

Abstract
Background
A Lebanese Maronite family presented with 13 relatives affected by various congenital heart defects (mainly atrial septal defects), conduction tissue anomalies and midline defects. No mutations were found in GATA4 and NKX2-5.
Methods and Results
A set of 399 poly(AC) markers was used to perform a linkage analysis which peaked at a 2.98 lod score on the long arm of chromosome 15. The haplotype analysis delineated a 7.7 meganucleotides genomic interval which included the alpha-cardiac actin gene (ACTC1) among 36 other protein coding genes. A heterozygous missense mutation was found (c.251T>C, p.(Met84Thr)) in the ACTC1 gene which changed a methionine residue conserved up to yeast. This mutation was absent from 1000 genomes and exome variant server database but segregated perfectly in this family with the affection status. This mutation and 2 other ACTC1 mutations (p.(Glu101Lys) and p.(Met125Val)) which result also in congenital heart defects are located in a region in close apposition to a myosin heavy chain head region by contrast to 3 other alpha-cardiac actin mutations (p.(Ala297Ser),p.(Asp313His) and p.(Arg314His)) which result in diverse cardiomyopathies and are located in a totally different interaction surface.
Conclusions
Alpha-cardiac actin mutations lead to congenital heart defects, cardiomyopathies and eventually midline defects. The consequence of an ACTC1 mutation may in part be dependent on the interaction surface between actin and myosin.
Introduction
The prevalence of congenital heart defects (CHD) is about 0.8% of live birth and higher in still birth. The etiology of CHD is complex and combines both environmental and genetic causes. However, there are already more than 50 genes associated with CHD in humans [1]. Familial cases of CHD were very useful to discover most of these CHD genes. Although these discoveries concern a very small percentage of CHD cases, they shed a new light on cardiac diseases because they demonstrated in several cases that mutations in a single gene can result in CHD, arrhythmia and/or cardiomyopathies. Thus, mutations in TBX20 [2,3], MYH7 [4,5], NKX2-5 [6,7], GATA4[8], and ACTC1 [9,10] lead to a variety of cardiac anomalies including CHD, arrhythmia and cardiomyopathies. It is also the case in Noonan syndrome—now part of a larger group of disease referred to as RASopathies—where CHD and hypertrophic cardiomyopathies are found [11,12]. This is not yet the case of all cardiac genes but it showed that genes which are important for cardiac development might also be important for cardiac function during adulthood [6,13]. It is not clear yet why mutations in a single gene results in such diverse cardiac anomalies. It could either be related to modifier genes when a single mutation results in various cardiac diseases or it could be due to diverse protein dysfunctions when different mutations in a single gene result in various cardiac anomalies.
In this study, we report on a large Lebanese family with 13 affected members suffering from various congenital heart defects, arrhythmia, and valvular and conduction anomalies. In addition, several cardiac patients have also midline defects. A missense mutation was found in the alpha-cardiac actin gene which cosegregated perfectly within the family. This mutation (p.(Met84Thr)) and 2 other mutations also responsible for CHD (p.(Glu101Lys) and p.(Met125Val)) mapped to a small domain of the actin protein in very close contact to the myosin heavy chain, suggesting that disruption of this interaction domain leads to an altered cardiac development.
Materials and Methods
Patients
Members of this Lebanese family were examined by a cardiologist (clinical examination, rest ECG and echocardiography) and by a geneticist (clinical examination). After signing an informed consent, a peripheral blood sample was obtained to extract DNA with a standard protocol. The study was approved by the ethical committee of Hôtel Dieu hospital, Beirut, Lebanon.
Genotyping and linkage analysis
Genotyping from all available DNA samples was performed at the Genotyping National Center (CNG, Evry). A panel of 399 microsatellites was tested (Life Technologies, Evry, France). Multipoint linkage analysis was prepared with easyLINKAGE [14] and performed with GeneHunter v2.1r5 [15] initially with a disease allele frequency of 0.01%, a fully penetrant disease and a 0% phenocopy rate and then with decreasing penetrance values to 85%. Haplotypes of the region of interest were prepared with GeneHunter. Genehunter output files were used to visualize haplotypes with the Haplopainter 1.043 software [16].
