

Abstract
L-DOPA therapy in Parkinson’s disease often results in side effects such as L-DOPA-induced dyskinesia (LID). Our previous studies demonstrated that defective desensitization of dopamine receptors caused by decreased expression of G protein-coupled receptor kinases (GRKs) plays a role. Overexpression of GRK6, the isoform regulating dopamine receptors, in parkinsonian rats and monkeys alleviated LID and reduced LID-associated changes in gene expression. Here we show that 2-fold lentivirus-mediated overexpression of GRK6 in the dopamine-depleted striatum in rats unilaterally lesioned with 6-hydroxydopamine ameliorated supersensitive ERK response to L-DOPA challenge caused by loss of dopamine. A somewhat stronger effect of GRK6 was observed in drug-naïve than in chronically L-DOPA-treated animals. GRK6 reduced the responsiveness of p38 MAP kinase to L-DOPA challenge rendered supersensitive by dopamine depletion. The JNK MAP kinase was unaffected by loss of dopamine, chronic or acute L-DOPA, or GRK6. Overexpressed GRK6 suppressed enhanced activity of Akt in the lesioned striatum by reducing elevated phosphorylation at its major activating residue Thr308. Finally, GRK6 reduced accumulation of ΔFosB in the lesioned striatum, the effect that paralleled a decrease in locomotor sensitization to L-DOPA in GRK6-expressing rats. The results suggest that elevated GRK6 facilitate desensitization of DA receptors, thereby normalizing of the activity of multiple signaling pathways implicated in LID. Thus, improving the regulation of dopamine receptor function via the desensitization mechanism could be an effective way of managing LID.
Keywords: Parkinson’s disease, L-DOPA-induced dyskinesia, GRK6, lentivirus, ERK1/2, p38, Akt, JNK, ΔFosB
Introduction
Parkinson’s disease (PD)1 is an age-related neurodegenerative movement disorder caused by degeneration of dopaminergic neurons that provide dopamine (DA) to the striatum. Unilateral DA depletion in rodents with neurotoxin 6-hydroxydopamine (6-OHDA) produces movement defects reminiscent of akinesia in PD and is often used as a model of PD. Loss of DA in the basal ganglia deregulates DA receptors eliciting a broad array of signaling abnormalities. Most notable abnormalities are striking responses of signaling pathways to acute dopaminergic stimulation that are weaker or nonexistent in the intact striatum (Brown et al., 2005; Bychkov et al., 2007; Gerfen, 2000; Gerfen et al., 2002; Murer and Moratalla, 2011; Santini et al., 2010; Sgambato-Faure et al., 2005), also see (Gurevich and Gurevich, 2008) and reference therein). It has been suggested that loss of DA causes DA receptors, particularly, the D1 subtype, to become supersensitive (Aubert et al., 2005; Gerfen et al., 2002; Guigoni et al., 2005a; Guigoni et al., 2007). However, the molecular mechanisms underlying such supersensitivity are still unclear.
DA replacement therapy with levodopa (L-DOPA) is the best pharmacological treatment available today. L-DOPA treatment effectively reverses akinesia in PD but eventually leads to severe motor side effects such as L-DOPA-induced dyskinesia (LID) (Cotzias et al., 1969; Stocchi et al., 1997). Surprisingly, the pathophysiology of LID, despite decades of research, is still poorly understood. The reason for this is that mechanisms of signaling alterations triggered by loss of DA and subsequent L-DOPA treatment remain obscure. In 6-OHDA-lesioned rodents, chronic treatment with L-DOPA desensitizes some supersensitive signaling responses, in parallel with the reversal of DA depletion-induced akinesia. E.g., chronic L-DOPA suppresses supersensitivity of the ERK pathway caused by loss of DA (Bezard et al., 2005; Brown et al., 2005; Bychkov et al., 2007; Kim et al., 2006; Santini et al., 2010). Chronic L-DOPA treatment is also known to augment lesion-induced supersensitivity of select pathways and/or further deregulate their activity (Bychkov et al., 2007; Santini et al., 2010; Sgambato-Faure et al., 2005). These signaling alterations correlate with progressively increasing frequency of L-DOPA-induced rotations (process known as behavioral sensitization to L-DOPA) (Ahmed et al., 2010; Ahmed et al., 2007; Bordet et al., 1997; Bychkov et al., 2007) and of abnormal involuntary movements (AIMs) (Ahmed et al., 2010; Cenci, 2007; Cenci and Konradi, 2010; Cenci et al., 1998; Cenci and Lundblad, 2007). These molecular mechanisms likely contribute to pathophysiology of LID.
DA receptors belong to the superfamily of G protein-coupled receptors (GPCRs). Upon stimulation, many GPCRs undergo desensitization via activation-dependent receptor phosphorylation by G protein-coupled receptor kinases (GRKs) followed by arrestin binding. Arrestin binding terminates further G protein signaling (DeWire et al., 2007; Gurevich and Gurevich, 2006a). GRKs and arrestins are the main regulators of GPCR signaling via receptor desensitization (Gurevich et al., 2012; Gurevich and Gurevich, 2006b). Simple modulation of GRK or arrestin expression has a profound effect on GPCR signaling (Bohn et al., 2003; Bohn et al., 1999; Gainetdinov et al., 2003; Iaccarino et al., 1998; Kim et al., 2001; Menard et al., 1997; Pan et al., 2003; Willets et al., 2004; Willets et al., 1999; Xu et al., 1997).
We have recently demonstrated that DA depletion in hemiparkinsonian rats significantly downregulates several GRK isoforms including GRK6 (Ahmed et al., 2010; Ahmed et al., 2007), which is believed to be the main GRK isoform desensitizing striatal DA receptors in vivo (Ahmed et al., 2010; Gainetdinov et al., 2003) see also (Gurevich et al., 2012) and references therein]. Furthermore, we demonstrated that lentivirus-mediated GRK6 overexpression alleviated, whereas knockdown exacerbated sensitization to L-DOPA and LID in the rat and monkey models of PD (Ahmed et al., 2010). The behavioral improvement was accompanied by amelioration of several molecular hallmarks of LID (Ahmed et al., 2010). We hypothesize that the origin of multiple signaling abnormalities in the DA-depleted striatum is inadequate desensitization of DA receptors that is either not normalized or further deregulated by subsequent chronic L-DOPA treatment. Defective receptor desensitization may partially stem from reduced expression of GRK isoforms in the DA depleted striatum that is not normalized by L-DOPA (Ahmed et al., 2007). Signaling abnormalities could then be propagated throughout the signaling network. The corollary of this hypothesis is that elevated expression of GRKs should rescue desensitization of DA receptors, thus ameliorating abnormal signaling via multiple pathways and improving behavior.
In our previous study (Ahmed et al., 2010), we examined the effect of overexpressed GRK6 on gene expression in hemiparkinsonian rats chronically treated with L-DOPA but did not examine the activity of signaling pathways. Here we document the activity of multiple signaling pathways and their responsiveness to L-DOPA challenge in drug-naïve and chronically L-DOPA-treated hemiparkinsonian rats overexpressing GRK6 in the lesioned striatum in comparison to control GFP-expressing rats. Dynamic signaling responses to acute L-DOPA, as compared to saline, challenge correspond to the peak of L-DOPA-induced behavior, or peak-dose LID, when L-DOPA-derived DA is present in the striatum. We determined how these responses were altered by chronic L-DOPA treatment, and whether GRK6 effects depend on the nature of the challenge or treatment. We report that, in agreement with our hypothesis, elevation of GRK6 in the lesioned striatum normalized the activity of multiple signaling pathways and suppressed accumulation of ΔFosB, a known correlate of LID (Cao et al., 2010; Darmopil et al., 2009; Pavon et al., 2006; Westin et al., 2007).
Materials and methods
Animals, surgery, and drug treatment
Adult Sprague-Dawley rats (Charles Rivers) were used for all experiments. The animals were housed at the Vanderbilt University’s animal facility with 12h/12h light/dark cycle and had free access to food and water. All procedures followed NIH guidelines and were approved by the institutional IACUC committee. Rats were deeply anesthetized with ketamine/xylazine (75/5 mg/kg i.p.) and mounted on stereotaxis. The 6-OHDA lesion was performed as previously described (Ahmed et al., 2007; Bychkov et al., 2007). Briefly, rats were treated with desimipramine (25 mg/kg i.p.) 30 min prior to infusion of 6-OHDA. 6-OHDA (8 μg in 4 μl of 0.05% ascorbic acid) was infused unilaterally into the medial forebrain bundle at coordinates A=−4.3 mm, L=1.2 mm, H =−8.5 mm. After 3 weeks, the animals were randomly assigned to one of the two experimental groups for chronic treatment, saline or L-DOPA (L-3,4-Dyhydroxyphenylalanine methyl ester hydrochloride in saline, 25 mg/kg s.c. twice daily) (Fig. 1).
Figure 1. Schematic representation of the experimental design.
