

Abstract
In a previous study we reported the presence of a large conductance K+ channel in the membrane of endoplasmic reticulum (ER) from rat hepatocytes. The channel open probability (Po) appeared voltage dependent and reached to a minimum 0.2 at +50 mV. Channel activity in this case was found to be totally inhibited at ATP concentration 2.5 mM, glibenclamide 100 µM and tolbutamide 400 µM. Existing evidence indicates an impairment of endoplasmic reticulum functions in ER stress condition. Because ER potassium channels have been involved in several ER functions including cytoprotection, apoptosis and calcium homeostasis, a study was carried out to consider whether the ER potassium channel function is altered in a high fat diet model of ER stress. Male Wistar rats were made ER stress for 2 weeks with a high fat diet. Ion channel incorporation of ER stress model into the bilayer lipid membrane allowed the characterization of K+ channel. Our results indicate that the channel Po was significantly increased at voltages above +30 mV. Interestingly, addition of ATP 7.5 mM, glibenclamide 400 µM and tolbutamide 2400 µM totally inhibited the channel activities, 3-fold, 4-fold and 6-fold higher than that in the control groups, respectively. Our results thus demonstrate a modification in the ER K+ channel gating properties and decreased sensitivity to drugs in membrane preparations coming from ER high fat model of ER stress, an effect potentially linked to a change in ER K+ channel subunits in ER stress condition. Our results may provide new insights into the cellular mechanisms underlying ER dysfunctions in ER stress.
Keywords: endoplasmic reticulum, potassium channel, ER stress, bilayer lipid membrane, hepatocytes
Introduction
The endoplasmic reticulum (ER) is a vast intracellular organelle responsible for protein synthesis, folding, maturation, quality control, trafficking, lipid synthesis, and drug detoxification. Various factors that interfere with ER function lead to accumulation of unfolded proteins, including oxidative stress, ischemia, disturbance of calcium homeostasis, and over expression of normal and/or incorrectly folded proteins. This process is called ER stress. To eliminate the toxic protein components, cells activate an adaptive mechanism that consists of a number of intracellular signaling pathways, collectively known as unfolded protein response (UPR) (Xu et al., 2005[62]; Ron and Walter, 2007[46]; Basseri and Austin, 2011[5]). However, during prolonged or overwhelming ER stress, UPR fails to restore the normal function of the ER, and apoptotic cascade will be activated (Paschen and Frandsen, 2001[39]; Rao et al., 2004[44]). The exact mechanism underlying the switch of the UPR from a prosurvival mechanism to a proapoptotic response is not clear.
Ion channels are present in membranes of intracellular organelles such as mitochondria (Bednarczyk, 2009[6]), nucleus (Fedorenko et al., 2010[14]), zymogen granules (Thevenod, 2002[54]), and Golgi complex (Thompson et al., 2002[55]). These channels are thought to play an important role in cellular processes such as compensation for electrical charges and regulation of pH (Edwards and Kahl, 2010[12]; Khodaei et al., 2014[26]), oxidative stress (Averaimo et al., 2010[2]; Malinska et al., 2010[31]), or acting as therapeutic targets (Fedorenko et al., 2010[14]). Potassium channels have also been found in endo/sarcoplasmic reticulum. Picard et al. (2002[41]) demonstrated the electro-pharmacological properties of sarcoplasmic reticulum (SR) K+ channels from human and sheep atrial cells (Picard et al., 2002[41]). Voltage-gated potassium channels were also identified in SR of diaphragm (Picher et al., 1996[42]) and frog skeletal muscle. Accordingly, we have shown a large conductance voltage-activated K+ channel in the RER (rough endoplasmic reticulum) membranes of hepatocytes (Sepehri et al., 2007[49]) which is regulated by intracellular ATP (Ghasemi et al., 2014[16]). It has been suggested that both SR/ER Cl- and K+ channels act as counter transport systems during rapid Ca2+ release and uptake to keep the electro-chemical force on Ca2+ ions by maintaining the ER membrane potential away from ECa (Gutman et al., 2005[18]; Li et al., 2006[29]) and protect ER luminal homeostasis.
Some evidence demonstrated that ER stress altered potassium channel function in cell membrane. For example, Kito et al. (2011[27]) demonstrated that ER stress induced up-regulation of inward rectifier K+ channel and facilitated cell death (Kito et al., 2011[27]). Furthermore, dysfunction of an ER-resident Ca2+ channel, inositol 1,4,5-trisphosphate receptor (IP3R), promotes cell death during ER stress (Higo et al., 2010[23]). The possibility that ER channels play a role in the ER stress is not clear. In this study, we show significant abnormalities in ER potassium channel in high fat diet model of ER stress. Our results provide new insights into the cellular mechanisms of ER stress complications.
Material and Methods
Sucrose, potassium chloride, ATP, gli-benclamide, tolbutamide, 2-amino-2-(hydroxymethyl)-1,3-propanediol (Trisma base) and 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid (HEPES) were purchased from Sigma (St. Louis, MO, USA) and n-decane was obtained from Merck (Darmstadt, Germany). Salt and solvent were analytical grade.
Lipid preparation
The ER membrane is relatively enriched in the neutralzwitterionic phospholipids having large polar headgroups such as L-α-phosphatidyl choline (van Meer et al., 2008[57]). Therefore, we formed bilayer lipid membrane by L-α-lecithin. L-α-phosphatidyl choline (L-α-lecithin) was extracted from fresh egg yolk by the procedure as described by Singleton et al. (1965[51]).
