

Abstract
Iron is critical to the survival of almost all living organisms. However, inappropriately low or high levels of iron are detrimental and contribute to a wide range of diseases. Recent advances in the study of iron metabolism have revealed multiple intricate pathways that are essential to the maintenance of iron homeostasis. Further, iron regulation involves processes at several scales, ranging from the subcellular to the organismal. This complexity makes a systems biology approach crucial, with its enabling technology of computational models based on a mathematical description of regulatory systems. Systems biology may represent a new strategy for understanding imbalances in iron metabolism and their underlying causes.
Keywords: hydroxyl radical, heme, phagocytose, constant decay rate, Petri nets, homeostasis, transferrin, erythroid bone marrow, erythrocytes, phagocytosis, plasma, continuous vs. discrete models
Introduction
Dysregulation of iron metabolism plays a role in a wide range of diseases [1], and understanding this role is crucial in the search for therapeutics. Fortunately, over the last decade, some key mechanisms involved in iron regulation have been uncovered, and a more complete picture of iron regulation is starting to emerge [2]. Complicating the matter, however, is the fact that iron regulation involves processes at scales ranging from the organism to subcellular compartments, each of which interacts with the others. At each scale, the control system uses several intertwined feedback loops that also cross scales. Thus, it is crucial that, on the one hand, we understand iron metabolism as a multi-scale control system, and, on the other hand, we move beyond a purely descriptive static characterization of this control system.
Systems biology provides an approach and tool set to address both of these requirements. Systems biology integrates individual components of a system by tying them together through their interactions. This is done through the use of computational models that are capable of synthesizing all the different interactions between components into a dynamical system that captures global dynamic behavior. In particular, such a dynamical system representation allows the integration of regulatory events at different scales. System dynamics can then be probed using the computational model as a virtual laboratory, for the purpose of formulating hypotheses that can then be validated in the laboratory.
This review begins with a description of iron metabolism at the systemic and intracellular scales and discusses some of the most important diseases involving dysregulation of iron. We then take a systems biology view and describe some of the computational models of iron metabolism at both scales. Our aim is to present several different approaches to the construction of computational models, and the advantages and disadvantages of the different methods.
Iron Metabolism
The earliest accounts of iron being present in blood date back to as early as the 18th century, but it was not until the late 1930s that the first accounts of iron metabolism at the molecular level emerged and not until 1958 that the first comprehensive review of iron absorption was published [3,4]. More recently, key findings have shaped our current view of iron metabolism. These include the discovery of TfR, the transferrin receptor, in the 1970s, the discovery of the IRE/IRP regulatory axis in the 1980s, and the discovery of HFE, the gene mutated in hereditary hemochromatosis, in 1996 [5–7]. Arguably, the most seminal finding in recent years was the discovery of the long-sought iron-regulatory hormone, hepcidin, and its target ferroportin, in the early 2000s [8–13]. It is now apparent that many iron-associated disorders are attributable to genetic malfunctions that affect the hepcidin-ferroportin axis. Nevertheless, our knowledge of iron biology remains incomplete.
The importance of iron to almost all living organisms is undeniable; iron is required for oxygen transport, energy production, DNA synthesis, and cellular respiration. For example, iron is a component of hemoglobin, an oxygen carrier that transports oxygen from the lungs to the peripheral tissue and then carries carbon dioxide back to the lungs. Likewise, iron is a constituent of myoglobin, an oxygen storage protein that provides oxygen to muscle tissue. At the same time, excess iron can be toxic due to the ability of iron to exist in various oxidation states. The ability of iron to redox cycle can facilitate the formation of hydroxyl or lipid radicals, which in turn can damage proteins, DNA, and lipids. To maintain iron homeostasis at both the systemic and the cellular levels, vertebrates have developed an elaborate machinery to control iron intake, storage, utilization, and recycling. Our understanding of diseases associated with iron depends on our knowledge of iron homeostasis.
Systemic Iron Homeostasis
An adult well-nourished human contains approximately 3–5 g of iron. Nearly 60% of this iron is incorporated into hemoglobin, with 10% in muscle myoglobin. The rest is stored in hepatocytes and reticuloendothelial macrophages. There is no known mechanism of iron excretion from the body. Roughly 1–2 mg of iron is lost daily through sweat, blood loss, sloughing of intestinal epithelial cells, and desquamation. To compensate for this loss, the body absorbs about 1–2 mg of dietary iron per day, but hemoglobin synthesis alone requires 20–25 mg of iron per day. To support hemoglobin synthesis and other metabolic processes, iron must be recycled and tightly regulated within the system. The circulating peptide hormone hepcidin together with its receptor ferroportin primarily maintain systemic iron homeostasis, whereas iron-regulatory proteins play a primary role in the control of intracellular iron homeostasis. Recently, an intracellular iron network consisting of 151 chemical species and 107 reactions and transport steps was identified [2]. Here, only key features are presented; for more details, comprehensive reviews, and current advances the reader is encouraged to consult the articles [2,14–19].