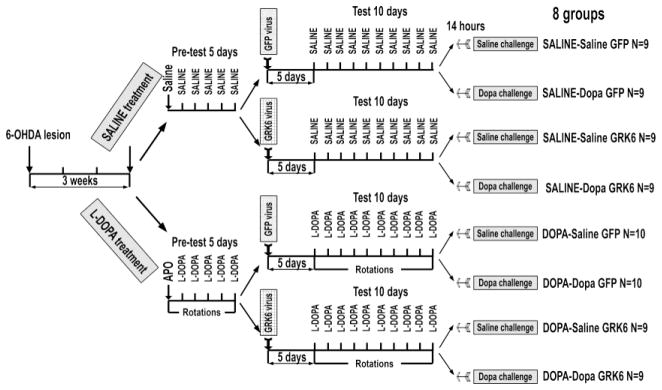
Rats received unilateral 6-OHDA injection into the medial forebrain bundle and were allowed to recover for 3 weeks before being randomly assigned to the SALINE or L-DOPA treatment groups. Rats from the L-DOPA group were pretested for apomorphine-induced rotations once and then for L-DOPA-induced rotations for 5 days. Following pretesting, rats of both SALINE and L-DOPA groups were assigned to GFP or GRK6 groups and received unilateral injections of respective lentiviruses into the lesioned dorsolateral caudate-putamen as described in Methods. Following 5-day recovery period, rats were tested for L-DOPA-induced rotations (L-DOPA group) or treated with saline (SALINE group) for 10 days. Approximately 14 h after the last injection, the rats of each group were randomly challenged with either saline or L-DOPA and sacrificed 45 min later. The design produced 8 experimental groups; N – number of rats in each group.
The rats assigned to the L-DOPA group were tested for rotational response to apomorphine (0.05 mg/kg s.c.) for 1 h using an automated rotometer (AccuScan Instruments, Columbus, OH) and then pre-tested for L-DOPA-induced rotations for 5 days, as described previously (Ahmed et al., 2010) (Fig. 1). Since even a single injection of a dopaminergic drug produces priming (Nadjar et al., 2009; Santini et al., 2010), the rats assigned to the saline treatment group were injected with saline instead of apomorphine and then with saline twice daily instead of L-DOPA, and exposed to the apparatus but did not display rotations. Since we found no clear relationship between the degree of DA depletion and rotation frequency or LID (Ahmed et al., 2010; Ahmed et al., 2007; Guigoni et al., 2005b), we did not exclude animals from the study based on apomorphine-induced rotations. Instead, apomorphine was used to maintain consistency in the experimental design with previous experiments (Ahmed et al., 2010). The consistency with the previous experiments was important, since we were interested in signaling mechanisms underlying anti-LID activity of GRK6 we have documented.
Upon completion of pre-testing phase, rats of saline group were randomly assigned to the GFP or GRK6 groups (Fig. 1). Animals expressing GFP were used as a control. For gene transfer, the lentiviral vector (Invitrogen, Carlsbad, CA) incorporating GFP or untagged wild type rat GRK6A under control of CMV promoter was used. Lentivirus was produced using 4-plasmid system in HEK293-FT cells and titered in HEK293A cells, as described previously (Ahmed et al., 2010). The rats were injected with the GFP or GRK6 [rat GRK6c isoform; equivalent to human GRK6A (Mushegian et al., 2012); accession # NP_001106183] lentivirus into the lesioned dorsolateral CPu under ketamine/xylazine anesthesia (75/5 mg/kg i.p.) at coordinates AP 0.2; ML 3.5; DV 5.7, as described (Ahmed et al., 2010). The rats were allowed to recover for 5 days, and then drug treatment resumed and continued for 10 days. The L-DOPA-treated rats were tested for rotations for 1 h every day after the morning injections starting 15 min after the drug injection. The saline-treated rats were injected with saline twice daily instead of L-DOPA, and exposed to the apparatus but did not display rotations. At the end of the 10-day treatment period, the GFP and GRK6 saline- and L-DOPA-treated groups were randomly split in two to receive either saline or L-DOPA challenge (25 mg/kg s.c) 14 h after the last injection (Fig. 1).
Tissue preparation
The rats were decapitated 45 min after the saline or L-DOPA challenge under isoflurane anesthesia, the brains were collected and rapidly frozen on dry ice. The brain area in the dorsolateral CPu surrounding the injection site in the lesioned striatum was outlined precisely on the cryostat-cut 100 μm-thick coronal sections, and the tissue was scraped into 200 μl of Lysis solution (Ambion, Austin, TX). The corresponding area approximately of the same size was collected from the intact striatum. Protein concentration in the samples was measured with Bradford reagent (Bio-Rad, Hercules, CA). Samples were stored at −80°C until needed.
Quantitative Western Blotting
To prepare samples for Western blotting, 100 μg of total protein was precipitated from Lysis buffer with 90% (v/v) methanol. The protein was then pelleted by centrifugation [10,000g, 10 min at room temperature (RT)], washed with 1 ml of 90% methanol, dried, and dissolved in sodium dodecyl sulfate sample buffer at the final concentration of 0.5 mg/ml. The samples (samples from different groups loaded in counterbalanced order) were run in 26-well Criterion cassettes (Bio-Rad, Hercules, CA). Electrophoresis and transfer onto Immobilon-P (Millipore, Bedford, MA, USA) membrane were performed essentially as we described previously (Ahmed et al., 2010; Ahmed et al., 2008; Bezard et al., 2005; Bychkov et al., 2007; Bychkov et al., 2010). To estimate the degree of dopamine denervation, we measured tyrosine hydroxylase (TH) concentration in dorsolateral CPu using rabbit polyclonal antibody (Chemicon, Temecula, CA) at 1:10,000. We used rabbit polyclonal antibodies (catalog #sc-566; Santa Cruz Biotechnology, Santa Cruz, CA) to quantify endogenous GRK6 (1:500). Mouse monoclonal anti-GFP antibody (JL-8 Clontech, 1:500) was used to quantify expressed GFP. The following antibodies to signaling proteins were used (all from Cell Signaling Technologies; catalog numbers given in parentheses) at dilutions recommended by the manufacturer: mouse monoclonal antibody to phospho-ERK1/2 (#9106), mouse monoclonal antibodies to phospho-p38 (#9216), rabbit polyclonal to phospho-Akt(Thr308) (#4056) and phospho-Akt(Ser473) (#9277), rabbit monoclonal to phospho-SAPK/JNK (#9251), rabbit polyclonal to ERK1/2 (#9102), Akt (#9272), p38 (#9212), rabbit monoclonal to SAPK/JNK1/2/3 (#9258). The antibodies uses were not isoform-specific detecting all isoforms of signaling proteins of interest. ΔFosB was detected with rabbit polyclonal antibody (#9890) that recognizes ΔFosB and Δ2ΔFosB [a double truncated FosB that lacks N-terminal Fos-homology domain; produced from ΔfosB mRNA by alternative translation initiation (Sabatakos et al., 2008)], but does not cross-react with FosB. Blots were incubated overnight at 4°C with appropriate primary antibodies followed by horseradish peroxidase-conjugated goat anti-rabbit or rabbit anti-mouse secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:10,000 dilution (1 h at RT) and SuperSignal enhanced chemiluminescence reagent WestPico (Pierce, Rockford, IL, USA). Upon development, the blots were exposed to X-ray film for appropriate periods of time.
Standards were used to help with band identification, ensure signal linearity for quantification, and normalize inter-blot variations. For GRK6, dilutions of standard Sf9 cells-expressed purified human GRK6 (Lodowski et al., 2006) kindly provided by Dr. John Tesmer (University of Michigan at Ann Arbor) in sample buffer were loaded onto each gel along with samples. Appropriate dilutions of GRK6 protein were loaded onto each gel alongside the samples to generate calibration curves. To determine absolute ERK1/2 concentrations, we used recombinant purified phospho-ERK2 or unphosphorylated ERK2 expressed in E. coli and purified as described (Waas et al., 2003) as standards. To identify bands corresponding to phosphorylated p38 and Akt at Thr308 or Ser473, we used lysates prepared from HEK293 cells stimulated with FGF (10 ng/mL) for 5 min. To identify phospho-JNK bands, we used lysates prepared from HEK293 cells stimulated with anisomycin (1 μM for 15 min).