Ethical considerations
All experiments were executed in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health Publication No. 80-23, revised 1996) and were approved by the Research and Ethics Committee of Shahid Beheshti University of Medical Sciences.
Induction of ER stress in rats
Six male Wistar rats, weighing 165–185 g, were allowed to acclimatize for 7 days in an environmentally controlled room at 22 ˚C with an alternating 12 h light/dark cycle and free access to normal laboratory food and water. After 1 week acclimation, the animals were randomly assigned to either a control or ER stress groups. Rats in the control group were continually fed only normal chow diet (composed of fat 12 %, protein 23 %, carbohydrate 65 % based on caloric content) and ER stress rats were fed with high fat diet (HFD; composed of fat 59 %, protein 15 %, carbohydrate 26 % based on caloric content) (Srinivasan et al., 2005[52]; Park et al., 2010[38]; Nivala et al., 2013[35]) and free access to water for two weeks. HFD prepared from mixing and blending of new intra abdominal bovine fat with normal rat meal in 1:3 ratios. Serum glucose, insulin, free fatty acid, triglycerides, total cholesterol levels and weight were checked on 0 and 14 days (before and after feeding with HFD, respectively). Animals were considered to be an ER stress animal model if they had ER stress features (Matveyenko et al., 2009[33]) such as high blood concentrations of glucose, insulin, free fatty acid, triglycerides and total cholesterol. After 2 weeks, the HFD fed rats were sacrificed and the liver excised for study.
Endoplasmic reticulum isolation
Hepatic endoplasmic reticulum vesicles from control and HFD fed rats were extracted by methods as described by Kan et al. (1992[25]) with minor modifications. Rats were lightly anesthetized by ether and livers were rapidly removed and homogenized in 50 ml cold sucrose (0.25 M) solution at 2850 rpm using a potter homogenizer (Potter-Elvehjem Homogenizer, Iran). The homogenate was centrifuged for 13 min at 8700 ×g. Then, the supernatant was centrifuged at 110000 ×g for 14 min at 4 °C (Beckman model J-21B, USA). The pellet was gently suspended in 9 ml of ice-cold sucrose 2 M, by glass homogenizer to obtain a homogenous suspension. Subsequently, in a sucrose gradient condition, the suspension was centrifuged at 300,000 ×g for 67 min, and the obtained sediment was dissolved in 20 ml of sucrose 0.25 mM + imidazole 3 mM + Na pyrophosphate 0.5 mM. The solution was then centrifuged three times at 140000 ×g for 40 min. The obtained sediment (ER microsomes) was dissolved in 1 ml sucrose 0.25 mM + imidazole 3 mM at a final concentration of 7 mg/ml. ER microsomes were stored in 10 µl aliquots in 250 mM sucrose/3 mM imidazole, pH 7.4 at -80 °C until being used.
Planar lipid bilayers and vesicle fusion
Experiments were performed by using black (bilayer) lipid membrane technique (Ries et al., 2004[45]). Planar phospholipid bilayers were formed in a 250 µm-diameter hole drilled in a Delrin partition, which separated two chambers, cis (cytoplasmic side) and trans (luminal side). Chambers contained 4 ml of KCl 200 mM/50 mM cis/trans. Under these conditions there will be a net movement of water across the bilayer from trans to cis face. Vesicles in the prefusion state will swell if water enters the lumen across the bilayer (Ashrafpour et al., 2008[1]). The pH on both sides was adjusted to 7.4 with Tris–HEPES. Planar phospholipid bilayers were painted using a suspension of L-α-lecithin in decane at a concentration of 25 mg/ml. An indication of the thickness of the bilayer membrane formed across the hole obtained by monitoring capacitance. A low frequency (1-10 Hz), low amplitude (5-20 mV peak to peak) triangular wave is used. Typical capacitance values ranged from 200 to 300 pF. Fusion of the vesicles was initiated mechanically by gently touching the bilayer from the cis face using a small stainless steel wire of 150 µm diameter, on the tip of which a small drop of the vesicle-containing solution was deposited.
Electrical recording and data analysis
BC-525D amplifier (Warner Instrument, USA) in the voltage clamp mode was used to amplify the current and control the voltage across the bilayer through Ag/AgCl electrodes. The cis electrode was set to a command voltage relative to the trans electrode, which was grounded. The recordings were filtered at 1 kHz with a four-pole Bessel low-pass filter, digitized at a sampling rate of 10 kHz and stored on a personal computer for off line analysis by PClamp10 (Axon Instruments Inc, USA). Unitary channel conductance was calculated from the current– voltage relationship. The channel open probability (Po) was calculated using the standard event detection algorithms in Pclamp10, on segments of continuous recordings lasting 50 s. The results are expressed as means ± standard error of the means (S.E.M.).
