Scholar]
- 53.Nenutil R, Smardova J, Pavlova S, Hanzelkova Z, Muller P, Fabian P, et al. Discriminating functional and non-functional p53 in human tumours by p53 and MDM2 immunohistochemistry. J Pathol. 2005;207:251–9. doi: 10.1002/path.1838. [DOI] [PubMed] [Google Scholar]
- 54.Nicolini A, Carpi A, Tarro G. Biomolecular markers of breast cancer. Front Biosci. 2006;11:1818–43. doi: 10.2741/1926. [DOI] [PubMed] [Google Scholar]
- 55.Nicolini A, Ferrari P, Cavazzana A, Carpi A, Berti P, Miccoli P. Conventional and new emerging prognostic factors in breast cancer: an update. Biomark Med. 2007;1:525–40. doi: 10.2217/17520363.1.4.525. [DOI] [PubMed] [Google Scholar]
- 56.Okorokov AL, Ponchel F, Milner J. Induced N- and C-terminal cleavage of p53: a core fragment of p53, generated by interaction with damaged DNA, promotes cleavage of the N-terminus of full-length p53, whereas ssDNA induces C-terminal cleavage of p53. EMBO J. 1997;16:6008–17. doi: 10.1093/emboj/16.19.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olivier M, Langerød A, Carrieri P, Bergh J, Klaar S, Eyfjord J, et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006;12:1157–67. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 58.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pani G, Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxid Redox Signal. 2011;15:1715–27. doi: 10.1089/ars.2010.3499. [DOI] [PubMed] [Google Scholar]
- 60.Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev. 2010;29:351–78. doi: 10.1007/s10555-010-9225-4. [DOI] [PubMed] [Google Scholar]
- 61.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 62.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 63.Ray G, Batra S, Shukla NK, Deo S, Raina V, Ashok S, et al. Lipid peroxidation, free radical production and antioxidant status in breast cancer. Breast Canc Res Treat. 2000;59:163–70. doi: 10.1023/a:1006357330486. [DOI] [PubMed] [Google Scholar]
- 64.Riethdorf S, Wikman H, Pantel K. Biological relevance of disseminated tumor cells in cancer patients. Int J Cancer. 2008;123:1991–2006. doi: 10.1002/ijc.23825. [DOI] [PubMed] [Google Scholar]
- 65.Rohaly G, Chemnitz J, Dehde S, Nunez AM, Heukeshoven J, Deppert W, et al. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell. 2005;122:21–32. doi: 10.1016/j.cell.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 66.Royds JA, Iacopetta B. p53 and disease: when the guardian angel fails. Cell Death Differ. 2006;13:1017–26. doi: 10.1038/sj.cdd.4401913. [DOI] [PubMed] [Google Scholar]
- 67.Salo DC, Pacifini RE, Davies KJN. Superoxide dismutase is preferentially degraded by a proteolytic stem from red blood cells following oxidative modification by hydrogen peroxide. Free Radic Biol Med. 1988;5:335–9. doi: 10.1016/0891-5849(88)90105-0. [DOI] [PubMed] [Google Scholar]
- 68.Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst. 2000;92:1926–34. doi: 10.1093/jnci/92.23.1926. [DOI] [PubMed] [Google Scholar]
- 69.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Staib F, Robles AI, Varticovski L, Wang XW, Zeeberg BR, Sirotin M, et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005;65:10255–64. doi: 10.1158/0008-5472.CAN-05-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stephen CW, Lane DP. Mutant conformation of p53. Precise epitope mapping using filamentous phage epitope library. J Mol Biol. 1992;225:577–83. doi: 10.1016/0022-2836(92)90386-x. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki M, Tarin D. Gene expression profiling of human lymph node metastases and matched primary breast carcinomas: clinical implications. Mol Oncol. 2007;1:172–80. doi: 10.1016/j.molonc.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- 74.Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat. 2007;102:143–55. doi: 10.1007/s10549-006-9317-6. [DOI] [PubMed] [Google Scholar]
- 75.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.UICC, Union for International Cancer Control. The TNM classification of malignant tumours. Geneva: UICC; 1997. [Google Scholar]
- 77.Varley JM, Attwooll C, White G, McGown G, Thorncroft M, Kelsey AM, et al. Characterization of germline TP53 splicing mutations and their genetic and functional analysis. Oncogene. 2001;20:2647–54. doi: 10.1038/sj.onc.1204369. [DOI] [PubMed] [Google Scholar]
- 78.Vojtesek B, Bartek J, Midgley CA, Lane DP. An immunochemical analysis of the human nuclear phosphoprotein p53. New monoclonal antibodies and epitope mapping using recombinant p53. J Immunol Methods. 1992;151:237–44. doi: 10.1016/0022-1759(92)90122-a. [DOI] [PubMed] [Google Scholar]
- 79.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 80.Vousden KH, Prives C. P53 and prognosis: new insights and further complexity. Cell. 2005;120:7–10. doi: 10.1016/j.cell.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 81.Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, van't Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A. 2003;100:15901–5. doi: 10.1073/pnas.2634067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 83.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 84.Yeh CC, Hou MF, Tsai SM, Lin SK, Hsiao JK, Huang JC, et al. Superoxide anion radical, lipid peroxides and antioxidant status in the blood of patients with breast cancer. Clin Chim Acta. 2005;361:104–11. doi: 10.1016/j.cccn.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 85.Zhong W, Oberly LW. Suppression of the malignant phenotype of human glioma cells by overexpression of MnSOD. Oncogene. 1997;14:481–90. doi: 10.1038/sj.onc.1200852. [DOI] [PubMed] [Google Scholar]
- 86.Zhou W, Muggerud AA, Vu P, Due EU, Sørlie T, Børresen-Dale A, et al. TP53 mutation is an early event in breast cancer progression. Cancer Res. 2009;69(Suppl 1):abstract no. 1047. [Google Scholar]