Isolation and characterization of the nuclear and cytosolic fractions
The Nuclei Pure Prep Nuclei Isolation Kit (Sigma, St. Louis, MO) was used to prepare nuclear and cytosolic fractions from the rat tissue. The intact and lesioned striatal from rats injected with either GFP or GRK6 lentivirus and chronically treated with L-DOPA as described above were dissected and homogenized in 8 ml of Lysis buffer and the nuclei were separated from the cytosolic structured by differential centrifugation according to the manufacturer’s instructions. Final nuclei preparations were dissolved in 200 ml of Nuclei Storage buffer from the kit. Protein concentration in the samples was measured with Bradford reagent (Bio-Rad, Hercules, CA). Proteins from the nuclear and cytosolic fractions were precipitated with methanol and dissolved in in sodium dodecyl sulfate sample buffer at the final concentration of 0.5 mg/ml. ΔFosB was detected with rabbit polyclonal antibody (Cell Signaling Technology, cat.#9890, 1:1000) as described above. Lentivirally-delivered GFP and GFP-tagged GRK6 were detected with mouse monoclonal anti-GFP antibody (JL-8 Clontech, 1:500). Caspase-3 detected with rabbit polyclonal antibody (Cell Signaling Technology, cat.#9662, 1:1000) and histone deacetylase 1 (HDAC1, Cell Signaling Technology, cat.#2062, 1:1000) were used as markers of the cytosolic and nuclear fractions, respectively.
Data Analysis
For Western blots, the gray values of the bands were measured on X-ray film using Versadoc imaging system (Bio-Rad, Hercules, CA). Calibration curves were fitted to linear equations using Prism 4.0 (GraphPad Software, San Diego, CA) to ensure that all the samples were in the linear range and to reduce blot-to-blot variability, as previously described (Ahmed et al., 2010; Ahmed et al., 2007; Bychkov et al., 2007; Bychkov et al., 2010; Bychkov et al., 2011; Bychkov et al., 2013; Bychkov et al., 2008). For the statistical analysis StatView software (SAS Institute, Cary, NC) was used. The rat rotational behavior was analyzed by two-way repeated measure ANOVA with Protein (GFP and GRK6) as between and Session as within group factor. Alternatively, rotations within each testing session were analyzed by two-way repeated measure ANOVA with Protein (GFP and GRK6) as between and Bin (5 min testing intervals) as within group factor. The experiment with signaling pathways essentially followed full factorial design with Protein (GFP versus GRK6), Treatment (SALINE versus L-DOPA), and Challenge (Saline and L-DOPA), in addition to within group factor Hemisphere (Intact versus Lesioned). The interaction of Protein with Hemisphere was limited, since GFP or GRK6 were only expressed in the lesioned striatum. The full analysis was rarely informative. Western blot data were analyzed separately for each Treatment by repeated measure ANOVA with Hemisphere as repeated measure factor and Protein and Challenge as between group factors (Table S1). Since the effect of GRK6 was confined to the lesioned striatum, separate analysis of the data in the lesioned hemisphere by three-way ANOVA with Protein, Treatment, and Challenge yielded most meaningful results. In some cases, a separate analysis of the L-DOPA-challenged groups by two-way ANOVA with Protein and Treatment was also performed. The comparison of values in the intact and lesioned striata within each experimental group was performed with paired Student’s t-test. Post hoc comparisons of means were performed in some cases using Student’s t-test (for two-group comparisons). In all cases the value of p<0.05 was considered significant.
Results
The purpose of these experiments was to examine the effect of the elevated GRK6 concentration, which has been shown previously to have anti-LID effect in both rat and monkey models of LID (Ahmed et al., 2010), on the activity of the MAP kinase and Akt pathways in the lesioned striatum of hemiparkinsonian rats as a function of chronic L-DOPA treatment and L-DOPA challenge. The measurements in corresponding intact striata were used to evaluate the degree of supersensitivity of signaling responses.
Expression of GRK6 in the lesioned striatum suppresses L-DOPA-induced rotations
The lesion of the medial forebrain bundle brought about a very extensive loss of dopaminergic fibers in the striatum as determined by quantitative Western blotting for TH (Fig. 2A). Less than 2% of the TH level in the intact striatum remained following the lesion, and the degree of depletion did not differ among experimental groups. Injection of the GRK6-GFP lentivirus to the lesioned dorsolateral caudatoputamen yielded widespread infection of the striatal neurons (Fig. 2B). Hemiparkinsonian rats treated with saline displayed no rotational behavior (not shown). However, rats treated with L-DOPA displayed contralateral rotations, and chronic treatment with L-DOPA resulted in progressively increasing frequency indicative of behavioral sensitization to L-DOPA (Fig. 2C). We have employed the experimental design we used previously (Ahmed et al., 2010) that included pre-testing. Before the virus injection, the rats assigned to GFP and GRK6 groups displayed similar rotations (Fig. 2B). However, after virus injection, we detected significantly lower rotation frequencies in rats overexpressing GRK6 as compared to control rats across all testing sessions (F(1,252)=7.136, p=0.0124) (Fig. 2B). The slope of increase in the rotation frequency was less steep in GRK6-overexpressing group, as demonstrated by significant interaction between the Protein factor (GRK6 versus GFP) and Session (treated as repeated measure) (Protein X Session F(9,252)=6.7 p<0.0001). GRK6-overexpressing rats demonstrated no increase in the rotation frequency after the first 10 min during the last testing session (Day 10), whereas control rats increased their rotations until 60 min (Protein p=0.0043; Protein x Bin interaction p=0.0025 by two-way repeated measure ANOVA with Protein as between group and Bin as within group factors). These data demonstrate that the striatal overexpression of GRK6 suppresses L-DOPA-induced rotational behavior, as we reported previously (Ahmed et al., 2010).
Figure 2. Overexpression of GRK6 in the motor striatum suppresses L-DOPA-induced contralateral rotations in the hemiparkinsonian rat.

(A) The expression of GRK6-GFP in the rat lesioned striatum (upper image) detected with anti-GFP antibody as described in Methods. The same section was co-stained with anti-tyrosine hydroxylase (TH) antibody (lower image) to show that only the lesioned side received the GRK6 lentivirus. CPu, caudatoputamen, NAC – nucleus accumbens, Sept, septum, Ctx, cortex. (B) Loss of TH in the lesioned striatum. Upper panel shows representative Western blot for TH. Standards were produced by serial dilutions of the tissue lysate from control striata of several rats. The values are expressed in μg of total striatal protein per lane. Lower panel – blot for β-actin following stripping and re-probing the original TH blot demonstrating equal loading. (C) The graph shows net contralateral L-DOPA-induced rotations registered for 1 h in rats pre-sensitized with L-DOPA for 5 days before injection of the lentivirus encoding GFP or GRK6 and then tested with L-DOPA for 10 days post-injection. * - p<0.05, ** - p<0.01 between the GRK6 and control GFP groups, post hoc Student’s unpaired t-test following two-way repeated measure ANOVA with Protein (GFP versus GRK6) as between and Session as repeated measure factors. Note that both groups had similar rotation frequencies before virus injection, whereas after the injection GRK6-expressing rats demonstrated reduced rotational behavior. (D) The graph representing the distribution of net contralateral rotations across 5-min bins during the first (Day 1) and last (Day 10) 60 min testing sessions. * - p<0.05, ** - p<0.01, * - p<0.001 between the GRK6 and corresponding control GFP groups, post hoc Student’s unpaired t-test following two-way repeated measure ANOVA with Proteins (GFP versus GRK6) as between group and Bin as repeated measure factors. (E) The upper panel shows the expression of GFP in the lesioned striatum of GFP-expressing animals of all experimental groups. The middle panel shows the expression of endogenous GRK6 in both intact and lesioned striata of GFP-expressing rats and in the intact striatum of GRK6-overexpressing rats of all experimental groups detected with anti-GRK6 antibody. Serial dilutions of purified recombinant human GRK6 were used as standards (left 6 lanes). In the lesioned striatum of rats injected with GRK6 virus total (endogenous plus exogenous) GRK6 was detected. The arrow points to the GRK6 band (the lower thicker band is nonspecific). Lower images in both panels show blots for β-actin following stripping and re-probing the original blots to demonstrate equal loading. Lower panel shows a higher magnification of a part of the GRK6 blot to emphasize the differences in the GRK6 levels between the intact and lesioned striata in the GFP-expressing rat and between the lesioned striata in the GFP- and GRK6-expressing rats. (F) The graph presenting quantification of the levels of endogenous and total GRK6 in the lesioned caudate-putamen in rats injected with lentivirus encoding GFP or GRK6, as percent of values of respective intact hemispheres (means±S.E.M.). N=9–10 rats per group. * - p<0.01, * - p<0.05 to respective intact hemisphere by one-way repeated measure ANOVA separately for each group; # - p<0.001 to respective lesioned hemisphere in GFP-expressing rats by Student’s unpaired t-test.
We measured the absolute level of expression of exogenous GRK6 in comparison to endogenous GRK6 as well as the concentration of endogenous GRK6 in control and lesioned hemispheres of GFP-expressing and GRK6-overexpressing rats. In agreement with our previous results (Ahmed et al., 2010; Ahmed et al., 2007), the concentration of endogenous GRK6 was significantly reduced following 6-OHDA lesion, and its expression was not restored by chronic L-DOPA treatment (Fig. 2C, D). Injection of the GRK6 lentivirus into the lesioned striatum resulted in the infection of approximately 70% of neurons in the dorso-lateral CPu (motor striatum) as judged by the GFP expression. The infection lead to approximately two-fold elevation of the GRK6 level in the lesioned striatum as compared to GFP-expressing rats, with no significant differences among experimental groups (90.85±4.61% average increase). The degree of infection and overexpression was similar to that achieved in the previous study (Ahmed et al., 2010).