Results
Blood factors concentrations and animal weights
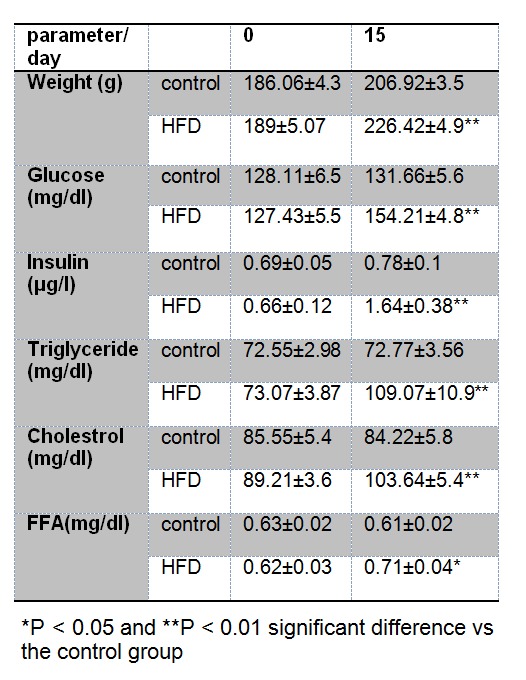
A number of studies have shown that HFD feeding of lab animals can increase weight and blood concentrations of glucose (Srinivasan et al., 2005[52]; Matveyenko et al., 2009[33]), insulin (Barnard et al., 1998[3]), cholesterol (Ishii et al., 2010[24]), triglyceride (Yuan et al., 2010[63]) and free fatty acids (Charbonneau et al., 2007[8]) and subsequently induces endoplasmic reticulum stress (Charbonneau et al., 2007[8]). In this study, two weeks HFD feeding of rats were significantly increased weight values by ~9 % (P < 0.01), serum concentrations of glucose by ~17 % (P < 0.03), insulin ~110 % (P < 0.03), triglyceride ~50 % (P < 0.01), cholesterol ~23 % (P < 0.01) and free fatty acids ~16 % (P < 0.01) respect to control group (Table 1(Tab. 1)). These results confirmed the validity of our HDF-induced ER stress rat model.
Table 1. Comparison of blood serum parameters and weight values in control and high fat diet (HFD) fed groups. Values are means ± SD for all factors.
Biophysical properties of the ion channel
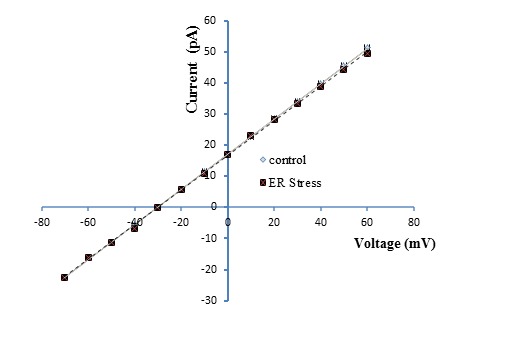
Our previous study revealed an ER membrane localized voltage-gated potassium channel with a conductance of 598 ± 20 pS (Sepehri et al., 2007[49]), but no study has been performed using an ER membrane preparation from a rat ER stress model. Examples of single channel recordings obtained upon fusion of ER vesicles in control and ER stress rats under asymmetrical condition (50 mM-trans/250 mM-cis) are presented in Figure 1A and B(Fig. 1), respectively. Clear current out-ward jumps could be observed at potentials more positive than 10 mV, with a reverse potential value estimated at -30 mV, confirming the cationic nature of the channel. The channel gating behavior was voltage dependent with decreased channel opening separated by longer silent periods at increasingly positive potential values in control but not ER stress model. In addition, the reverse potential close to -34 mV shows unidirectional reconstitution of the channel into lipid bilayer membrane. As illustrated in Figure 2(Fig. 2), the current–voltage (I–V) relation was linear in control and ER stress rats and the slope conductances were 569 pS and 560 pS, respectively with negative reversal potentials close to -30 mV, which attest its cationic selectivity under these conditions. There were no significant differences in channel conductance and current amplitude between control and ER stress rats. The effect of voltage on the channel activity was investigated by measuring the channel open probability (Po) as a function of voltage. As seen Po estimates for the ER stress model reached values close to ~0.88 at +60 mV with already the same values ~0.8 at -60 mV, while the control potassium channel Po decreased from ~0.89 at 60 mV to a minimum of ~0.12 (n = 5) at +60 mV (Table 1(Tab. 1)). These results confirmed that channels coming from ER stress rat preparations present an overall ~1.7 to ~9 increase in Po relative to non-ER stress preparations at +40 to +60 mV, respectively.
Figure 1. Single channel recordings as a function of voltages. (A) Single channel recordings in 200/ 50 mM KCl (cis/trans) gradient after reconstitution of liver rough endoplasmic reticulum membrane vesicles in planar lipid bilayer at potentials ranging from -60 to +60 mV in control and (B) ER stress condition. PO is open probability of each trace. Closed levels are indicated by –.
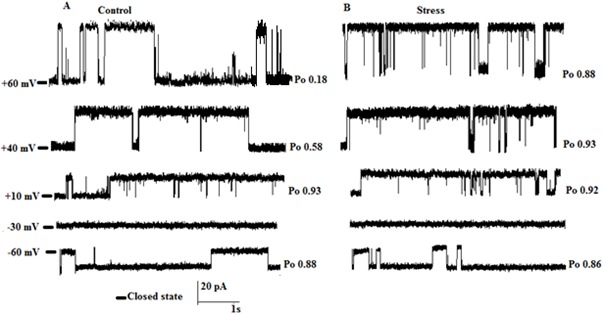
Figure 2. Single channel current voltage relationship for the ER K+ channel from high fat diet model of ER stress compared to control. There was no significant effect in conductance in the ER stress group (569 ± 12.5 pS) relative to control (565 ± 15.5 pS). Data points are mean ± SEM, obtained from 6 experiments.