DNA sequencing
The sequence of NKX2-5 (ENST00000329198) and GATA4 (ENST000335135) exons was obtained by Sanger sequencing. The 6 coding exons of the ACTC1 gene (ENST00000290378) were amplified by PCR with 60ng of genomic DNA, 1.5 mM of MgCl2, 0.5 μM of forward and reverse primers, 0.2 mM of dNTPs and 1 U of Taq Platinum DNA polymerase (Invitrogen, San Diego, CA, USA) with appropriate buffer. PCR products were purified with the NucleoFast 96 PCR Clean up kit (Macherey-Nagel). Sanger sequencing was done with 0.8 μL BigDye terminator V1.1 or V3.1 with the appropriate buffer, 0.5μM of each primer and 1μL of PCR product denatured at 95° for 1 minute, then 25 cycles at 95° for 1 min, 50° for 1 min and 60° for 4 min 30 s. X-terminator product purification was done before it was sequenced with an Applied Biosystems 3730 DNA Analyzer. Sequence analysis was carried out with SeqScape v2.5 software. Variant analysis was carried out with Visual Alamut 2.6.1 Software. The variant was submitted to LOVD 3.0 shared installation (http://databases.lovd.nl/shared/) and received the variant ID # 0000064762.
Variants prediction methods
In order to evaluate the deleterious consequence of putative variants, the following prediction software were used: Align GVGD [17], SIFT [18], Mutation Taster [19] and PolyPhen2 [20]. Grantham score [21] was obtained. The interspecies conservation of amino-acid was evaluated and the presence of variants was searched in 1000 genomes (http://www.1000genomes.org/) and Exome Variant Server (http://evs.gs.washington.edu/EVS/).
Structural interpretation of the actin and myosin variants
The interpretation of the structural consequences of specific actin and myosin variants was performed by analyzing the recently published Rigor Actin-Tropomyosin-Myosin complex (PDB accession codes 4A7F, 4A7H, 4A7L and 4A7N), reported as the first subnanometer-resolution structure of the actin-tropomyosin-myosin complex in the rigor (nucleotide-free) state determined by cryo-EM [22]. Images were prepared using the Molmol software [23].
Results
Clinical analysis
This Lebanese family belongs to the Christian Maronite community. It had 13 members (Fig 1) with congenital heart defect (CHD): 7 isolated atrial septal defects (ASD) (II:2, II:3, II:5, III:6, III:7, III:10 and IV:5), 2 ASD and pulmonary stenosis (PS) (III:2 and IV:2), 1 ASD and aortic stenosis (AS) (IV:8), 1 ASD and mitral regurgitation (MR) and stenosis (MR) (III:5), 1 ASD and Ebstein anomaly (IV:3) and 1 membranous ventricular septal defect (VSD) (IV:7). In addition, one family member had a Wolff-Parkinson-White syndrome (WPW) and had complained since childhood about short bouts of undocumented paroxysmal tachycardia (III:6), one affected relative had sinus bradycardia (III:10), and one had atrial fibrillation at the age of 40 years (II:5). Beside cardiac anomalies, 6 relatives had a midline defect: 5 cases of pectus excavatum (II:5, III:10, IV:2, IV:3 and IV:8), 1 case of kyphoscoliosis (III:10), 3 cases had hypertelorism (IV:2, IV:3 and IV:8), and 1 case had cleft lip and diastema between superior incisors (IV:2). All other family members had neither cardiac nor midline anomaly. The founder of the family (I:1) died at the age of 66 years of chronic heart failure although he had neither coronary disease nor left ventricle obstruction. He had a sister (I:3) who had a cleft lip and palate and died of heart failure at age of 60. This pedigree was reported earlier [24] (OMIM #603642: atrial septal defect, secundum, with various cardiac and noncardiac defects).
Fig 1. Pedigree of the family with recurrent cardiopathies.

Squares (males) and circle (females), crossed symbols (deceased), empty symbols (unaffected) and filled symbols (affected), arrow (proband). AF: atrial fibrillation; AR: aortic regurgitation; AS: aortic stenosis; ASD-OS: atrial septal defect ostium secundum type; LAH: left anterior hemiblock; LV: left ventricle; PAH: pulmonary artery hypertension; PS: pulmonary stenosis; RBBB: right bundle branch block; SB: sinus bradycardia; VSD: ventricular septal defect; WPW: Wolff Parkinson White.