Expression of GRK6 in the lesioned striatum suppressed supersensitivity of the MAP kinase pathways
ERK1/2 activity
GPCRs regulate activity of the ERK pathway via multiple mechanisms (Goldsmith and Dhanasekaran, 2007). Acute L-DOPA challenge or chronic L-DOPA treatment had no significant effects in the intact striatum (Fig. 3A, B). Acute challenge with L-DOPA induced a strong ERK response in the DA-depleted striatum (Fig. 3A, B), both in saline- and L-DOPA-treated rats (Fig. 3A, B and Table S1), in agreement with previous reports (Bychkov et al., 2007; Gerfen et al., 2002; Santini et al., 2010; Santini et al., 2007). Accordingly, the analysis revealed highly significant effect of Hemisphere across the dataset as well as separately in saline-treated or L-DOPA-treated rats (p<0.0001) (Table S1). Since GRK6 was only expressed in the lesioned striatum, the analysis of its effect was only meaningful for the lesioned striatum. Thus, we have conducted a separate three-way ANOVA analysis in the lesioned striatum with Protein, Challenge, and Treatment as main factors. In the lesioned striatum, only L-DOPA challenge evoked a response but not saline, indicating that there was no change in the basal ERK signaling (Fig. 3A, B). Chronic L-DOPA treatment reduced the ERK1/2 response in the lesioned striatum to L-DOPA challenge (p=0.0083 for Treatment by three-way ANOVA). There was also significant Protein x Challenge interaction (p=0.0139, Table S1) indicating that the effect of GRK6 is only evident in L-DOPA-challenged rats. Thus, we analyzed the data for these groups separately by two-way ANOVA with Protein and Treatments as main factors. No effects of the factors were seen on saline-challenged rats. Upon L-DOPA challenge, the overall pERK1/2 levels were significantly lower in GRK6-expressing rats than in the GFP control upon L-DOPA-challenge across treatments (p=0.028). Although the difference appeared more evident in drug-naive than in L-DOPA-treated rats (Fig. 3A, B), Protein x Treatment interaction was not significant (p=0.2), indicating the GRK6 effect in both treatment groups. The comparison of GFP- and GRK6 expressing rats yielded significant difference only in the saline-treated L-DOPA-challenged group, which confirmed a trend towards a stronger effect of GRK6 in drug-naïve as compared to chronically L-DOPA-treated animals. No changes in the total level of ERK1/2 were detected (Fig. 3B and Table S1).
Figure 3. Overexpression of GRK6 in the striatum reduces ERK activation induced by acute L-DOPA.

(A) Western blot showing activation of ERK1/2 in the lesioned striatum of rats expressing GFP (control) or GRK6, chronically treated with saline or L-DOPA and challenged with either saline or L-DOPA (upper panel). Lower blot shows total ERK1/2 expression. (B) Quantification of Western blots of ERK1/2 activation by acute L-DOPA challenge in rats expressing GFP or GRK6 in the lesioned striatum and chronically treated with either saline (drug-naïve) of L-DOPA for 10 days. Rats were challenged with either L-DOPA or saline 45 min prior to sacrifice. N=9–10 rats per group. * - p<0.001 ** - p<0.01, to intact striatum, paired Student’s t-test; a –p<0.05 – to GFP-expressing animals by two-way ANOVA separately in the lesioned striatum in L-DOPA-challenged groups across treatments; # - p<0.05 to the GFP-expressing SALINE-treated L-DOPA-challenged group, lesioned striatum, group, unpaired Student’s post hoc t-test.
The p38 activity
The p38 MAP kinase, similarly to JNK, is activated by various stress factors (Cuadrado and Nebreda, 2010; Huang et al., 2009) and plays a role in fundamental physiological processes (Cuadrado and Nebreda, 2010; Huang et al., 2009; Keshet and Seger, 2010). Recently, a role for p38 signaling in regulating behavioral responses to the GPCR stimulation has been demonstrated (Bruchas et al., 2007; Bruchas et al., 2006). Similarly to ERK1/2, the level of phospho-p38 in the intact striatum did not change following L-DOPA challenge or chronic treatment. In the lesioned striatum, an elevated level of phospho-p38 as compared to the intact striatum was detected following saline challenge indicating enhanced basal activity of the p38 pathway associated with the loss of DA. L-DOPA challenge caused a significant increase in p38 phosphorylation as compared to saline challenge (p=0.0471 by three-way ANOVA separately in the lesioned striatum) (Fig. 4A, B and Table S1). Chronic L-DOPA treatment had no significant effect on p38 phosphorylation in the lesioned striatum (p=0.51 by three-way ANOVA). GRK6 expression suppressed p38 phosphorylation in the lesioned striatum across treatments and challenges (p=0.0463 by three-way ANOVA), and the interactions of the Protein factor with Challenge or Treatment were not significant (p=0.61 and 0.65, respectively) suggesting even effect of the GRK6 overexpression across all conditions (Fig. 4B and Table S1). Direct pairwise comparison of p-p38 levels in the lesioned striatum of the GFP- and GRK6-expressing animals yielded significant difference only in the saline-treated L-DOPA-challenged group (Fig. 4B) suggesting a trend towards a stronger effect of GRK6 in drug-naïve as compared to chronically L-DOPA-treated rats, similarly to that observed with pERK1/2. Interestingly, in contrast to ERK1/2, the 6-OHDA lesion upregulated the expression of p38 (Fig. 4A, C), whereas other factors had no effect on p38 expression. Increased phosphorylation of p38 in the lesioned striatum was not entirely due to elevated expression, since the ratio of phospho-p38 to total p38 significantly increased in the lesioned hemisphere across experimental conditions (p<0.001) (Fig. 4D and Table S1). There was no overall effect of Treatment or Challenge on the phospho-p38/total p38 ratio in the lesioned striatum. The overexpression of GRK6 significantly reduced the phospho-p38/total p38 ratio across groups (p=0.0375 by three-way ANOVA; no significant interactions) (Fig. 4D and Table S1) but only the difference between GFP- and GRK6-expressing saline-treated L-DOPA-challenged groups was significant in pairwise comparison suggesting that GRK6 tends to have a stronger effect in drug-naïve animals.
Figure 4. Overexpression of GRK6 in the striatum reduces p38 phosphorylation induced by acute L-DOPA.

(A) Western blots showing activation of p38 in the lesioned striatum of rats expressing GFP (control) or GRK6, chronically treated with saline or L-DOPA, and challenged with either saline or L-DOPA (upper panel). Lower blot shows total p38 expression. (B) Western blots showing activation of p38 in the lesioned striatum of rats expressing GFP (control) or GRK6, chronically treated with saline or L-DOPA and challenged with either saline or L-DOPA. (C) Quantification of the Western blot data for total p38 expression. (D) Ratio of phospho-p38 to total p38 calculated for each animal quantified across experimental groups. N=9–10 rats per group. * - p<0.001, ** - p<0.01, * - p<0.5, to intact striatum, paired Student’s t-test; a – p<0.05 – to GFP-expressing animals by three-way ANOVA separately in the lesioned striatum across treatments and challenges; # - p<0.05 to the GFP-expressing SALINE-treated L-DOPA-challenged group, lesioned striatum, group, unpaired Student’s post hoc t-test.
The JNK1/2/3 activity
We have examined the activity of the third member of the MAP kinase family, JNK, in the condition of DA depletion and L-DOPA treatment. We were unable to detect phosphorylated or unphosphorylated JNK3 isoform. The ubiquitous JNK1 and JNK2 are each represented by two shorter (46 kDa) isoforms derived by alternative splicing, JNK1α1, JNK1β1, JNK2α1, and JNK2β1, and two longer (54 kDa) isoforms, JNK1α2, JNK1β2, JNK2α2, and JNK2β2 (Gupta et al., 1996). The striatal phospho-JNK1/2 was mostly represented by p46 isoforms, whereas total levels of p54 and p46 isoforms were similar (Fig. 5). We found that, in contrast to ERK1/2 and p38, the activity of JNK was unaffected by the loss of DA, chronic L-DOPA treatment, or L-DOPA challenge (Fig. 5 and Table S1). The GRK6 expression also did not alter the level of pJNK1/2 (Fig. 5 and Table S1). Similarly to pJNK, we found no differences in the level of total JNK among experimental conditions (Fig. 5 and Table S1).
Figure 5. Overexpression of GRK6 in the striatum has no effect on the phosphorylation or expression of JNK isoforms in hemiparkinsonian rats.