Pharmacological properties of the ion channel
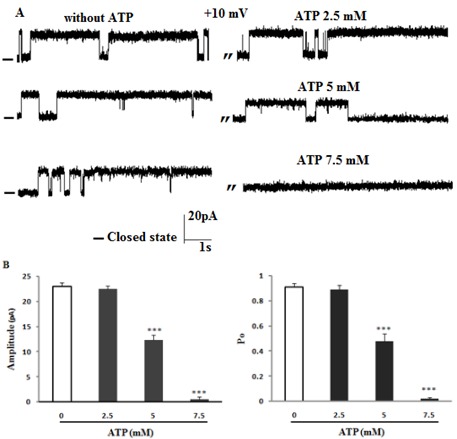
In a previous work we have reported an ATP-sensitive potassium channel blocked by ATP and glibenclamide (Ashrafpour et al., 2008[1]; Ghasemi et al., 2014[16]). Experiments were thus performed to determine the nature of the potassium channel observed from ER stress rat preparations. As seen in Figure 3A and B(Fig. 3) the addition of 2.5 mM of ATP to cis compartment did not alter neither the channel conducting nor gating behavior in ER stress rats. In contrast, addition of 5 mM ATP decreased channel Po (P < 0.001) and current amplitude (P < 0.001) and 7.5 mM ATP (3-fold higher than that in the control group) totally blocked the channel activities at +10 mV (n = 3).
Figure 3. The effect of ATP on single channel activity in ER stress condition. (A) Representative recordings of channel currents with and without cis addition of ATP at different concentrations. Data on the current amplitude and Po of reconstituted channels in the absence or presence of ATP are summarized in panel B at +10 mV. There was significant decrease in Po value and current amplitude in the presence of ATP demonstrating that the channel that was incorporated is less ATP-sensitive. Data are mean ± SEM (n = 3). Closed levels are indicated by –.
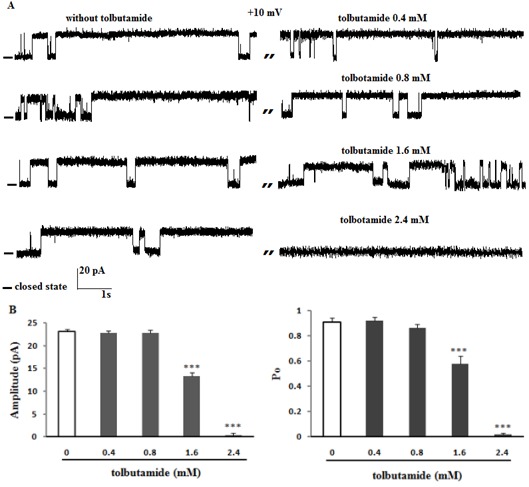
To further characterize this channel, the effect of glibenclamide and tolbutamide as well-known sulfonylurea to block ATP-sensitive K+ channel was examined on channel activities. Figure 4A and B(Fig. 4) shows that the addition of 100 and 200 µM glibenclamide on the cis side had no significant effect on channel amplitude and Po at +10 mV (n = 4). Potassium single channel recordings after addition of 300 µM glibenclamide to the cis face (n = 3) decreased current amplitude (P < 0.001) and channel Po (P < 0.001) at +10 mV. Glibenclamide (400 µM; 4-fold higher than that in the control group) totally blocked channel activities (Figure 4A and B(Fig. 4)). Figure 5A and B(Fig. 5) shows that the K+ current was completely inhibited by addition of tolbutamide (2.4 mM; 6-fold higher than that in the control group ) to cis compartment at +10 mV (n = 4) (P < 0.001), while after addition of 1.6 mM tolbutamide the current amplitude and channel open probability decreased significantly (P < 0.001). Tolbutamide 0.4 and 0.8 mM had no effect on channel activities (Figure 5A and B(Fig. 5)).
Figure 4. The effect of glibenclamide on channel gating behavior. (A) Single-channel recordings of ER K+ channels from high fat model of ER stress in 200/50 mM KCl; cis/trans conditions with and without cis addition of glibenclamide at +10 (n = 4). Data on the current amplitude and Po of reconstituted channels in the absence or presence of glibenclamide are summarized in panel B. There was significant decrease in Po value and current amplitude in the presence of glibenclamide, indicating that the channel is less sensitive to glibenclamide in ER stress condition. Closed levels are indicated by –.
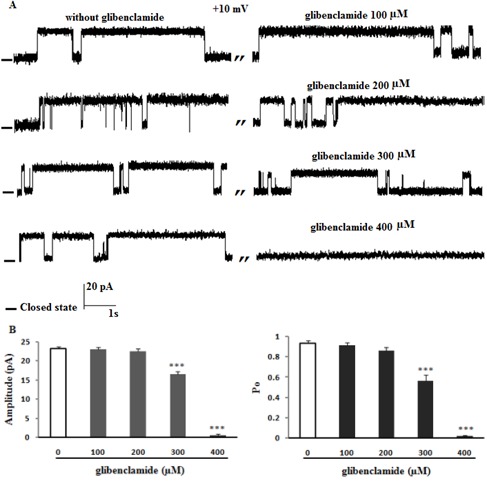
Figure 5. The effect of tolbutamide on channel activity at +10 mV. (A) Representative recordings of ER K+ channel from high fat diet model of ER stress with and without tolbutamide to cis face. Data on the current amplitude and Po of reconstituted channels in the absence or presence of tolbutamide are summarized in panel B. There was significant decrease in Po value and current amplitude in the presence of tolbutamide, showing that the channel is less tolbutamide-sensitive. Data are mean ± SEM (n = 4). Closed levels are indicated by –.