Linkage analysis and gene mutation identification
The screening of the NKX2-5 and GATA4 genes in the proband DNA (IV:8) discovered no mutations. A set of 399 poly(AC) markers was used to genotype all available DNA of this family (13 affected and 9 unaffected individuals) (Fig 1). After a parametric analysis, we found that there were only 3 peaks above 0 (S1A Fig). The tallest peak reached a lod score of 2.98. It is located on chromosome 15. This result was robust to re-analysis with decreased penetrance values (to 85%). With non parametric parameters, there was a peak nearly exclusive on chromosome 15 (S1B Fig). Haplotypes of the chromosome 15 region obtained from linkage analysis demonstrated that all affected relatives had the same allele on the marker D15S1007 which is located at genomic position 33,545,560–33,545,736 (GRCh38) on the long arm of chromosome 15 at the q14 band (S2 Fig). Moreover, a recombination between D15S165 and D15S1007 on the centromeric side (individual II:3) and one between D15S1007 and D15S1012 on the telomeric side (individual IV:8) gave the limits of the genomic interval for the causal mutation (D15S165 to D15S1012). This genomic interval of about 7,747,000 nucleotides includes 37 coding genes among which the ACTC1 gene is found. This gene encodes alpha cardiac actin 1. Since this gene appeared as the best candidate, the 6 coding exons were sequenced. A single heterozygous variant was found in the 3rd exon: c.251T>C changing the Methionine at position 84 to a Threonine. This variant is absent in 1000 genomes and in the Exome Variant Server. The Met84 is highly conserved across species to yeast and the physico-chemical properties of Methionine and Threonine are significantly different (Grantham score of 81 on a range of 0 to 215). Three software (GVGD, SIFT, Mutation Taster) predicted that it is a disease causing variant but PolyPhen2 predicted the variant to be benign. This variant was found in all affected individuals and was absent from all unaffected relatives (Fig 1). Taken together these data, we concluded that this variant is the causal mutation.
3D structure analysis
The Met84 residue is found within a surface exposed helix of the globular part of F-Actin. Interestingly, as observed in all the previously published F-actin fiber structures, there is no evidence of a native actin-actin contact surface involving residue 84 in actin filaments reconstructions (Fig 2A). Looking closely at the 3D structure of globular actin, this mutation appeared to reside in a region of the actin filament in extremely tight apposition to the myosin head (Fig 2B). Furthermore, two other well-characterized mutations of actin (p.(Glu101Lys) and p.(Met125Val)), which both also resulted mainly in atrial septal defects [9,25,26] are located in the same subdomain (Fig 2B). Disease-causing mutations disrupt both electrostatic and hydrophobic contacts, thereby directly perturbing the interaction between actin, tropomyosin and myosin. All published ACTC1 mutations in humans are summarized on Table 1. Note that in several publications, the amino acid position was given after subtraction of the first two residues that are removed during actin maturation (the original reported Met123Val mutation should actually be described as Met125Val). Interestingly, ACTC1 mutations resulting in congenital heart defects (essentially atrial septal defects) are restricted to the first half of the protein (from residue Met84 to residue Met178 [27]). Beyond residue Met178, all reported ACTC1 mutations result in diverse cardiomyopathies [10,28–31] with an unusual prevalence of non-compaction of the apex or hypertrophied apex.
Fig 2. Molecular representation of the PDB 4A7L complex, displaying an actin fiber (blue and green monomers, with ADP molecules in purple) with myosin heads (brown monomers).