Western blot images showing activation of JNK1/2 in the lesioned striatum of rats expressing GFP (control) or GRK6, chronically treated with saline or L-DOPA and challenged with either saline or L-DOPA (upper panel). Lower blot shows total JNK1/2 expression.
Expression of GRK6 modulated the activity of the Akt pathway
Phosphorylation of Akt at Thr308
Akt phosphorylation at Thr308 confers almost full activation (Alessi et al., 1996; Hers et al., 2011). We have previously shown that the Akt pathway is rendered supersensitive upon DA depletion, and this supersensitivity is supported by chronic L-DOPA treatment (Bychkov et al., 2007). Similarly to p38, the level of pAkt(Thr308) was slightly elevated in the lesioned hemisphere even without L-DOPA challenge, resulting in highly significant effect of Hemisphere in both saline- and L-DOPA-treated rats (p<0.0001) (Fig. 6A, B and Table S1). However, in contrast to ERK1/2, the effect of L-DOPA challenge on Akt phosphorylation at Thr308 in the lesioned striatum was not desensitized by chronic L-DOPA treatment (Treatment p=0.19 by three-way ANOVA). The effect of Challenge remained highly significant (p=0.003) without Challenge x Treatment interaction (p=0.5). There was significant Protein x Challenge interaction (p=0.039, Protein effect p=0.1) suggesting the dependence of the GRK6 effect on the L-DOPA challenge. The separate analysis of the L-DOPA-challenged groups by two-way ANOVA with Protein and Treatment as main factors yielded significant effect of Protein (p=0.0146), no effect of Treatment (p=0.18), and no Protein x Treatment interaction (p=0.46). Thus, the expression of GRK6 in the lesioned striatum significantly reduced the level of Akt(Thr308) phosphorylation in both saline- and L-DOPA-treated rats challenged with L-DOPA. (Fig. 6A, B and Table S1).
Figure 6. Overexpression of GRK6 in the lesioned striatum reduces phosphorylation of Akt at Thr308.

(A) Western blots showing activation of Akt at Thr308 (upper panel) and Ser473 (middle panel) in the lesioned striatum of rats expressing GFP (control) or GRK6, chronically treated with saline or L-DOPA and challenged with either saline or L-DOPA. Lower blot shows total Akt expression. (B) Quantification of Western blots of Akt phosphorylation at Thr308 and Ser473 following acute challenge with L-DOPA or saline in rats expressing GFP or GRK6 in the lesioned striatum and treated with either saline (drug-naïve) of L-DOPA for 10 days. N=9–10 rats per group. * - p<0.001, ** - p<0.01, * - p<0.05 to intact striatum, paired Student’s t-test; a – p<0.05 – to GFP-expressing animals by two-way ANOVA separately in the lesioned striatum in L-DOPA-challenged groups across treatments.
Phosphorylation of Akt at Ser473
Phosphorylation of Akt at the second activating residue, Ser473, yields a much smaller increase in the Akt activity than phosphorylation at Thr308 (5-fold versus 100-fold) (Alessi et al., 1996; Hers et al., 2011). Akt phosphorylation at Ser473 facilitates phosphorylation at Thr308 and may affect not only Akt activity, but also its substrate specificity (Hers et al., 2011; Jacinto et al., 2006; Sarbassov et al., 2005). In contrast to Akt phosphorylation at Thr308, Akt phosphorylation at Ser473 was not significantly altered by Protein, Treatment, or Challenge. The only significant effect revealed by the three-way repeated measure ANOVA analysis of the dataset was that of Hemisphere both in saline- and L-DOPA-treated rats (p<0.001) due to elevated level of Akt(Ser473) phosphorylation in the lesioned striatum in all experimental groups (Fig. 6A, B and Table S1). The expression of GRK6 in the lesioned striatum had no effect on the level of Akt phosphorylation at Ser473. There were no significant changes in the total concentration of Akt in the experimental groups (Fig. 6A and Table S1).
Overexpression of GRK6 suppressed accumulation of ΔFosB
We have examined the accumulation of transcription factor ΔFosB, a known marker of signaling distortions caused by chronic L-DOPA (Cao et al., 2010; Pavon et al., 2006). ΔFosB has been shown to slowly accumulate in the DA-depleted striatum in the course of chronic L-DOPA treatment in parallel with the development of LID (Cao et al., 2010; Pavon et al., 2006). We have measured the level of ΔFosB in the lesioned striatum in saline- and L-DOPA-treated rats expressing GFP or GRK6 and challenged with saline or L-DOPA. The intact striatum was not examined in all rats, since it displayed essentially no ΔFosB immunoreactivity (Fig. 7A). The ΔFosB levels in the lesioned striatum were also quite low in the saline-treated groups regardless of the challenge or GFP/GRK6 expression (Fig. 7A, B). However, chronic L-DOPA treatment dramatically upregulated ΔFosB concentration (F(1,64)=162, p<0.0001 by three-way ANOVA with Protein, Treatment, and Challenge analyzed separately in the lesioned hemisphere). Acute L-DOPA challenge had no effect (p=0.33). The expression of GRK6 in the lesioned striatum of rats chronically treated with L-DOPA significantly suppressed the ΔFosB accumulation irrespective of the challenge (p=0.0075; Fig. 7A, B).
Figure 7. Overexpression of GRK6 in the lesioned striatum reduces L-DOPA-induced accumulation of ΔFosB.

(A) Representative Western blots showing ΔFosB accumulation in the lesioned striatum of rats expressing GFP (control) or GRK6, chronically treated with saline or L-DOPA and challenged with either saline or L-DOPA. Note that there is no ΔFosB expression in the intact striatum regardless of treatment or challenge. Arrows point to the two ΔFosB isoforms. Lower panel – Western blot for β-actin produced by stripping the original ΔFosB blot and reprobing with β-acting antibody. (B) Quantification of Western blots of ΔFosB expression in the lesioned striatum following acute challenge with L-DOPA or saline in rats expressing GFP or GRK6 treated with either saline (drug-naïve) of L-DOPA for 10 days. N=9–10 rats per group. * - p<0.05 to GFP-expressing L-DOPA-treated groups across challenges by two-way ANOVA with Protein and Challenge. (C) Detection of ΔFosB accumulation in the nuclei as compared to the cytosolic fractions of the intact and lesioned striata of rats expressing GFP or GRK6-GFP, chronically treated with saline or L-DOPA. Nuclear and cytosolic fractions were isolated as described in Methods. Representative Western blot of nuclear and cytosolic (7.5 μg protein per lane) were probed for ΔFosB, and then blots were stripped and re-probed for caspase-3 and histone deacetylase 1 (HDAC1) to demonstrate equal loading of the samples in both fractions. Lentivirally-delivered GFP and GRK6-GFP were detected with anti-GFP antibody on a separate blot (seen only in the cytosolic fraction as expected).
In order to determine whether accumulation of ΔFosB in chronically L-DOPA-treated animals occurs in the nucleus, we have used a separate cohort of rats that received injections of GFP or GRK6-GFP and were chronically treated with L-DOPA for 10 days in the regular regiment as described in Methods. We found that most of the accumulation of the ΔFosB immunoreactivity in the lesioned striatum is detected in the nuclear fraction (Fig. 7C).
Discussion
We have previously reported that overexpression of GRK6 in the DA-depleted striatum of hemiparkinsonian rats or parkinsonian monkeys alleviated LID (Ahmed et al., 2010). Overexpressed GRK6 also reduced the degree of upregulation of dynorphin and enkephalin mRNA and the increase in the D3 DA receptor concentration (Ahmed et al., 2010). These molecular changes are well-known markers of LID, although their causal connection with LID development has never been convincingly demonstrated. Furthermore, in GRK6 expressing rats, we observed normalized internalization and trafficking of the D1 DA receptor known to be defective in LID (Ahmed et al., 2010; Guigoni et al., 2007). The data suggested that improved behavior and normalized molecular markers in GRK6-overexpressing animals were likely due to improved functioning of the receptor desensitization machinery. In this study, we set to examine whether overexpression of GRK6 in the lesioned striatum in hemiparkinsonian rats would normalize the function of the signaling pathways known to be deregulated following DA depletion and/or chronic L-DOPA treatment. Our data show that facilitation of DA receptor desensitization in the DA depleted striatum via increased availability of GRK6 ameliorated signaling supersensitivity.