Iron Absorption
Inorganic, nonheme iron is available in many foods, e.g., eggs and vegetables, and is absorbed by duodenal enterocytes. Ferrireductase, Cybrd1 (DcytB), reduces nonheme iron to Fe2+ before it is transported through the cellular membrane by the divalent metal transporter 1, DMT1 (SLC11A2) [20–24]. The absorption of heme iron, found in red meats, is not fully understood. Once heme iron is absorbed it is transported into the cytosol and released by heme oxygenase 1 (HO1) [25]. Excess intracellular iron is stored in the storage protein ferritin. Ferritin oxidizes and sequesters excess ferrous iron into a ferrihydrite mineral core [26,27]. Iron sequestered in the ferritin of enterocytes is lost after a few days through the sloughing of intestinal epithelial cells. Dietary cytosolic iron is exported into the plasma by the basolateral iron exporter ferroportin (Fpn, SLC40A1) [8,9,11]. Export of iron from enterocytes into the circulation requires the ferroxidase hephaestin (HEPH), a multicopper oxidase, that oxidises Fe2+ to Fe3+ [28]. In the plasma, Fe3+ circulates bound to transferrin (Tf), a glycoprotein that has two binding sites for ferric iron and maintains iron in a soluble form. The discovery of transferrin as a plasma iron transporter dates back to 1946 [29]. Transferrin has two important functions: it limits the formation of toxic radicals and delivers iron to cells. In healthy humans, about 1/3 of transferrin is saturated with iron. Iron concentrations in healthy adults are approximately 14 – 32 μmol/L, with virtually all circulating iron bound to Tf. In conditions of iron overload, non-transferrin-bound-iron (NTBI) accumulates. NTBI is thought to contribute substantially to the pathology associated with iron overload (Table 10.1) [17].
Table 10.1.
Levels of transferrin saturation
| Transferrin saturation (%) | Clinical Interpretation |
|---|---|
| < 16% | Iron deficiency |
| 16%–45% | Normal levels |
| 45%–60% | Signs of iron overload |
| > 60% | Iron overload |
Iron Utilization, Recycling, and Storage
The principal consumer of iron is the erythroid bone marrow, and most of that iron comes from internal recycling by tissue macrophages, predominantly splenic macrophages. Erythroblasts acquire iron via a ubiquitous protein expressed on the cell surface, transferrin receptor 1, TfR1. Through receptor-mediated endocytosis TfR1 transfers iron-loaded Tf (Holo-Tf) into acidified endosomes where iron dissociates from transferrin with the assistance of six transmembrane epithelial antigen of the prostate (STEAP) proteins and exits the endosome via DMT1 [30]. Transferrin and transferrin receptor are recycled back to the cell surface. Iron is imported into mitochondria from intracellular compartments by the inner membrane protein mitoferrin 1 to form heme, the majority of which is then used for hemoglobin production [31]. Since excess heme is toxic and can lead to apoptosis, mechanisms must be in place to maintain heme at appropriate levels. It has been proposed that feline leukemia virus subgroup C cellular receptor (FLVCR) and ATP binding cassette protein G2 (ABCG2) export excess heme, although this is not completely understood [32,33].
Macrophages recapture iron from senescent and damaged erythrocytes by first phagocytosing erythrocytes and then catabolizing heme using heme oxygenase, to release iron. Ferrous iron is exported into the plasma via the iron exporter ferroportin (SLC40A1) and unused iron is stored in macrophages, mainly in ferritin [15,17,34]. Another major storage site for iron is the liver; the majority of iron entering the liver is stored in ferritin and can be mobilized when required by the body. Hepatocytes acquire Holo-Tf through two receptors, TfR1 and TfR2, but TfR2 is believed to act mainly as a transferrin saturation “sensor” and has much lower affinity for Holo-Tf than TfR1 [35–37]. Most importantly, when serum iron levels surpass the transferrin binding capacity, the liver becomes the major storage site for non-transferrin-bound-iron (NTBI) [15]. The mechanism by which hepatocytes acquire NTBI is not completely understood; one candidate for uptake of NTBI is zinc transporter Zip14 (SLC39A) [38]. Other tissues, such as heart and pancreas represent sites of iron accumulation in iron overload, and are also proposed to have mechanisms for NTBI uptake.
Regulation of Systemic Iron Homeostasis
To avoid iron overload or deficiency, an organism must maintain an internal equilibrium of iron and make iron available only when and where it is needed. The circulating peptide hormone hepcidin is a key molecule that regulates systemic iron homeostasis. It is predominantly produced by the liver, although studies indicate that other tissues also generate hepcidin [12,13]. Hepcidin levels are modified in response to physiological stimuli that affect iron homeostasis, such as iron overload, hepatic iron stores, inflammation, iron deficiency, erythropoietic activity, and hypoxia. Higher levels of hepcidin reduce iron absorption and vice versa.
Hepcidin modulates serum iron levels and controls transferrin saturation by inhibiting iron release from duodenal enterocytes, macrophages, and hepatocytes (Fig 10.1). More precisely, hepcidin regulates iron efflux by binding to the iron exporter ferroportin, triggering its internalization and degradation in lysosomes [39]. The mechanism is facilitated by Janus kinase 2 (Jak2) that binds to the ferroportin-hepcidin complex, phosphorylates ferroportin, and targets ferroportin for degradation [40]; an ubiquitin-mediated pathway of ferroportin degradation has also been described [41,42].