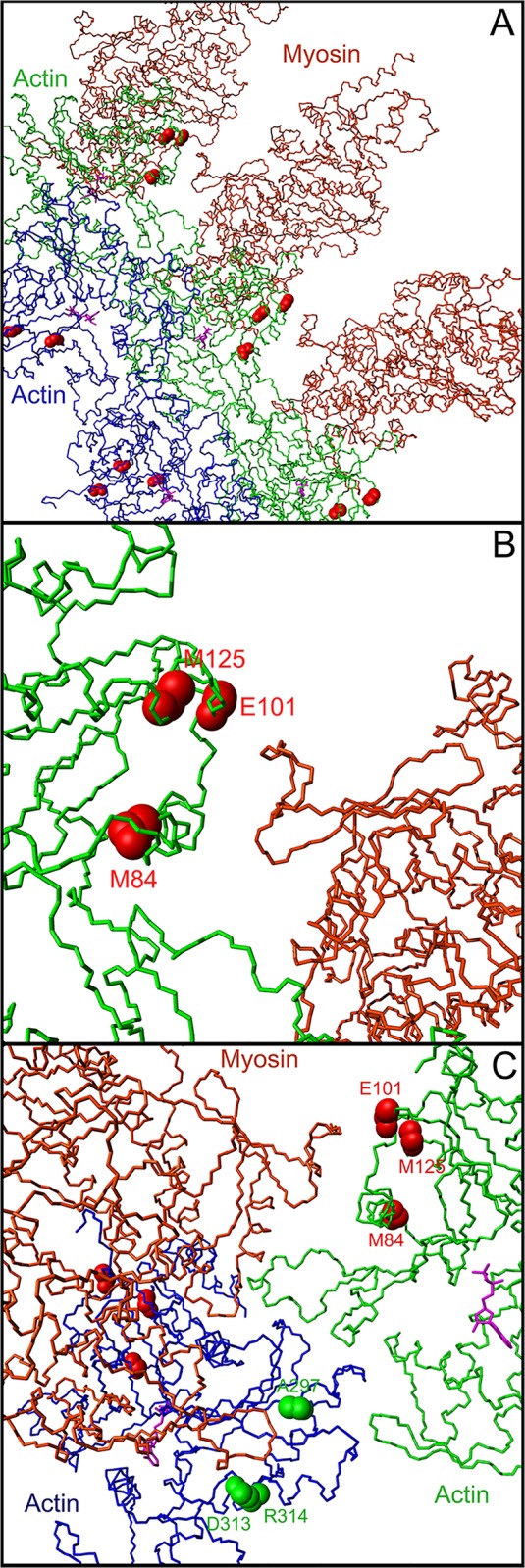
Actin and myosin amino acid numbers are according to human numbering (A) Backbone representation of an actin fiber in complex with 3 myosin heads, showing secondary structure elements. The backbone atoms of the 3 amino acids altered by mutations causing atrial septal defects (residues 84, 101, and 125) are shown by red spheres on every actin monomer. (B) Close up on the interaction between the region spanning residues 84, 101 and 125 of an F-actin monomer and a very close loop of the myosin head. The actin monomer is shown in green, the myosin head is shown in brown. The interatomic distances measured in the complex between residues 84, 101 and 125 and the myosin surface typically range from 3 to 10 Å. The 562–571 region of the myosin head makes numerous contacts with the surface of the actin filament, and interacts closely with residues 84, 101 and 125 on the surface of actin. (C) Close up showing one myosin head interacting with the region 84, 101, and 125 region of the actin monomer, and the 297, 313 and 314 region of an adjacent actin monomer. The 562–571 region of the myosin head closely interacts with residues whose mutation leads to atrial septal defects (84, 101, and 125, in red), whereas the 367–365 region (human numbering) of the same myosin head interacts directly with an adjacent actin monomer (residues 297, 313, 314, in green), whose mutation leads to cardiomyopathies. The orientation of the actin monomers in panel A and B is similar whereas the molecules in panel C have been rotated for a better view of the interaction with residues 297, 313, and 314.
Table 1. Reported cases with ACTC1 gene mutations.