Several signaling pathways that are not normally activated by DA (or L-DOPA) show strong activation by L-DOPA or other dopaminergic agonists following DA depletion. The ERK Map kinase is the best-studied pathway in the context of DA depletion and LID mechanisms. The supersensitivity of the ERK pathway to dopaminergic stimulation following DA depletion has been reported by many laboratories, and elevated ERK activity has been linked to LID development (Alcacer et al., 2012; Bezard et al., 2005; Bychkov et al., 2007; Gerfen et al., 2002; Santini et al., 2012; Santini et al., 2010; Santini et al., 2007). However, the exact role of the ERK deregulation in the LID development is still unclear. Moreover, chronic treatment with L-DOPA required for LID development desensitizes the ERK response, making it more likely that high ERK activity is associated with priming rather than with LID expression per se (Bezard et al., 2005; Bychkov et al., 2007; Gerfen et al., 2002; Santini et al., 2010). Since priming is an important component of LID pathophysiology, reducing the ERK hyperactivity may have an anti-LID effect. Successful attempt has been made to ameliorate LID by controlling the activity of the neuron-specific ERK1/2 activator Ras-GRF1 factor (Fasano et al., 2010). Here we found that, expression of GRK6 in the lesioned striatum significantly suppressed ERK1/2 activation in response to L-DOPA challenge. We also found, in agreement with previous reports (Bezard et al., 2005; Brown et al., 2005; Bychkov et al., 2007; Kim et al., 2006; Santini et al., 2010), that chronic L-DOPA treatment reduced the degree of ERK supersensitivity to L-DOPA challenge. The effect of GRK6 was strong in drug-naïve animals but diminished with chronic L-DOPA treatment. In fact, GRK6 brought down the ERK1/2 response in drug-naïve animals to the level of the desensitized ERK1/2 response in chronically L-DOPA-treated animals.
In contrast to the ERK, the p38 pathway has never been reported to be activated by L-DOPA in the DA-depleted striatum. The role of the p38 pathway in general has not previously attracted attention in connection to LID, although evidence exists as to the role of the p38 pathway in neuronal death in PD (Wang et al., 2012). We detected elevated basal activity of p38 kinase in the lesioned striatum and found that p38, similarly to ERK1/2, was rendered supersensitive to L-DOPA challenge by DA depletion. In saline-treated animals, elevated level of phospho-p38 in the lesioned striatum was reflective of increased p38 expression, since phospho-p38 to total p38 ratio was unchanged. In L-DOPA-treated animals, the ratio was elevated to a comparable degree in saline- and L-DOPA challenged rats, suggesting increased basal activity of the p38 pathway in the course of chronic L-DOPA treatment. Thus, elevated p38 activity may be associated not only with peak-dose LID, as is the case with ERK1/2, but with the dyskinetic state itself. In contrast to ERK, chronic L-DOPA did not significantly desensitize the p38 response to L-DOPA challenge. The overexpression of GRK6 reduced the degree of the p38 supersensitivity across experimental groups, including reducing the basal activity in the L-DOPA-treated rats, but the effect was most noticeable in drug-naïve L-DOPA-challenged animals.
We have previously reported that loss of DA in the striatum resulted in a sustained elevation of the responsiveness of the Akt pathway to dopaminergic stimulation, which was not desensitized by chronic L-DOPA (Bychkov et al., 2007). Here we confirmed these findings and further demonstrated that hyperphosphorylation of Akt at the main activating residue Thr308 in response to L-DOPA challenge in both drug-naïve and L-DOPA-treated rats was ameliorated by overexpression of GRK6, similarly to the effect seen on ERK1/2 and p38 activation. Therefore, our data demonstrate that by overexpressing GRK6, which presumably facilitated desensitization of DA receptors in the lesioned striatum, it was possible to partially normalize signaling via at least three pathways, ERK, p38, and Akt, all of which became supersensitive to DA upon DA depletion.
Enhanced ERK response to DA stimulation in the DA-depleted striatum has been linked to the D1 receptor supersensitivity (Gerfen et al., 2002; Santini et al., 2009; Santini et al., 2007; Westin et al., 2007). The elevated GRK6 likely reduced the ERK activation by L-DOPA via facilitation of the D1 receptor desensitization and normalization of the D1 intracellular trafficking and regulation (Ahmed et al., 2010). The supersensitivity of the p38 or Akt pathways has not yet been attributed to a specific receptor. However, it is reasonable to conclude that they are also normalized via improved receptor desensitization. GRKs are believed to have rather broad receptor specificity, although detailed information for individual GRK isoforms is lacking (for review see (Gurevich et al., 2011)). GRK6 has been shown to preferentially regulate the D2 DA receptor in vivo (Gainetdinov et al., 2003). Our previous data demonstrated that in the conditions of DA depletion GRK6 effectively modulates the signaling and trafficking of the supersensitive D1 receptor (Ahmed et al., 2010). There is no information whether GRK6 regulates other GPCRs expressed by striatal neurons and involved in the movement control and/or LID, such as adenosine A1 (Xiao et al., 2011) and A2A (Armentero et al., 2011; Fredduzzi et al., 2002), serotonin 5-HT1A (Bibbiani et al., 2001; Dupre et al., 2008; Dupre et al., 2011; Dupre et al., 2013; Iderberg et al., 2013) and 5-HT1B (Iderberg et al., 2013; Zhang et al., 2008), or opioid (Chen et al., 2005; Cox et al., 2007; Henrya et al., 2001) receptors in vitro or in vivo.
The concentration of GRK6, along with other GRK isoforms, is reduced following the loss of DA (Ahmed et al., 2010; Ahmed et al., 2007), which could contribute to faulty receptor desensitization and enhanced signaling. Although the degree of reduction in case of GRK6 is not large (25–40%), it could be quite functionally significant in view of the demand created by huge surge of DA produced from L-DOPA. Furthermore, hemizygous GRK6 knockout mice expressing approximately 50% of GRK6 display full phenotype when treated with psychostimulant drugs (Gainetdinov et al., 2003) suggesting that the concentration of GRK6 is critically important for the normal functioning of the DA receptors, particularly in the conditions of the dopaminergic overload. In the MPTP-lesioned monkey, the concentration of GRK6 is not reduced by loss of DA (Bezard et al., 2005), but still additional GRK6 ameliorates LID and normalizes the D1 receptor trafficking and signaling (Ahmed et al., 2010), suggesting that the GRK6 level is insufficient to meet the demand at the peak-dose LID conditions. Exogenous GRK6 likely compensates for insufficient level of endogenous GRKs facilitating receptor desensitization and normalizing the receptor responsiveness to dopaminergic challenge.
Chronic L-DOPA treatment appeared to diminish the normalizing effect of GRK6 on signaling, most obviously in case of ERK. This was a somewhat unexpected finding. It seems that GRK6 suppressed acute signaling effects of L-DOPA, thus alleviating peak-dose LID, but not the long-term L-DOPA effects that predispose the animals to LID. This conclusion is consistent with the fact that overexpression of GRK6 diminished locomotor sensitization to L-DOPA, but does not abolish it. Sensitization still occurs, albeit at a reduced rate. The reduction is possibly due to weakened priming effect of every dose of L-DOPA. Chronic L-DOPA has no further effect on the GRK6 concentration reduced by DA depletion [(Ahmed et al., 2007), see also present work]. Therefore, GRK6 overexpression should be expected to normalize signaling even following chronic L-DOPA treatment, which was only partially the case. It is conceivable that overexpressed GRK6 facilitates acute desensitization of DA receptors, whereas upon chronic L-DOPA additional factors are introduced either not under GRK6 control or affected by GRK6 in a different manner. Weakened priming effect of every single L-DOPA dose accumulated over the course of the chronic treatment eventually may result in reduced gene expression associated with LID that we have reported previously (Ahmed et al., 2010). Reduced potency of every dose of the drug may also be the cause of lower accumulation of transcription factor ΔFosB in the lesioned striatum of GRK6-overexpressing rats observed in this study. ΔFosB has been shown to directly contribute to the development of LID (Berton et al., 2009; Cao et al., 2010; Engeln et al., 2014). A lower level of ΔFosB in GRK6-overexpressing rats is consistent with the reduced rate of behavioral sensitization and LID in these animals. We also show that the increase in the ΔFosB concentration in the lesioned striatum occurs in the nuclei fraction, and this accumulation is blunted by GRK6.
LID remains an unmet medical need in the management of PD severely affecting the efficacy of treatment and the patients’ quality of life. Advanced degeneration of dopaminergic innervation observed shortly after diagnosis of PD (Kordower et al., 2013) makes the improvement of existing symptomatic therapies, in addition to the development of neuroprotective therapies, critical. In recent years, investigations uncovered numerous signaling alterations associated with LID [reviewed in (Cenci and Konradi, 2010; Gurevich and Gurevich, 2008; Murer and Moratalla, 2011)]. Reduced supersensitivity of the ERK pathway and reduced accumulation of ΔFosB have been shown to lead to amelioration of LID (Berton et al., 2009; Cao et al., 2010; Fasano et al., 2010). However, the role of most signaling pathways deregulated by loss of DA and/or chronic L-DOPA in LID simply has not been explored. Furthermore, many signaling pathways that operate in the striatum have never been investigated in connection with PD or LID. Thus, the sheer number of signaling pathways potentially involved in LID makes an informed choice of proper targets for anti-LID therapy difficult. It also might be necessary to normalize most of them to achieve sustained anti-LID benefits, and it is unclear how to accomplish that. The root cause of signaling changes following DA depletion and chronic L-DOPA treatment seems to be abnormal signaling via DA receptors. Managing signaling at the receptor level may be a more effective way of normalizing multiple pathways and bringing about anti-LID benefits than targeting each pathway separately. The data presented here together with our earlier observations (Ahmed et al., 2010) demonstrate that facilitation of receptor desensitization leads to improvement in the function of multiple signaling pathways, amelioration of molecular abnormalities associated with LID, together with reduction of LID at the behavioral level. The results have further support the notion that GRK6 is an attractive target for anti-LID therapy and that it may offer multiple therapeutic benefits unattainable by directly targeting signaling pathways.