Fig 10.1. Systemic Iron Homeostasis.
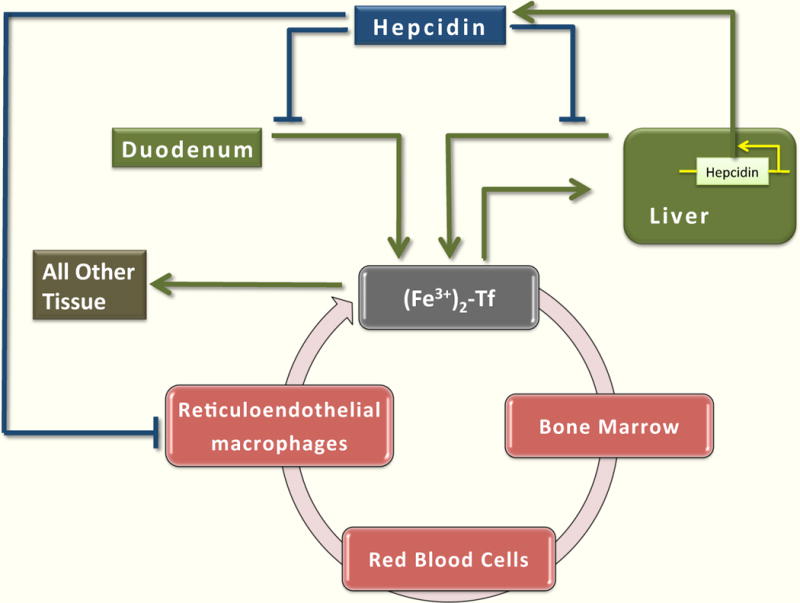
Inorganic, nonheme iron is absorbed by duodenal enterocytes. In the plasma, iron circulates bound to transferrin (Tf). The principal consumer of iron is the erythroid bone marrow, and most of that iron comes from internal recycling by tissue macrophages, predominantly splenic macrophages. Liver is the major storage site of iron. Iron entering the liver is stored in ferritin and can be mobilized when required by the body. Some iron is incorporated in other tissues. Hepcidin regulates systemic iron homeostasis by inhibiting iron release from duodenal enterocytes, macrophages, and hepatocytes.
Transcriptional Regulation of Hepcidin
Expression of hepcidin in the liver is primarily affected by transcriptional mechanisms mediated by the bone morphogenetic protein (BMP) family of transcription factors and other signaling components, which are members of the TGF-β family of ligands [43]. Recent studies suggest that the principal regulator of hepcidin is BMP6, which is increased in response to hepatic iron stores [44,45]. BMP binds to its receptor (BMP-R) and co-receptor hemojuvelin (HJV), a glycosylphosphatidylinositol-linked protein [43]. This interaction induces the phosphorylation of R-SMAD proteins and subsequent formation of active transcription complexes involving the co-regulator SMAD4, which bind to BMP responsive elements in the hepcidin promoter [46]. The membrane receptor neogenin (NEO1) enhances BMP signaling and hepcidin expression, perhaps by stabilizing HJV [47,48]. The transmembrane serine protease TMPRSS6 cleaves HJV, inactivates it, and consequently inhibits production of hepcidin [49].
Another mechanism for hepcidin regulation involves hemochromatosis proteins (HFEs). HFE has been suggested to act as a switch between two sensors of holo-Tf, TfR1 and TfR2. In this model, high concentrations of holo-Tf displace HFE from TfR1 and permit the interaction of HFE with TfR2. The HFE/TfR2 complex then promotes hepcidin transcription through an unknown mechanism [50–52].
Hepcidin expression is also induced by the inflammatory cytokine interleukin-6 (IL-6) and other cytokines by activating STAT3, signal transducer and activator of transcription 3 [53–55]. STAT3 binds to specific sequences in the HAMP promoter. Cytokine-mediated induction of hepcidin is thought to contribute to the hypoferremia that frequently accompanies chronic infections, acute inflammation and cancer [56].
Despite the substantial progress that has been made in defining key players in hepcidin regulation, the identification of critical components involved in hepcidin signaling and their functional relationships is far from complete. Mechanisms of hepcidin regulation mentioned above are depicted in Fig 10.2.
Fig 10.2. Transcriptional regulation of Hepcidin.