| Familial/de novo | Cardiac Disease | Cardiac rythm | Evolution | Other features | Mutation | ENST00000290378 NM_005159.4 | ENST00000290378 NM_005159.4 | Reference |
|---|---|---|---|---|---|---|---|---|
| Familial (13x) | 12x ASD, 1x VSD, 2x PS, 1x AR, 1x AS, 1x Ebstein | 2x WPW, 1x AF, 1x SB | CHF | 5x midline defects | p.(Met84Thr) | p.(Met84Thr) | c.251T>C | Current study |
| Familial (2x) | 1x ASD, 1x VSD? | - | - | - | 17bp del starting AA 86 | p.(Pro72Hisfs*18) | c.215_231del | Matsson et al. 2008 |
| Familial (18x) | 5x hypertrophied and 3x trabeculated LV apex, 11x HCM, 4x MR, 2x AR | 3x AF, 3x AV block or SB, 2x short PR interval | AF, CHF | - | p.(Glu99Lys) | p.(Glu101Lys) | c.301G>A | Arad M et al. 2005 |
| Familial (94x) | 22x apical HCM, 23x LVNC, 8x ASD, 1x VSD, 3x RCM | 3x AF | 1x CHF, 1x graft, 5x SD | - | p.(Glu99Lys) | p.(Glu101Lys) | c.301G>A | Monserrat L et al. 2007 |
| Familial (9x) | 6x HCM, 2x trabeculated and 5x hypertrophied LV apex, 1x ASD | 1x VT, 2x VF | 2x SD | - | p.(Glu99Lys) | p.(Glu101Lys) | c.301G>A | Olson T al. 2000 |
| Familial (20x) | 20x ASD | - | - | - | p.(Met123Val) | p.(Met125Val) | c.373A>G | Matsson H et al. 2008 |
| de novo (17 mo) | HCM, hypertrophied LV apex | PM | - | - | p.(Pro164Ala) | p.(Pro166Ala) | c.496C>G | Olson et al. 2000. |
| Familial (6x) | 6x ASD | - | - | - | p.(Met178Leu) | p.(Met178Leu) | c.532A>T | Greenway at al. 2014 |
| Familial (3x) | 2x HCM | - | 1x SD | - | p.(Ala232Val) | p.(Ala232Val) | c.695C>T | Van Driest et al. 2003 |
| Familial (18x) | 13x HCM, 5x MR, 2x AR | 1x AF, 1x WPW, 1x VF | 2x DCM | - | p.(Ala295Ser) | p.(Ala297Ser) | c.889G>T | Mogensen et al. 1999 |
| Familial (3x) | 2x RCM, 1x RMC/DCM | - | 1x graftn | - | p.(Asp313His) | p.(Asp313His) | c.937G>C | Kaski et al. 2008 |
| Familial (4x) | 3x DCM | - | - | - | p.(Arg312His) | p.(Arg314His) | c.941G>A | Olson et al. 1998 |
| de novo (8 yo) | HCM, hypertrophied LV apex | VF, IAD | - | - | p.(Ala331Pro) | p.(Ala333Pro) | c.997G>C | Olson et al. 2000 |
| Familial (4x) | 4x DCM | - | - | - | p.(Glu361Gly) | p.(Glu363Gly) | c.1088A>G | Olson et al. 1998 |
In the first column (familial/sporadic), the number of reported cases is notified (for instance: 13x: 13 reported cases). In de novo mutations, the age at diagnosis is stated. Mutation: mutation description as reported in the original article; Columns “ENST00000290378, NM_005159.4”: current description at the protein and DNA levels of the same mutation as in the column “Mutation”. AF: Atrial Fibrillation, AR: Aortic Regurgitation, AS: Aortic Stenosis, ASD: Atrial Septal Defect, AV: Atrioventricular, CHF: Cardiac Heart Failure, DCM: Dilated Cardiomyopathy, HCM: Hypertrophic Cardiomyopathy, IAD: Implantable Automatic Defibrillator, LV: Left Ventricle, LVNC: Left Ventricular Non Compaction, mo: months old, MR: Mitral Regurgitation, PM: Pace Maker, PS: Pulmonary Stenosis, RCM: Restrictive Cardiomyopathy, SB: Sinus Bradycardia, SD: Sudden Death, VF: Ventricular Fibrillation, VSD: Ventricular Septal Defect, VT: Ventricular Tachycardia, WPW: Wolff Parkinson White, yo: years old.
Discussion
The list of genes involved in congenital heart defects (CHD) is growing [1]. At the same time, the frontier between CHD and cardiomyopathies (CM) is becoming blurred. There are reports on familial cases with relatives being affected with CM or CHD or both. In addition, the list of genes which can lead to CM and/or CHD is also growing [2–8,11,12]. The actin gene is one of these genes. ACTC1 mutations were first identified in a series of dilated CM [30]. Other mutations can result in apical hypertrophic CM, while others are associated with CHD and in particular atrial septal defects (ASD). In this report, mutation carriers of the p.(Met84Thr) missense suffer from a variety of CHD (ASD, Ebstein anomaly, VSD) but also from valvular anomalies (aortic and pulmonary stenosis, mitral regurgitation and stenosis), conduction tissue anomalies (sinus bradycardia, WPW syndrome), and CM (hypertrophic CM in one patient and presumably dilated CM in the founder and his sister). Thus, alpha cardiac actin 1 mutations can result in a variety of cardiopathies even within a single family suggesting that modifying factors might modulate the expressivity of ACTC1 mutation.