These results demonstrated that the sensitivity of channel obtained from ER stress rat preparations significantly decreased to ATP and sulfonylurea drugs.
Discussion
The endoplasmic reticulum (ER) is the synthesis and folding site for membrane and secretory proteins, and is responsible for several important cellular functions, including Ca2+ storage and cell signaling. Disruption in homeostasis and accumulation of unfolded and misfolded protein in the ER lumen causes ER stress (Scheuner et al., 2005[47]). If the function of the ER is severely impaired, genes and pathway leading to cell death and/or inhibition of survival are also activated (Rao et al., 2004[44]). The cellular signals involved in ER stress-mediated cell death and apoptosis are complex and yet not fully understood (Breckenridge et al., 2003[7]; Rao et al., 2004[44]). The present findings demonstrate that ER K+ channel activity is increased in fatty acid-induced ER stress model, with no effect on conductance, altered voltage-dependent gating properties, including increased Po at depolarizing potentials. Furthermore, our results demonstrate an altered sensitivity of the channel to inhibitory effects of ATP and sulfonylurea drugs. A modification in drug sensitivity could contribute to the observed changes in ER K+ gating properties while being partly responsible for ER impairment. Existing evidence shows various factors lead to ER stress. For example, it has been shown that elevated plasma free fatty acids (FFAs) can induce beta cell (Cnop et al., 2007[10]) and hepatic ER stress (Nivala et al., 2013[35]). There is evidence, including our current work, that already during the second week after introduction of high-fat diet, body weight, glucose, FFAs, triglyceride and cholesterol increased significantly more in the high-fat diet-fed animal than in the normal diet-fed animal (Winzell and Ahren, 2004[61]; Ishii et al., 2010[24]; Nivala et al., 2013[35]). The mechanism behind ER stress-induced lipoapoptosis is complicated and could involve several factors; including depletion of ER calcium levels (Gwiazda et al., 2009[19]). It has also been proposed that FFAs, such as palmitate, reduce ER-to-Golgi trafficking, contributing to ER stress through protein overload in the ER lumen (Preston et al., 2009[43]). Furthermore, we observed high fat diet-increased plasma glucose. Hyperglycemia leads to considerable generation of reactive oxygen species (ROS), which is responsible for the oxidative stress (Basha et al., 2012[4]). It is still unclear whether the ER stress response can be initiated as a direct response to the increasing load on protein synthesis and maturation due to hyperglycemia or due to the hyperglycemia-associated oxidative stress. The possibility that ER stress response also can lead to excessive ROS formation cannot be ruled out. Furthermore, we observed increased BiP and CHOP protein expressions in high-fat diet-fed rat (article is being prepared for submission). This observation is supported by van der Kallen et al. (2009[56]) who showed hepatocytes from rats on a high saturated fat diet were characterized by increased CHOP protein (Wang et al., 2006). CHOP is expressed at a very low level under physiological conditions but its expression level significantly increases in the presence of severe or persistent ER stress (Wang and Ron, 1996[59]). There is some evidence to show that ER stress affects plasma membrane ion channel regulation (Hambrock et al., 2006[20]; Kito et al., 2011[27]).
Ion channels are present in membranes of intracellular organelles (Sepehri et al., 2007[49]; Ashrafpour et al., 2008[1]; Fahanik-Babaei et al., 2011[13]).These channels are thought to play an important role in cellular processes such as compensation of electrical charges and formation of pH (Szewczyk, 1998[53]), oxidative stress (Malinska et al., 2010[31]), or acting as therapeutic targets (Peixoto et al., 2010[40]). Since, our previous studies have relatively well described the biophysical properties of a big K+ channel which was inhibited by ATP and glibenclamide (Sepehri et al., 2007[49]; Ashrafpour et al., 2008[1]; Ghasemi et al., 2014[16]), our focus has turned to the biophysical and pharmacological analysis of this channel in ER stress condition.