Supplementary Material
Highlights.
Signaling was examined in drug-naïve and DOPA-treated hemiparkinsonian rats
We document the activity of signaling pathways upon striatal overexpression of GRK6
GRK6 reduced pERK in the lesioned striatum in response to L-DOPA challenge
GRK6 also reduced supersensitive p38, and Akt responses
GRK6 reduced accumulation of ΔFosB upon chronic L-DOPA treatment
Acknowledgments
We are grateful to Dr. John Tesmer (University of Michigan at Ann Arbor) for purified GRK6. We thank Mr. Yoni Carl for assistance with rat surgeries and virus preparation. The study was supported by NIH grants NS065868 and R21DA030103 (to EVG) and by GM059802 and Welch Foundation (F-1390) (to KND).
Footnotes
Abbreviations: 6-OHDA - 6-hydroxydopamine; DA – dopamine; GFP - green fluorescent protein; GPCRs - G protein-coupled receptors; GRK - G protein-coupled receptor kinase; LID – L-DOPA-induced dyskinesia; PD - Parkinson’s disease.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed MR, Berthet A, Bychkov E, Porras G, Li Q, Bioulac BH, Carl YT, Bloch B, Kook S, Aubert I, Dovero S, Doudnikoff E, Gurevich VV, Gurevich EV, Bezard E. Lentiviral overexpression of GRK6 alleviates L-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci Transl Med. 2010;2:28ra28. doi: 10.1126/scitranslmed.3000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MR, Bychkov ER, Gurevich VV, Benovic JL, Gurevich EV. Altered expression and subcellular distribution of GRK subtypes in the dopamine-depleted rat basal ganglia is not normalized by L-DOPA treatment. J Neurochem. 2007;104:1622–1636. doi: 10.1111/j.1471-4159.2007.05104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MR, Gurevich VV, Dalby KN, Benovic JL, Gurevich EV. Haloperidol and clozapine differentially affect the expression of arrestins, receptor kinases, and extracellular signal-regulated kinase activation. J Pharmacol Exper Ther. 2008;325:276–83. doi: 10.1124/jpet.107.131987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcacer C, Santini E, Valjent E, Gaven F, Girault JA, Hervé D. Gα(olf) mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci. 2012;32:5900–5910. doi: 10.1523/JNEUROSCI.0837-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Armentero MT, Pinna A, Ferré S, Lanciego JL, Müller CE, Franco R. Past, present and future of A(2A) adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol Ther. 2011;132:280–299. doi: 10.1016/j.pharmthera.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert I, Guigoni Hakansson K, Li Q, Dovero S, Barthe N, Bioulac BH, Gross CE, Fisone G, Bloch B, Bezard E. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol. 2005;57:17–26. doi: 10.1002/ana.20296. [DOI] [PubMed] [Google Scholar]
- Berton O, Guigoni C, Li Q, Bioulac BH, Aubert I, Gross CE, Dileone RJ, Nestler EJ, Bezard E. Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol Psychiatry. 2009;66:554–561. doi: 10.1016/j.biopsych.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis. 2005;18:323–335. doi: 10.1016/j.nbd.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Bibbiani F, Oh JD, Chase TN. Serotonin 5-HT1A agonist improves motor complications in rodent and primate parkinsonian models. Neurology. 2001;57:1829–1834. doi: 10.1212/wnl.57.10.1829. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Sotnikova TD, Medvedev IO, Lefkowitz RJ, Dykstra LA, Caron MG. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J Neurosci. 2003;23:10265–73. doi: 10.1523/JNEUROSCI.23-32-10265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ridray S, Carboni C, Diaz J, Sokoloff P, Schwartz JC. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Nat Acad Sci USA. 1997;94:3363–3367. doi: 10.1073/pnas.94.7.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci. 2005;22:247–256. doi: 10.1111/j.1460-9568.2005.04190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007;27:11611–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov E, Ahmed MR, Dalby KN, Gurevich EV. Dopamine depletion and subsequent treatment with L-DOPA, but not the long-lived dopamine agonist pergolide, enhances activity of the Akt pathway in the rat striatum. J Neurochem. 2007;102:699–711. doi: 10.1111/j.1471-4159.2007.04586.x. [DOI] [PubMed] [Google Scholar]
- Bychkov E, Ahmed MR, Gurevich EV. Sex differences in the activity of signalling pathways and expression of G-protein-coupled receptor kinases in the neonatal ventral hippocampal lesion model of schizophrenia. Int J Neuropsychopharmacol. 2010;17:1–15. doi: 10.1017/S1461145710000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov E, Ahmed MR, Gurevich VV, JLB, Gurevich EV. Reduced expression of G protein-coupled receptor kinases in schizophrenia but not in schizoaffective disorder. Neurobiol Dis. 2011;44:248–258. doi: 10.1016/j.nbd.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov E, Zurkovsky L, Garret M, Ahmed MR, Gurevich EV. Distinct cellular and subcellular distribution of G protein-coupled receptor kinase and arrestin isoforms in the striatum. PLoS One. 2013;7:e48912. doi: 10.1371/journal.pone.0048912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov ER, Gurevich VV, Joyce JN, Benovic JL, Gurevich EV. Arrestins and two receptor kinases are upregulated in Parkinson’s disease with dementia. Neurobiol Aging. 2008;29:379–396. doi: 10.1016/j.neurobiolaging.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Yasuda T, Uthayathas S, Watts RL, Mouradian MM, Mochizuki H, Papa SM. Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J Neurosci. 2010;30:7335–7343. doi: 10.1523/JNEUROSCI.0252-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Konradi C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res. 2010;183:209–233. doi: 10.1016/S0079-6123(10)83011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Björklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr Protoc Neurosci. 2007;9 doi: 10.1002/0471142301.ns0925s41. [DOI] [PubMed] [Google Scholar]
- Chen L, Togasaki DM, Langston JW, Di Monte DA, Quik M. Enhanced striatal opioid receptor-mediated G-protein activation in L-DOPA-treated dyskinetic monkeys. Neuroscience. 2005;132:409–420. doi: 10.1016/j.neuroscience.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism by chronic treatment with L-dopa. N Engl J Med. 1969;280:337–345. doi: 10.1056/NEJM196902132800701. [DOI] [PubMed] [Google Scholar]
- Cox H, Togasaki DM, Chen L, Langston JW, Di Monte DA, Quik M. The selective kappa-opioid receptor agonist U50,488 reduces L-dopa-induced dyskinesias but worsens parkinsonism in MPTP-treated primates. Exp Neurol. 2007;205:101–107. doi: 10.1016/j.expneurol.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry. 2009;66:603–613. doi: 10.1016/j.biopsych.2009.04.025. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Ann Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Dupre KB, Eskow KL, Barnum CJ, Bishop C. Striatal 5-HT1A receptor stimulation reduces D1 receptor-induced dyskinesia and improves movement in the hemiparkinsonian rat. Neuropharmacology. 2008;55:1321–1328. doi: 10.1016/j.neuropharm.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre KB, Ostock CY, Eskow Jaunarajs KL, Button T, Savage LM, Wolf W, Bishop C. Local modulation of striatal glutamate efflux by serotonin 1A receptor stimulation in dyskinetic, hemiparkinsonian rat. Exp Neurol. 2011;229:288–299. doi: 10.1016/j.expneurol.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre KB, Ostock CY, George JA, Eskow Jaunarajs KL, Hueston CM, Bishop C. Effects of 5-HT receptor stimulation on D1 receptor agonist-induced striatonigral activity and dyskinesia in hemiparkinsonian rats. ACS Chem Neurosci. 2013;4:747–760. doi: 10.1021/cn300234z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeln M, Bastide MF, Toulmé E, Dehay B, Bourdenx M, Doudnikoff E, Li Q, Gross CE, Boué-Grabot E, Pisani A, Bezard E, POF Selective Inactivation of Striatal FosB/ΔFosB-Expressing Neurons Alleviates L-Dopa-Induced Dyskinesia. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.07.007. In press. [DOI] [PubMed] [Google Scholar]
- Fasano S, Bezard E, D’Antoni A, Francardo V, Indrigo M, Qin L, Doveró S, Cerovic M, Cenci MA, Brambilla R. Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc Natl Acad Sci USA. 2010;107:21824–21829. doi: 10.1073/pnas.1012071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredduzzi S, Moratalla R, Monopoli A, Cuellar B, Xu K, Ongini E, Impagnatiello F, Schwarzschild MA, Chen JF. Persistent behavioral sensitization to chronic L-DOPA requires A2A adenosine receptors. J Neurosci. 2002;22:1054–1062. doi: 10.1523/JNEUROSCI.22-03-01054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, Torres GE, Kim KM, Lefkowitz RJ, Caron MG, Premont RT. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron. 2003;38:291–303. doi: 10.1016/s0896-6273(03)00192-2. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. Dopamine-mediated gene regulation in models of Parkinson’s disease. Ann Neurol. 2000;47(Suppl):S42–S50. [PubMed] [Google Scholar]