In addition to cardiac signs, several patients showed midline anomalies (diastema of the upper incisors, cleft lip, hypertelorism, kyphoscoliosis and pectus excavatum). Midline defects are very rare and the occurrence of 5 cases (6 cases if the grand-aunt I:3 is included) in a single family cannot be fortuitous. In addition, each relative with a midline defect actually has more than one midline defect sign. The co-segregation of midline defects with cardiac anomalies and the Met84Thr mutation suggests that cardiac and midline defect could be secondary to the ACTC1 Met84 mutation. The co-occurrence of cardiac and midline defects was never reported previously in ACTC1 mutation carriers. Alternatively, it is possible that a second mutation in another gene is present in this minimal genomic region. Actually, there exists a single gene among the 36 genes—excluding the ACTC1 gene–(S1 Table) which can lead to midline defects. This gene, SLC12A6, leads to mild midline defects similar to the ones reported in this study. However, it also leads to mental disability, complete or partial agenesis of the corpus callosum, and severe peripheral neuropathy [32]. This severe disease is autosomal recessive, so we can rule out this type of inheritance in this Lebanese family because individuals with midline defect share only one parental haplotype as evidenced by linkage analysis. This report prompts cardiologists to pay attention to midline anomalies in familial ASD. It is possible that cardiologists have overlooked midline defects and failed to report this type of anomaly in previous ACTC1 mutation reports.
The actin p.(Met84Thr) mutation, which was identified in this work, as well as the previously identified p.(Glu101Lys) [9,25] and p.(Met125Val) [26] mutations, result mainly in ASD. Those 3 mutations occur in a small spatially well-defined region, which is on a surface exposed region of F-actin in very close contact with myosin monomers, as observed in the most accurate muscular fiber reconstruction published to date. The importance of this region for the Actin-Myosin interaction is supported by previous mutagenesis studies demonstrating that altering the charge of residue Glu101 by histidine substitution reduces in vitro motility by five-fold [33] whereas the p.(Met125Val) substitution showed a significantly reduced affinity for myosin [26]. Another argument to support the importance of the tight actin-myosin interaction is found by carefully analyzing the position of residues 297 [29], 313 [28] and 314 [30] of actin in the 4A7L actin-myosin complex. Mutations on those residues have been known to lead to various cardiomyopathies and they also appear to make extremely close contact with the adjacent myosin monomer, but using a totally different interaction surface compared to residues 84 (current study), 101 [9,10,25] and 125 [26] (Fig 2C).
The human ACTC1 gene produces a protein with 94% homology to the gamma actin gene (ACTG1). Mutations in this latter gene were associated with dominant progressive deafness [34], a disease that displays sensorineural hearing loss beginning in the twenties in the high frequencies and steadily progressing to include all frequencies. Although the two non-muscular actin genes (ACTG1 and ACTB) are expressed concomitantly in all mammalian cells, the auditory hair cell is one of the rare cell types where the predominant isoform is gamma actin (ACTG1). In auditory hair cells, the gamma actin protein is found in stereocilia, the cuticular plate, and adherens junctions. One of the six mutations found in the initial report is a threonine to isoleucine change at position 89, a position very close to the congenital cardiac disease ACTC1 mutations. Although the gamma actin residue at the corresponding site is not a threonine but a valine, it is interesting to comment on the changes induced by this p.(Thr89Ile) missense variant. It was tested in the yeast Saccharomyces cerevisiae because yeast actin is 91% identical to human gamma actin and the Thr89 residue is conserved in both species. The Thr89Ile mutation resulted in a higher population of cells with fragmented and/or depolarized cables and uniform distribution of patches in both mother cell and bud in comparison to wild-type [35]. However, the in vitro ability of purified Thr89Ile mutant actin to polymerize was not grossly modified suggesting an altered in vivo interaction with one or more of the numerous acting-binding proteins known to control actin cytoskeletal function and dynamics. It was actually the case since the p.(Thr89Ile) variant F-actin is much more susceptible to cofilin disassembly than wild type actin [36]. Cofilin severs F-actin and sequesters actin monomers. This result suggests that the Thr89 residue is involved in non-actin protein interaction and that mutations in this region destabilize protein interaction with cofilin in a presumably similar way as the cardiac actin Met84 mutation might destabilize the actin/myosin interaction.