The single ER K+ channel data presented here provide for the first time direct evidence for an increase in channel open probability (~9-fold increase compared with control group at +60 mV). We showed in this work that the I-V curve derived for the ER K+ channel of early ER stress preparations presents the same properties which observed with channels from non-ER stress preparations where the I-V relationships remained strictly linear under the same ionic conditions (Sepehri et al., 2007[49]; Ghasemi et al., 2014[16]). We cannot exclude, however, the possibility those ER K+ ion permeation properties can functionally impair during prolonged or overwhelming ER stress. Our results provide evidence for differences in channel open probability with channels coming from ER stress vesicle preparations characterized by an increased open probability at depolarizing potentials, in contrast to control ER K+ channels which showed the most significant decrease being observed at depolarizing potentials (above +30 mV). In line with this study, it has been reported that ER stress causes ion channel activation in cell membrane (Kito et al., 2011[27]). Saturated fatty acids such as palmitate and stearate in high fat diet are known inducers of ER stress (Wei et al., 2006[60]; Guo et al., 2007[17]) and saturated fatty acids incorporation into hepatic ER lipids can alter ER membrane fluidity and subsequently ion channel activity (Cnop, 2012[10]). Interestingly, our previous work described that ER K+ channel inactivation (Po reaches to 0) was observed in diabetic rats at voltages above +30 mV (Ghasemi et al., 2014[16]). It is possible that diabetic induces severe ER stress resulting in channel activities impairment while early FFA-induced ER stress causes increase channel open probability to trigger the UPR branches (an adaptive cellular response to the disturbance of normal ER functions). Existing evidence to show that under diabetic conditions, oxidative stress and ER stress are induced in various tissues (van der Kallen et al., 2009[56]) and vice versa (Pan et al., 1994[37]). Upon ER stress, excessive unfolded proteins accumulate in the ER lumen, resulting in the over expression and dissociation of BiP from the ER stress transducers (Haze et al., 2001[22]), which triggers activation of the UPR branches (Schroder and Kaufman, 2005[48]). However, during pro-longed or overwhelming ER stress, UPR fails to restore the normal function of the ER, and apoptotic cascade will be activated (Rao et al., 2004[44]). CHOP, a transcription factor, is thought to be important to the cell death process (Oyadomari et al., 2002[36]; Silva et al., 2005[50]). In accordance with this, CHOP over expression has been shown to sensitize cells to the toxicity of ER stress (McCullough et al., 2001[34]). We observed more increased CHOP over expression in diabetic condition while more increased BiP over expression in FFA-induced ER stress was observed (data not shown). We cannot currently rule out the possibility that the increased channel activation coming from ER stress preparation could be linked to the UPR. To resolve this issue, further studies will be needed.
Another aspect of the present work concerns the pharmacological profile of the ER K+ channels in ER stress preparations, compared with non-ER stress vesicles.
Our results provide also clear indications of decreased drug sensitivities for channels obtained from ER stress preparations relative to control. Figure 3(Fig. 3) demonstrates that addition of 2.5 mM of ATP to the cytoplasmic side (cis chamber) did not affect channel activity while higher concentration of ATP (7.5 mM) completely blocked channel activity. Our previous studies showed that lower doses of ATP (500 µM-2.5 mM) blocked the channel activity in control condition (Ashrafpour et al., 2008[1]; Ghasemi et al., 2014[16]). In addition, the channel sensitivity to sulphonylurea compounds appeared to be reduced. Figures 4(Fig. 4) and 5(Fig. 5) demonstrate that addition of 400 µM of glibenclamide and 2.4 mM of tolbutamide to cis chamber completely blocked channel activity, respectively. There is some evidence that show the KATP channel is inhibited by sulfonylurea (Kuum et al., 2012[28]). According to this result and our previous experiments, this endoplasmic reticulum cationic channel may be a KATP channel (Hambrock et al., 2006[20]). Very recently, our study also showed the presence of Kir6.2 subunit which may be co-expressed with SUR1 or SUR2B and SUR2A subunits in ER KATP channel structure (Ghasemi et al., 2014[15]). The KATP channel serves as a sensor of the cellular metabolic state and couples the metabolism of the cell with the membrane potential, primarily by sensing intracellular ATP levels (Matsuo et al., 2003[32]). It is an octamer composed of 4 Kir6 subunits and 4 SUR subunits (Matsuo et al., 2003[32]). SURs likely work in concert to regulate KATP channel activity (Linton, 2007[30]) and many of drugs act through the SUR subunit. In this regard, we found that there is ~4-6 folds decrease in sensitivity of the channel to sulphonylurea drugs in ER stress K channel preparations compared to control. As SURs was proposed to determine the sensitivity of the channel to sulphonylurea drugs, for example SUR1 exhibits high affinity for several sulfonylureas whereas SUR2 shows lower affinity for sulfonylureas (Hambrock et al., 2002[21]), it is possible that the observed decrease in drug sensitivity and increase in channel Po partly accounts for the altered SURs expression in ER stress rats compared to control group. Given that Ca+2 regulations by the ER is prominent in cellular apoptosis (Dolai et al., 2011[11]), these data may suggest that ER K+ channel coming from ER stress vesicles contribute to a defect in this regulatory process may cause a progressive loss of cell function and the generation of a cellular pathological state.
To the best of our knowledge, we have presented the first compelling evidence of a physical and pharmacological alteration of ER K+ channel in FFA-induced ER stress in rat hepatocytes. These changes may account for the profound abnormalities in channel gating properties, including increased open probability and decreased drug sensitivities. Results from this study may help us better understand the mechanisms that underlie channel dysfunction in ER stress, and moreover may provide new insights into the development of approaches for the management of disease.