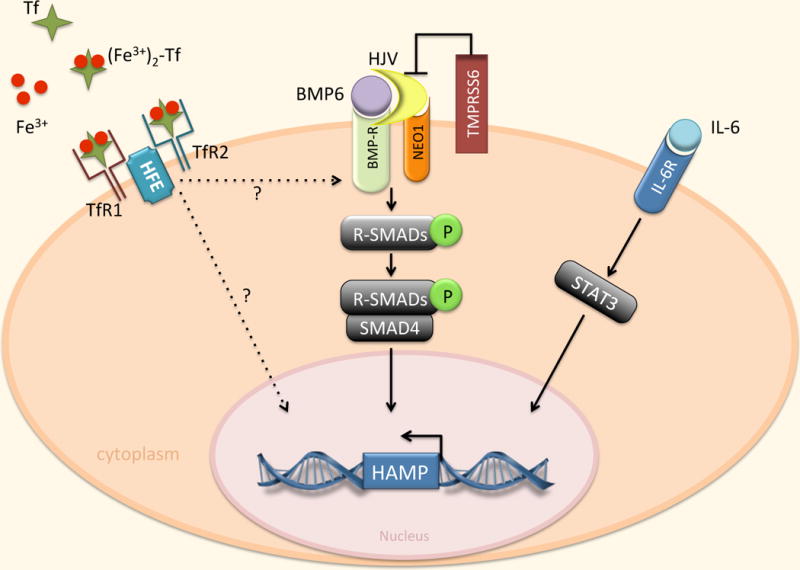
Regulation of hepcidin by BMP/SMAD and IL-6/STAT3 pathways. Expression of hepcidin in the liver is mainly affected by transcriptional mechanisms mediated by the BMP family, primarily BMP6. BMP binds to its receptor (BMPR) in conjunction with the co-receptor HJV. This interaction induces the phosphorylation of R-SMAD proteins which interact with the common mediator SMAD4, bind specific sequences in the hepcidin promoter, and trigger hepcidin gene (HAMP) transcription. NEO1 may enhance BMP signaling by interacting with HJV. TMPRSS6 negatively regulates hepcidin by cleaving HJV. Hepcidin expression is also induced by IL-6 through activation of STAT3. STAT3 binds to specific sequences in the HAMP promoter. TfR2 and HFE are also involved in hepcidin activation through mechanisms that are incompletely defined.
Intracellular Iron Homeostasis
Free ferrous iron can be toxic, since it contributes to the formation of the hydroxyl radical through the Fenton reaction. Hence, intracellular iron must be maintained as meticulously as systemic iron. The regulatory mechanism that coordinates intracellular iron uptake, utilization, storage and excretion is centered on the iron regulatory proteins (IRPs) and utilizes iron responsive elements (IREs). What follows is a brief description of the mechanism for a “generic cell” that encompasses pathways that have been consistently observed in many cell types (Fig 10.3). Further details can be found in [2].
Fig 10.3. Intracellular iron homeostasis of a generic cell.
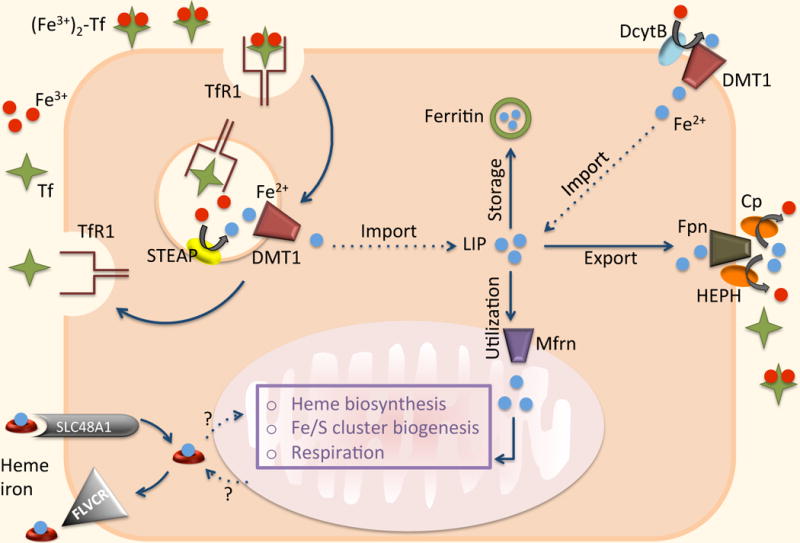
Cells acquire iron predominantly via TfR1. Ferric iron is reduced to Fe2+ by the transmembrane family of metalloreductases (STEAP). DMT1 then facilitates the transport of ferrous iron from the endosomes into the cytoplasm. In some cells, e.g. enterocytes, DMT1 participates in the transport of extracellular iron. DcytB reduces nonheme iron to Fe2+ before it is transported through the cellular membrane. Following egress from the endosome, iron enters the so-called “labile iron pool” (LIP). Ferroportin (Fpn) is believed to be the only ferrous iron exporter. It has been suggested that dietary heme iron can enter through SLC48A1 and be exported via FLVCR. A mechanism by which heme is moved in and out of mitochondria (the major site of iron utilization) is poorly understood. Ferrous iron is imported into mitochondria for incorporation into bioactive heme by the SLC transporter mitoferrin (Mfrn). Iron that is not exported or utilized is stored in ferritin (Ft).