In conclusion, we reported a novel ACTC1 gene mutation which resulted in various congenital heart defects and arrhythmia. This family study suggested that ACTC1 mutation could also lead to midline defects. Finally, we provided evidence to possibly explain the pleiotropic consequences of ACTC1 gene mutations by pointing to particular molecular domains where actin and myosin heavy chain are in close contact. Depending on the domain, ACTC1 mutation can lead rather to congenital heart defects or to cardiomyopathies.
Supporting Information
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We are very indebted to family members who participated to this study. PB is very grateful to Jean-Louis FEBVRE and foundation Renaud FEBVRE for their longstanding and very efficient support. PB dedicates this study to Prof. Edouard Stephan who passed away some years ago. He is the cardiologist who contributed to uncover this family. Maxime BOUVAGNET is warmly thanked for improving the edition of this manuscript.
Data Availability
All relevant data are within the paper and its supporting information files. The disease causing variant was declared in a public repository under the variant number: 0000058960 in the LOVD database (http://databases.lovd.nl/shared/variants).
Funding Statement
Projet de Recherche Hospitalier de Recherche (PHRC 2008) of the Ministère des Affaires Sociales et de la Santé, France. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Andersen TA, Troelsen Kde L, Larsen LA. Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci. 2014;71: 1327–52. 10.1007/s00018-013-1430-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81: 280–91. doi:S0002-9297(07)61194-X [pii] 10.1086/519530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qian L, Mohapatra B, Akasaka T, Liu J, Ocorr K, Towbin JA, et al. Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc Natl Acad Sci U A. 2008;105: 19833–8. doi:0808705105 [pii] 10.1073/pnas.0808705105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Budde BS, Binner P, Waldmuller S, Hohne W, Blankenfeldt W, Hassfeld S, et al. Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta-myosin heavy chain gene. PLoS One. 2007;2: e1362 10.1371/journal.pone.0001362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Postma AV, van Engelen K, van de Meerakker J, Rahman T, Probst S, Baars MJ, et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ Cardiovasc Genet. 2011;4: 43–50. doi:CIRCGENETICS.110.957985 [pii] 10.1161/CIRCGENETICS.110.957985 [DOI] [PubMed] [Google Scholar]
- 6. Costa MW, Guo G, Wolstein O, Vale M, Castro ML, Wang L, et al. Functional characterization of a novel mutation in NKX2-5 associated with congenital heart disease and adult-onset cardiomyopathy. Circ Cardiovasc Genet. 2013;6: 238–47. doi:CIRCGENETICS.113.000057 [pii] 10.1161/CIRCGENETICS.113.000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ouyang P, Saarel E, Bai Y, Luo C, Lv Q, Xu Y, et al. A de novo mutation in NKX2.5 associated with atrial septal defects, ventricular noncompaction, syncope and sudden death. Clin Chim Acta. 2011;412: 170–5. doi:S0009-8981(10)00622-4 [pii] 10.1016/j.cca.2010.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X, et al. GATA4 loss-of-function mutation underlies familial dilated cardiomyopathy. Biochem Biophys Res Commun. 2013;439: 591–6. doi:S0006-291X(13)01494-0 [pii] 10.1016/j.bbrc.2013.09.023 [DOI] [PubMed] [Google Scholar]
- 9. Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28: 1953–61. doi:ehm239 [pii] 10.1093/eurheartj/ehm239 [DOI] [PubMed] [Google Scholar]
- 10. Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32: 1687–94. doi: 10.1006/jmcc.2000.1204 S0022-2828(00)91204-5 [pii] [DOI] [PubMed] [Google Scholar]
- 11. Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135: 703–6. doi:S0022-3476(99)70088-0 [pii] [DOI] [PubMed] [Google Scholar]
- 12. Sznajer Y, Keren B, Baumann C, Pereira S, Alberti C, Elion J, et al. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007;119: e1325–31. doi:peds.2006-0211 [pii] 10.1542/peds.2006-0211 [DOI] [PubMed] [Google Scholar]