References
- 1.Ashrafpour M, Eliassi A, Sauve R, Sepehri H, Saghiri R. ATP regulation of a large conductance voltage-gated cation channel in rough endoplasmic reticulum of rat hepatocytes. Arch Biochem Biophys. 2008;471:50–56. doi: 10.1016/j.abb.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Averaimo S, Milton RH, Duchen MR, Mazzanti M. Chloride intracellular channel 1 (CLIC1): Sensor and effector during oxidative stress. FEBS Lett. 2010;584:2076–2084. doi: 10.1016/j.febslet.2010.02.073. [DOI] [PubMed] [Google Scholar]
- 3.Barnard RJ, Roberts CK, Varon SM, Berger JJ. Diet-induced insulin resistance precedes other aspects of the metabolic syndrome. J Appl Physiol. 1998;84:1311–1315. doi: 10.1152/jappl.1998.84.4.1311. [DOI] [PubMed] [Google Scholar]
- 4.Basha B, Samuel SM, Triggle CR, Ding H. Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Exp Diabetes Res. 2012;2012:481840. doi: 10.1155/2012/481840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basseri S, Austin RC. Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem Res Int. 2011;2012:841362. doi: 10.1155/2012/841362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bednarczyk P. Potassium channels in brain mitochondria. Acta Biochim Pol. 2009;56:385–392. [PubMed] [Google Scholar]
- 7.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 8.Charbonneau A, Unson CG, Lavoie JM. High-fat diet-induced hepatic steatosis reduces glucagon receptor content in rat hepatocytes: potential interaction with acute exercise. J Physiol. 2007;579:255–267. doi: 10.1113/jphysiol.2006.121954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cnop M. High fat feeding exacerbates endoplasmic reticulum stress and beta cell demise. Eur J Lipid Sci Technol. 2012;114:229–32. [Google Scholar]
- 10.Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, et al. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem. 2007;282:3989–3997. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- 11.Dolai S, Pal S, Yadav RK, Adak S. Endoplasmic reticulum stress-induced apoptosis in Leishmania through Ca2+-dependent and caspase-independent mechanism. J Biol Chem. 2011;286:13638–13646. doi: 10.1074/jbc.M110.201889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards JC, Kahl CR. Chloride channels of intracellular membranes. FEBS Lett. 2010;584:2102–2111. doi: 10.1016/j.febslet.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahanik-Babaei J, Eliassi A, Jafari A, Sauve R, Salari S, Saghiri R. Electro-pharmacological profile of a mitochondrial inner membrane big-potassium channel from rat brain. Biochim Biophys Acta. 2011;1808:454–460. doi: 10.1016/j.bbamem.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Fedorenko O, Yarotskyy V, Duzhyy D, Marchenko S. The large-conductance ion channels in the nuclear envelope of central neurons. Pflugers Arch. 2010;460:1045–1050. doi: 10.1007/s00424-010-0882-5. [DOI] [PubMed] [Google Scholar]
- 15.Ghasemi M, Khodaee AN, Khodagholi F, Eliassi A. A molecular study on endoplasmic reticulum K channels of hepatocytes in rat. Physiol Pharmacol. 2014;[in press] [Google Scholar]
- 16.Ghasemi M, Khodaei N, Salari S, Eliassi A, Saghiri R. Gating behavior of endoplasmic reticulum potassium channels of rat hepatocytes in diabetes. Iran Biomed J. 2014;18:165–172. doi: 10.6091/ibj.1308.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo W, Wong S, Xie W, Lei T, Luo Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab. 2007;293:E576–E586. doi: 10.1152/ajpendo.00523.2006. [DOI] [PubMed] [Google Scholar]
- 18.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 19.Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009;296:E690–E701. doi: 10.1152/ajpendo.90525.2008. [DOI] [PubMed] [Google Scholar]
- 20.Hambrock A, de Oliveira Franz CB, Hiller S, Osswald H. Glibenclamide-induced apoptosis is specifically enhanced by expression of the sulfonylurea receptor isoform SUR1 but not by expression of SUR2B or the mutant SUR1(M1289T) J Pharmacol Exp Ther. 2006;316:1031–1037. doi: 10.1124/jpet.105.097501. [DOI] [PubMed] [Google Scholar]
- 21.Hambrock A, Loffler-Walz C, Quast U. Gliben-clamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br J Pharmacol. 2002;136:995–1004. doi: 10.1038/sj.bjp.0704801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haze K, Okada T, Yoshida H, Yanagi H, Yura T, Negishi M, et al. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J. 2001;355:19–28. doi: 10.1042/0264-6021:3550019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M, et al. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron. 2010;68:865–878. doi: 10.1016/j.neuron.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Ishii Y, Ohta T, Sasase T, Morinaga H, Hata T, Miyajima K, et al. A high-fat diet inhibits the progression of diabetes mellitus in type 2 diabetic rats. Nutr Res. 2010;30:483–491. doi: 10.1016/j.nutres.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Kan FW, Jolicoeur M, Paiement J. Freeze-fracture analysis of the effects of intermediates of the phosphatidylinositol cycle on fusion of rough endoplasmic reticulum membranes. Biochim Biophys Acta. 1992;1107:331–341. doi: 10.1016/0005-2736(92)90420-q. [DOI] [PubMed] [Google Scholar]
- 26.Khodaei N, Ghasemi M, Eliassi A, Saghiri R. Cytoplasmic acidification reduces potassium channel activities in endoplasmic reticulum of rat hepatocytes. Physiol Pharmacol. 2014;[in press] [Google Scholar]
- 27.Kito H, Yamazaki D, Ohya S, Yamamura H, Asai K, Imaizumi Y. Up-regulation of K(ir)2.1 by ER stress facilitates cell death of brain capillary endothelial cells. Biochem Biophys Res Commun. 2011;411:293–298. doi: 10.1016/j.bbrc.2011.06.128. [DOI] [PubMed] [Google Scholar]