Iron Import
Mammalian cells acquire iron predominantly via transferrin receptor 1 (TfR1). Following binding of Holo-Tf to TfR1, Tf-bound Fe is taken up by receptor-mediated endocytosis into acidified endosomes where ferric iron is reduced to Fe2+ by the transmembrane family of metalloreductases (STEAP) [30]. The divalent metal transporter 1, DMT1, then facilitates the transport of ferrous iron from the endosomes into the cytoplasm. In some cells, e.g. enterocytes, DMT1 is also located on the cell surface and participates in the transport of extracellular iron. It is worth pointing out that the role of STEAP proteins has been studied and well defined in hepatocytes, macrophages, erythroid cells, and erythroblasts, while their role in peripheral tissues requires further investigation [14]. Following egress from the endosome, iron enters the so-called “labile iron pool” (LIP), a cytosolic pool of weakly bound iron available for a variety of interactions with other molecules.
It has been suggested that dietary heme iron is transported by the heme carrier protein 1 (SLC46A1) [57], but another study demonstrated that SLC46A1 is an important folate transporter [58,59]. A year later SLC48A1 was identified as a possible candidate for heme import [60]. Some cells, like macrophages, acquire heme indirectly by phagocytosing erythrocytes and catabolizing heme to release iron. Hepatocytes have multiple mechanisms of iron entry, including TfR2 and a possible transporter for non-transferrin-bound-iron (NTBI), zinc transporter Zip14 (SLC39A) [38].
Iron Export
While there is no known mechanism for iron excretion from the body, there is a well-organized and controlled regulation of iron excretion from cells. Ferroportin, located on the plasma membrane, is expressed in a wide variety of human tissue types and is believed to be the only ferrous iron exporter [8,9,11,34]. It requires coordinated efforts of ferroxidases (ceruloplasmin and/or hephaestin) to assist iron oxidation and loading onto transferrin. As mentioned above, cells also export iron in the form of heme through FLVCR and ABCG2 [32,33].
Iron Utilization and Storage
The major site of iron utilization is the mitochondrion, where iron is used in synthesis of heme and iron-sulfur (Fe/S) cluster prosthetic groups, but the understanding of the mechanism by which iron is moved inside the cell is still incomplete. Iron is imported into the mitochondrion for incorporation into bioactive heme by the SLC transporter mitoferrin (SLC25A37) [31]. Intracellular heme regulates its own production through delta aminolevulinate synthase (ALAS) and its degradation by inducing heme oxygenase (HO1) [61,62]. After heme is synthesized it is exported via an unknown mechanism into the cytosol for integration into proteins.
Ferrous iron that is not exported or utilized is stored in ferritin, a cytosolic protein whose main function is to oxidize and sequester excess ferrous iron into a ferrihydrite mineral core. Ferritin is a 24-subunit polymer comprised of heavy (ferritin H) and light (ferritin L) polypeptide chains in variable ratios. The subunit composition of ferritin depends on cell type and physiological status [27]. Each ferritin protein can accrue as many as 4500 iron atoms. Since free iron can promote formation of reactive oxygen species, ferritin is crucial to preventing iron-mediated cell damage by keeping excess iron in a non-reactive form.
Intracellular Iron Regulation
Intracellular iron homeostasis is regulated post-transcriptionally by the iron regulatory proteins IRP1 (ACO1) and IRP2 (IREB2) in response to changing iron levels. For a comprehensive review the reader is encouraged to consult [7,18].
IRP1 and IRP2 exert their effects by binding to iron responsive elements (IREs), cis-regulatory hairpin structures that are present in the untranslated regions (UTRs) of mRNAs involved in iron metabolism. The mRNAs encoding ferritin, ferroportin, ALAS2, mitochondrial aconitase (ACO2), and hypoxia-inducible factor 2α (HIF2α) contain a single IRE in their 5′UTRs. The mRNA encoding TfR1 contains multiple IREs within the 3′ UTR, whereas the mRNAs encoding DMT1, cell division cycle 14 homolog A (Cdc14A), hydroxyacid oxidase 1 (HAO1), and MRCKα contain a single IRE in their 3′UTRs.
When intracellular iron levels are low, iron regulatory proteins bind to IREs with high affinity. Binding of IRP’s to 5′ UTR IREs inhibits the translation of ferritin and ferroportin, while binding to the 3′ UTR IREs results in the stabilization of mRNA of the iron importer TfR1, thus increasing cytoplasmic iron levels. In iron-replete cells, the regulatory effect of IRPs stops: IRP2 is targeted for degradation and IRP1 acquires a completed iron sulfur cluster that impedes IRE binding (Fig 10.4). The role of IRP regulation in the mechanisms and functions of other IRE-containing mRNAs has been less thoroughly studied.
Fig 10.4.