- 13. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437: 270–4. doi:nature03940 [pii] 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- 14. Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21: 405–7. doi: 10.1093/bioinformatics/bti009 bti009 [pii] [DOI] [PubMed] [Google Scholar]
- 15. Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58: 1347–63. [PMC free article] [PubMed] [Google Scholar]
- 16. Thiele H, Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21: 1730–2. doi: 10.1093/bioinformatics/bth488 bth488 [pii] [DOI] [PubMed] [Google Scholar]
- 17. Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43: 295–305. doi:jmg.2005.033878 [pii] 10.1136/jmg.2005.033878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4: 1073–81. doi:nprot.2009.86 [pii] 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 19. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7: 575–6. doi:nmeth0810-575 [pii] 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- 20. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7: 248–9. doi:nmeth0410-248 [pii] 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185: 862–4. [DOI] [PubMed] [Google Scholar]
- 22. Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150: 327–38. doi:S0092-8674(12)00707-6 [pii] 10.1016/j.cell.2012.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14: 51–5, 29–32. doi:0263785596000094 [pii] [DOI] [PubMed] [Google Scholar]
- 24. Megarbane A, Stephan E, Kassab R, Ashoush R, Salem N, Bouvagnet P, et al. Autosomal dominant secundum atrial septal defect with various cardiac and noncardiac defects: a new midline disorder. Am J Med Genet. 1999;83: 193–200. [pii] [DOI] [PubMed] [Google Scholar]
- 25. Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, Ho CY, et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005;112: 2805–11. doi:112/18/2805 [pii] 10.1161/CIRCULATIONAHA.105.547448 [DOI] [PubMed] [Google Scholar]
- 26. Matsson H, Eason J, Bookwalter CS, Klar J, Gustavsson P, Sunnegardh J, et al. Alpha-cardiac actin mutations produce atrial septal defects. Hum Mol Genet. 2008;17: 256–65. doi:ddm302 [pii] 10.1093/hmg/ddm302 [DOI] [PubMed] [Google Scholar]
- 27. Greenway SC, McLeod R, Hume S, Roslin NM, Alvarez N, Giuffre M, et al. Exome sequencing identifies a novel variant in ACTC1 associated with familial atrial septal defect. Can J Cardiol. 2014;30: 181–7. doi:S0828-282X(13)01711-X [pii] 10.1016/j.cjca.2013.12.003 [DOI] [PubMed] [Google Scholar]
- 28. Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94: 1478–84. doi:hrt.2007.134684 [pii] 10.1136/hrt.2007.134684 [DOI] [PubMed] [Google Scholar]
- 29. Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103: R39–43. 10.1172/JCI6460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280: 750–2. [DOI] [PubMed] [Google Scholar]
- 31. Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108: 445–51. doi: 10.1161/01.CIR.0000080896.52003.DF 01.CIR.0000080896.52003.DF [pii] [DOI] [PubMed] [Google Scholar]
- 32. Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu J, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32: 384–92. doi: 10.1038/ng1002 ng1002 [pii] [DOI] [PubMed] [Google Scholar]
- 33. Johara M, Toyoshima YY, Ishijima A, Kojima H, Yanagida T, Sutoh K. Charge-reversion mutagenesis of Dictyostelium actin to map the surface recognized by myosin during ATP-driven sliding motion. Proc Natl Acad Sci U A. 1993;90: 2127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, et al. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet. 2003;73: 1082–1091. 10.1086/379286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bryan KE, Wen K-K, Zhu M, Rendtorff ND, Feldkamp M, Tranebjaerg L, et al. Effects of human deafness gamma-actin mutations (DFNA20/26) on actin function. J Biol Chem. 2006;281: 20129–20139. 10.1074/jbc.M601514200 [DOI] [PubMed] [Google Scholar]
- 36. Bryan KE, Rubenstein PA. Allele-specific effects of human deafness gamma-actin mutations (DFNA20/26) on the actin/cofilin interaction. J Biol Chem. 2009;284: 18260–18269. 10.1074/jbc.M109.015818 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its supporting information files. The disease causing variant was declared in a public repository under the variant number: 0000058960 in the LOVD database (http://databases.lovd.nl/shared/variants).