- 28.Kuum M, Veksler V, Liiv J, Ventura-Clapier R, Kaasik A. Endoplasmic reticulum potassium-hydrogen exchanger and small conductance calcium-activated potassium channel activities are essential for ER calcium uptake in neurons and cardiomyocytes. J Cell Sci. 2012;125:625–633. doi: 10.1242/jcs.090126. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Um SY, McDonald TV. Voltage-gated potassium channels: regulation by accessory subunits. Neuroscientist. 2006;12:199–210. doi: 10.1177/1073858406287717. [DOI] [PubMed] [Google Scholar]
- 30.Linton KJ. Structure and function of ABC transporters. Physiology (Bethesda) 2007;22:122–130. doi: 10.1152/physiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- 31.Malinska D, Mirandola SR, Kunz WS. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010;584:2043–2048. doi: 10.1016/j.febslet.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Matsuo M, Ueda K, Ryder T, Ashcroft F. The sulfonylurea receptor: an ABCC transporter that acts as an ion channel regulator. In: Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. ABC proteins: from bacteria to man. San Diego, CA: Academic Press; 2003. pp. 551–575. [Google Scholar]
- 33.Matveyenko AV, Gurlo T, Daval M, Butler AE, Butler PC. Successful versus failed adaptation to high-fat diet-induced insulin resistance: the role of IAPP-induced beta-cell endoplasmic reticulum stress. Diabetes. 2009;58:906–916. doi: 10.2337/db08-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nivala AM, Reese L, Frye M, Gentile CL, Pagliassotti MJ. Fatty acid-mediated endoplasmic reticulum stress in vivo: differential response to the infusion of Soybean and Lard Oil in rats. Metabolism. 2013;62:753–760. doi: 10.1016/j.metabol.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan DA, Hulbert AJ, Storlien LH. Dietary fats, membrane phospholipids and obesity. J Nutr. 1994;124:1555–1565. doi: 10.1093/jn/124.9.1555. [DOI] [PubMed] [Google Scholar]
- 38.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paschen W, Frandsen A. Endoplasmic reticulum dysfunction - a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem. 2001;79:719–725. doi: 10.1046/j.1471-4159.2001.00623.x. [DOI] [PubMed] [Google Scholar]
- 40.Peixoto PM, Ryu SY, Kinnally KW. Mitochondrial ion channels as therapeutic targets. FEBS Lett. 2010;584:2142–2152. doi: 10.1016/j.febslet.2010.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Picard L, Cote K, Teijeira J, Greentree D, Rousseau E. Sarcoplasmic reticulum K(+) channels from human and sheep atrial cells display a specific electro-pharmacological profile. J Mol Cell Cardiol. 2002;34:1163–1172. doi: 10.1006/jmcc.2002.2041. [DOI] [PubMed] [Google Scholar]
- 42.Picher M, Decrouy A, Rousseau E. Conducting and voltage-dependent behaviors of potassium ion channels reconstituted from diaphragm sarcoplasmic reticulum: comparison with the cardiac isoform. Biochim Biophys Acta. 1996;1279:93–103. doi: 10.1016/0005-2736(95)00239-1. [DOI] [PubMed] [Google Scholar]
- 43.Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia. 2009;52:2369–2373. doi: 10.1007/s00125-009-1506-5. [DOI] [PubMed] [Google Scholar]
- 44.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 45.Ries RS, Choi H, Blunck R, Bezanilla F, Heath JR. Black lipid membranes: visualizing the structure, dynamics, and substrate dependence of membranes. J Phys Chem. 2004;108B:16040–16049. [Google Scholar]
- 46.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 47.Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11:757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 48.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 49.Sepehri H, Eliassi A, Sauve R, Ashrafpour M, Saghiri R. Evidence for a large conductance voltage gated cationic channel in rough endoplasmic reticulum of rat hepatocytes. Arch Biochem Biophys. 2007;457:35–40. doi: 10.1016/j.abb.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singleton WS, Gray MS, Brown ML, White JL. Chromatographically homogeneous lecithin from egg phospholipids. J Am Oil Chem Soc. 1965;42:53–56. doi: 10.1007/BF02558256. [DOI] [PubMed] [Google Scholar]
- 52.Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–320. doi: 10.1016/j.phrs.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Szewczyk A. The intracellular potassium and chloride channels: properties, pharmacology and function (review) Mol Membr Biol. 1998;15:49–58. doi: 10.3109/09687689809027518. [DOI] [PubMed] [Google Scholar]
- 54.Thevenod F. Ion channels in secretory granules of the pancreas and their role in exocytosis and release of secretory proteins. Am J Physiol Cell Physiol. 2002;283:C651–C672. doi: 10.1152/ajpcell.00600.2001. [DOI] [PubMed] [Google Scholar]
- 55.Thompson RJ, Nordeen MH, Howell KE, Caldwell JH. A large-conductance anion channel of the Golgi complex. Biophys J. 2002;83:278–289. doi: 10.1016/S0006-3495(02)75168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Kallen CJ, van Greevenbroek MM, Stehouwer CD, Schalkwijk CG. Endoplasmic reticulum stress-induced apoptosis in the development of diabetes: is there a role for adipose tissue and liver? Apoptosis. 2009;14:1424–1434. doi: 10.1007/s10495-009-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 59.Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 60.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 61.Winzell MS, Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53(Suppl 3):S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- 62.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan HD, Kim SJ, Quan HQ, Huang B, Chung SH. Ginseng leaf extract prevents high fat diet-induced hyperglycemia and hyperlipidemia through AMPK activation. J Ginseng Res. 2010;34:369–375. [Google Scholar]