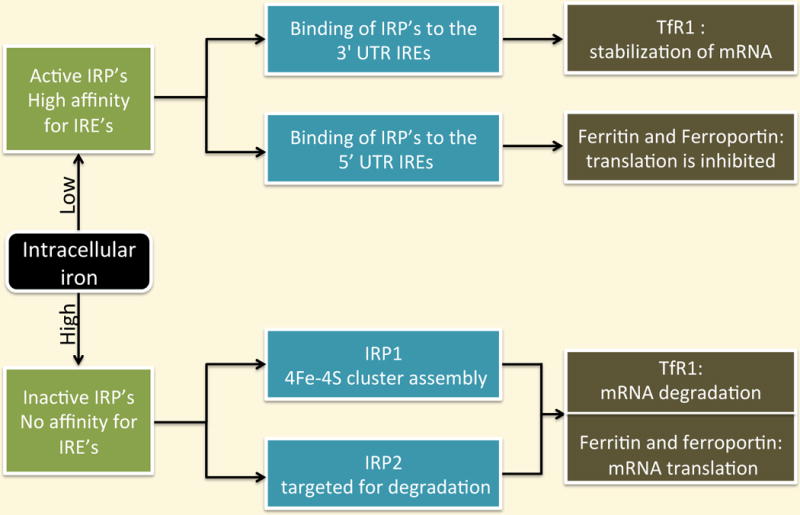
Intracellular iron regulation
Diseases of Iron Metabolism
Iron is required for oxygen transport, energy production, DNA synthesis and cellular respiration. Accordingly, inappropriately low or high levels of iron are detrimental and lead to a wide range of diseases.
Iron overload/deficiency is either hereditary or acquired. Levels of iron can be altered by the presence of mutated genes, diet that contains inappropriate amounts of iron (either insufficient or excessive), transfusion of red blood cells, iron injections, excessive blood loss, decreased intake or intestinal absorption of iron, and hemolysis.
Iron Overload
Excess iron leads to iron deposition in vital organs such as the liver, heart, pancreas, and endocrine glands. This propagates the formation of hydroxyl or lipid radicals, which damage proteins, DNA, cellular membranes, and can lead to cell death. Left untreated, chronic iron overload increases the risk of liver cirrhosis, cancer, hypogonadism, arthritis, cardiac arrhythmia, heart failure, retinal degeneration, diabetes mellitus, neurodegenerative diseases (Alzheimer’s, Parkinson’s, Huntington’s), and premature death. Treatments for iron overload include phlebotomy and iron chelation therapy [63].
Hereditary Hemochromatosis
Hemochromatosis is the most common genetic iron overload disorder and results from mutations in several genes, all of which affect the ferroportin/hepcidin regulatory axis (Table 10.2) [14,16,17]. The main characteristic of this disorder is increased absorption of dietary iron and its accumulation in the liver, heart, pancreas, endocrine glands, tissue and joints, where it causes injury and organ dysfunction, as described above. To date, researchers have identified five mutated genes associated with hemochromatosis, which can be grouped further into two cases: hepcidin deficiency and hepcidin resistance [16].
Table 10.2.
Genes involved in iron-related disorders
| Disorder | Genes | Protein | Protein function |
|---|---|---|---|
| Hemochromatosis | HFE | HFE | Involved in transcriptional regulation of hepcidin |
| Hemochromatosis | TfR2 | Transferrin receptor 2 | Holo-Tf sensor; at high Tf levels, HFE interaction with TfR2 is increased promoting hepcidin expression |
| Juvenile hemochromatosis | HJV | Hemojuvelin | Involved in transcriptional regulation of hepcidin; BMP co-receptor |
| Juvenile hemochromatosis | HAMP | Hepcidin | Modulates serum iron levels; regulates iron efflux by binding to the iron exporter ferroportin, triggering its internalization and degradation |
| Hemochromatosis (Hepcidin resistance) | SLC40A1 | Ferroportin | Iron exporter |
| Aceruloplasminemia | CP | Ceruloplasmin | Ferroxidase |
| Hypotransferrinemia | TF | Transferrin | Glycoprotein with two binding sites for ferric iron |
| IRIDA | TMPRSS6 | Matriptase-2 | Cleaves HJV, inactivates it and, consequently, inhibits production of hepcidin |
Hepcidin deficiency
Mutations in the genes encoding HFE, TfR2, hemojuvelin (HFE2, HJV), and hepcidin (HAMP) cause hemochromatosis by inactivating the pathway that up-regulates hepcidin. The most common and mild form of hereditary hemochromatosis (HH) is due to a missense mutation of the HFE gene, which is incompletely penetrant, and is influenced by environmental and other genetic factors [5,64]. A severe and less common form of HH, with extremely low or absent hepcidin levels, is juvenile hemochromatosis (mutations in HJV or HAMP genes) that leads to hypogonadism, heart failure, and death [65–67]. Another rare form of HH but less severe is caused by a mutation in the TfR2 gene [68].
Hepcidin resistance
Missense mutations in the gene encoding ferroportin obstruct hepcidin binding and result in insensitivity of ferroportin to regulation by hepcidin, leading to hepatocyte iron accumulation and high plasma iron [69].
Aceruloplasminemia
Aceruloplasminemia is a disorder caused by mutations in the gene encoding ceruloplasmin (Cp), a ferroxidase involved in the loading of Fe onto Tf following its release from cells [70,71]. Low serum ceruloplasmin levels and accumulation of iron in neural and glial cells of the brain, pancreatic islet cells, and hepatocytes characterize this disorder. The clinical outcome is retinal degeneration, diabetes mellitus, cerebellar ataxia, dementia, and neurologic diseases.