- Gerfen CR, Miyachi S, Paletzki R, Brown P. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2 kinase. J Neurosci. 2002;22:5042–5054. doi: 10.1523/JNEUROSCI.22-12-05042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Aubert I, Li Q, Gurevich VV, Benovic JL, Ferry S, Mach U, Stark H, Leriche L, Hakansson K, Bioulac BH, Gross CE, Sokoloff P, Fisone G, Gurevich EV, Bloch B, Bezard E. Pathogenesis of levodopa-induced dyskinesia: focus on D1 and D3 dopamine receptors. Parkinsonism Relat Disord. 2005a;11(Suppl 1):S25–9. doi: 10.1016/j.parkreldis.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Doudnikoff E, Li Q, Bloch B, Bezard E. Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol Dis. 2007;26:452–463. doi: 10.1016/j.nbd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Dovero S, Aubert I, Qin L, Bioulac BH, Bloch B, Gurevich EV, Gross CE, Bezard E. Levodopa-induced dyskinesia in MPTP-treated macaque is not dependent of the extent and pattern of the nigrostrial lesion. Eur J Neurosci. 2005b;22:283–287. doi: 10.1111/j.1460-9568.2005.04196.x. [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006a;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Dopamine receptors and the treatment of Parkinson’s disease. In: Neve KA, editor. Dopamine Receptors. Humana Press; Portland, OR: 2008. pp. 525–584. [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol Ther. 2011;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharm Ther. 2006b;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrya B, Foxb SH, Crossmanb AR, Brotchieb JM. μ- and δ-Opioid Receptor Antagonists Reduce Levodopa-Induced Dyskinesia in the MPTP-Lesioned Primate Model of Parkinson’s Disease. Exp Neurol. 2001;171:139–146. doi: 10.1006/exnr.2001.7727. [DOI] [PubMed] [Google Scholar]
- Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1215–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009;48:161–169. doi: 10.1016/j.cyto.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Rockman HA, Shotwell KF, Tomhave ED, Koch WJ. Myocardial overexpression of GRK3 in transgenic mice: evidence for in vivo selectivity of GRKs. Am J Physiol. 1998;275:1298–1306. doi: 10.1152/ajpheart.1998.275.4.H1298. [DOI] [PubMed] [Google Scholar]
- Iderberg H, Rylander D, Bimpisidis Z, Cenci MA. Modulating mGluR5 and 5-HT1A/1B receptors to treat l-DOPA-induced dyskinesia: effects of combined treatment and possible mechanisms of action. Exp Neurol. 2013;250:116–124. doi: 10.1016/j.expneurol.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- Kim DS, Palmiter RD, Cummins A, Gerfen CR. Reversal of supersensitive striatal dopamine D1 receptor signaling and extracellular signal-regulated kinase activity in dopamine-deficient mice. Neuroscience. 2006;137:1381–1388. doi: 10.1016/j.neuroscience.2005.10.054. [DOI] [PubMed] [Google Scholar]
- Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem. 2001;276:37409–37414. doi: 10.1074/jbc.M106728200. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodowski DT, Tesmer VM, Benovic JL, Tesmer JJ. The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem. 2006;281:16785–93. doi: 10.1074/jbc.M601327200. [DOI] [PubMed] [Google Scholar]
- Menard L, Ferguson SS, Zhang J, Lin FT, Lefkowitz RJ, Caron MG, Barak LS. Synergistic regulation of beta2-adrenergic receptor sequestration: intracellular complement of beta-adrenergic receptor kinase and beta-arrestin determine kinetics of internalization. Mol Pharmacol. 1997;51:800–808. [PubMed] [Google Scholar]
- Murer MG, Moratalla R. Striatal Signaling in L-DOPA-Induced Dyskinesia: Common Mechanisms with Drug Abuse and Long Term Memory Involving D1 Dopamine Receptor Stimulation. Front Neuroanat. 2011;5:51. doi: 10.3389/fnana.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian A, Gurevich VV, Gurevich EV. The Origin and Evolution of G Protein-Coupled Receptor Kinases. PLoS One. 2012;7:e33806. doi: 10.1371/journal.pone.0033806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadjar A, Gerfen CR, Bezard E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: a feature inherent to the treatment or the disease? Prog Neurobiol. 2009;87:1–9. doi: 10.1016/j.pneurobio.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- Pavon N, Martin AB, Mendialdua A, Moratalla R. ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry. 2006;59:64–74. doi: 10.1016/j.biopsych.2005.05.044. [DOI] [PubMed] [Google Scholar]
- Sabatakos G, Rowe GC, Kveiborg M, Wu M, Neff L, Chiusaroli R, Philbrick WM, Baron R. Doubly truncated FosB isoform (Δ2ΔFosB) induces osteosclerosis in transgenic mice and modulates expression and phosphorylation of Smads in osteoblasts independent of intrinsic AP-1 activity. J Bone Moner. 2008;23:584–595. doi: 10.1359/JBMR.080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Alcacer C, Cacciatore S, Heiman M, Hervé D, Greengard P, Girault JA, Valjent E, Fisone G. L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem. 2009;108:621–633. doi: 10.1111/j.1471-4159.2008.05831.x. [DOI] [PubMed] [Google Scholar]
- Santini E, Feyder M, Gangarossa G, Bateup HS, Greengard P, Fisone G. Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism. J Biol Chem. 2012;287:27806–27812. doi: 10.1074/jbc.M112.388413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Sgambato-Faure V, Li Q, Savasta M, Dovero S, Fisone G, Bezard E. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS One. 2010;5:e12322. doi: 10.1371/journal.pone.0012322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault J-A, Herve D, Greengard P, Fisone G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. 2007;27:6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sgambato-Faure V, Buggia V, Gilbert F, Levesque D, Benabid AL, Berger F. Coordinated and spatial upregulation of arc in striatonigral neurons correlates with L-dopa-induced behavioral sensitization in dyskinetic rats. J Neuropathol Exp Neurol. 2005;64:936–947. doi: 10.1097/01.jnen.0000186922.42592.b7. [DOI] [PubMed] [Google Scholar]
- Stocchi F, Nordera G, Marsden CD. Strategies for treating patients with advanced Parkinson’s disease with disastrous fluctuations and dyskinesias. Clin Neuropharmacol. 1997;20:95–115. doi: 10.1097/00002826-199704000-00001. [DOI] [PubMed] [Google Scholar]
- Waas WF, Rainey MA, Szafranska AE, Dalby KN. Two rate-limiting steps in the kinetic mechanism of the serine/threonine specific protein kinase ERK2: a case of fast phosphorylation followed by fast product release. Biochemistry. 2003;42:12273–12286. doi: 10.1021/bi0348617. [DOI] [PubMed] [Google Scholar]
- Wang G, Pan J, Chen SD. Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson’s disease. Progr Neurobiol. 2012;98:207–221. doi: 10.1016/j.pneurobio.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry. 2007;62:800–810. doi: 10.1016/j.biopsych.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willets JM, Nash MS, Challiss RA, Nahorski SR. Imaging of muscarinic acetylcholine receptor signaling in hippocampal neurons: evidence for phosphorylation-dependent and -independent regulation by G-protein-coupled receptor kinases. J Neurosci. 2004;24:4157–62. doi: 10.1523/JNEUROSCI.5506-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willets JM, Parent JL, Benovic JL, Kelly E. Selective reduction in A2 adenosine receptor desensitization following antisense-induced suppression of G protein-coupled receptor kinase 2 expression. J Neurochem. 1999;73:1781–1789. doi: 10.1046/j.1471-4159.1999.0731781.x. [DOI] [PubMed] [Google Scholar]
- Xiao D, Cassin JJ, Healy B, Burdett TC, Chen JF, Fredholm BB, Schwarzschild MA. Deletion of adenosine A1 or A(2A) receptors reduces L-3,4-dihydroxyphenylalanine-induced dyskinesia in a model of Parkinson’s disease. Brain Res. 2011;1367:310318. doi: 10.1016/j.brainres.2010.08.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Zhang X, Andren PE, Greengard P, Svenningsson P. Evidence for a role of the 5-HT1B receptor and its adaptor protein, p11, in l-DOPA treatment of an animal model of Parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2163–2168. doi: 10.1073/pnas.0711839105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.