Other diseases related to iron overload and degenerative neurologic conditions are Hallervorden-Spatz disease and Friedreich’s ataxia [63].
Hypotransferrinemia/Atransferrinemia
Practically undetectable plasma levels of transferrin characterize hypotransferrinemia [72]. Deficiency in transferrin allows non-transferrin bound iron (NTBI) to accumulate and deposit in the liver and other organs, leading to the accumulation of iron to toxic levels. On the other hand, reduction in Tf-bound iron impairs erythropoiesis in the bone marrow, which strictly depends on Tf-bound iron. Hypotransferrinemic patients also have a severe hepcidin deficiency, implying that transferrin is somehow involved in the regulation of hepcidin [16].
Transfusional Siderosis
Repeated blood transfusions are a life-saving therapy in many conditions, but multiple transfusions can also lead to toxicity and chronic iron overload. Transfusions are used in patients with beta thalassemia, sickle cell anemia, bone marrow failure (aplastic anemia, myelodysplastic syndrome, red blood cell aplasia), and patients receiving aggressive cancer therapy. Each unit of transfused blood contains 200–250 mg of iron, which is more than a hundred times the amount absorbed daily from the diet (1–2 mg). At first, iron accumulates in reticuloendothelial macrophages and later in parenchymal tissue cells of the liver, pancreas, heart, and endocrine tissue, where it can lead to cardiomyopathy and other iron overload disorders [63].
Iron Deficiency
Iron deficiency is the major cause of anemia and a public health problem worldwide. Since roughly two-thirds of total body iron is used in hemoglobin synthesis, deficiency in iron will affect the production of healthy red blood cells.
Iron-Deficiency Anemia
Approximately three billion people worldwide suffer from iron-deficiency anemia due to decreased dietary iron intake, poor absorption, and increased need for iron, which can result from blood loss, gastrointestinal bleeding, blood donations, pregnancy, and cancer of the esophagus, stomach, or colon. Children and women are at much greater risk. Iron-deficiency can result in premature birth, poor growth development and cognitive skills, and also affects the nervous system. Patients may experience symptoms associated with anemia that include chronic fatigue, poor exercise tolerance, headaches, and problem concentrating [14,63].
Left untreated, iron-deficiency anemia can lead to complications such as irregular heartbeat, angina, and heart attack, low birth weight, high risk of infection (childhood), and delayed growth (childhood) [63]. Changes in diet and iron supplements can treat minor iron deficiency, while severe cases may require transfusion of red blood cells, intravenous iron, or iron injections.
Iron-Refractory Iron-Deficiency Anemia (IRIDA)
Iron-refractory iron-deficiency anemia is triggered by a rare mutation in the gene TMPRSS6, which encodes matriptase-2 expressed in the liver. This mutation leads to reduced activity of TMPRSS6 and consequently high hepcidin levels. As a result, iron absorption from the intestine and iron release from macrophages is inhibited, causing severe iron deficiency [16,17].
Anemia of Chronic Inflammation
Anemia of chronic inflammation, also called anemia of chronic disease (ACD), is a systemic iron disorder and occurs in association with malignancy, chronic infections, trauma, inflammatory disorders, and organ failure [56]. Iron stores in ACD are not exhausted, but iron is sequestered in macrophages. In addition, absorption of iron is reduced and hemoglobin synthesis is inhibited. The decrease in serum iron is a consequence of hepcidin increase in response to inflammation, which seems to be an attempt to restrict iron availability to invading microorganisms and tumor cells [73]. Hepcidin production is induced by the inflammatory cytokine interleukin-6 (IL-6), bacterial pathogens, and lipopolysaccharide [74]. Anemia of chronic inflammation is considered to be mild-to-moderate anemia and treatment is usually focused on the underlying disorder.
Iron Homeostasis and Cancer
Dysregulation of iron metabolism in cancer is well known, and it has been argued for years that excess iron and increased cancer incidence go hand-in-hand [75], although this is not always observed [76]. Links between excess iron and cancer are also suggested by the efficacy of dietary iron deprivation [77] and iron chelators [78] in cancer therapy. In the early 1980’s it was observed that levels of transferrin receptor 1 (TfR1) are elevated in cancer, and use of TfR1 as a targeting ligand in the design of anti-cancer drugs was proposed [79,80].
More recently, it was observed that hepcidin and ferroportin are expressed in epithelial cells of peripheral tissue, such as the breast, where they exhibit the same regulatory interaction as in macrophages and liver cells [81]. Levels of hepcidin were increased and levels of ferroportin were decreased in breast cancer cell lines when compared to non-malignant breast cells, and this was correlated with an increased labile iron pool in malignant cells. Data collected from breast cancer patients also showed reduced levels of ferroportin in malignant compared to non-malignant breast tissue. Experimentally induced overexpression of ferroportin reduced tumor growth of breast cancer xenografts, implying a direct relationship between intracellular iron and tumor growth. Importantly, low levels of ferroportin in the tissues of breast cancer patients were associated with poor clinical outcome and reduced survival. Subsequent work determined that elevated TfR1 and reduced HFE (which would also be expected to elevate iron in tumor tissue) similarly predict poor survival in breast cancer patients [82].
Although cancer is of course more than an iron disorder, these findings indicate a clear and direct relationship between iron and cancer. Clarifying the precise nature of this relationship will require further study.
A Systems Biology Approach to Iron Metabolism
The complexity of iron regulation in mammals makes a systems biology approach crucial, with its enabling technology of computational models based on a mathematical description of the iron homeostasis control system. Before discussing specific mathematical models we briefly summarize the role that models play in systems biology (Fig 10.5) [83]. As discussed earlier, the primary role of mathematical models is to discover new biology. This is typically done in two steps. The first step, model building, consists of the description of the pertinent biology in mathematical terms. There are a variety of mathematical formalisms one can use for this purpose. Which one to choose for a given problem depends in large part on the type of information available and on the type of questions one would like to answer. A common approach is through a system of differential equations. In the case of a metabolic network, for instance, there will be one differential equation per molecular species in the network, viewing it as a biochemical reaction network. The equation for a given species describes the rate of change of the species in terms of the quantities of other species it depends on, together with a collection of kinetic parameters. Alternatively, one can view the network in terms of a collection of logical rules that govern the “decision making” of the node based on the states of the other molecular species. Rather than varying continuously, the species might take on categorical values, such as low or high.
Fig 10.5. The construction of a mathematical model.
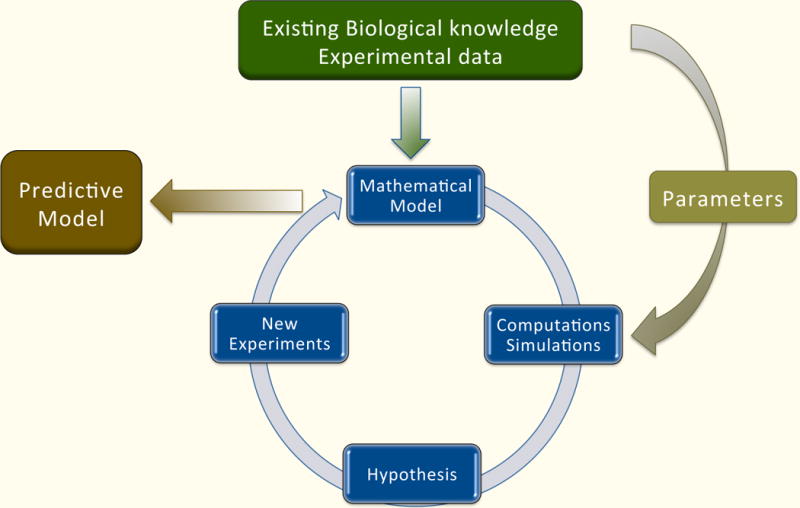
The starting point is a formulation of the problem and specific questions that the model will answer. Biological knowledge about components of the system, its structure, interactions, and any available experimental results must be gathered. Different types of experimental data are analyzed and integrated. This information is then used to construct a mathematical model. Since different models emphasize different features, the choice of mathematical model depends on the questions being asked. Its structure will also depend on the system description: organismal, cellular, or molecular. Some systems will have unknown biological parameters and will require detailed information about kinetic constants or time course data in order to estimate model parameters. Various computational techniques are used to assess if the model is in accordance with experimental results, and if not, hypotheses underlying the model need to be refined, and different types of experiments might be proposed. This iterative process is repeated and the model is refined until it is accurately describes the relevant aspects of the system.
The second step is model analysis, which, in many cases, relies on simulation. That is, the mathematical model is implemented as a computational algorithm in a computer. Simulation of the model consists of the choice of an initial concentration of all the species in the network, from which the time evolution of the model is calculated. Observations might include which steady state is reached from this initialization or whether the simulation is robust with respect to small changes in the model parameters. The result of these simulations is an understanding of how the model behaves under certain perturbations of interest. If the model correctly captures the key features of the biology, then an understanding of the model can lead to a more targeted investigation of the actual network, leading to new biological insights. Below we discuss some published computational models related to iron regulation.
Models of Iron Homeostasis
Biological systems can be described mathematically as dynamical systems, in terms of functional relationships between the variables, which govern the temporal evolution of the system. Some biological data is best modeled by systems in which the model components take on discrete states, while others require continuously varying system states. Dynamic models of iron homeostasis that are presented in this section are of two different types: continuous and discrete. The models summarized in Diseases of Iron Metabolism section above are all continuous in the form of ordinary differential equations (ODE), whereas the model in the Transfusional Siderosis Section above is discrete and is based on the theory of Petri nets.
Generally speaking, differential equations describe the temporal change of state variables, e.g., the change in concentration of a molecular component. To put this in the context of iron metabolism, let duodenal enterocytes represent a compartment and let y denote the concentration of labile iron in this compartment. Then the rate of change of y, dy/dt, describes how the level of iron changes over time. If it is constant then the differential equation will be dy/dt=0. Since iron is brought into the cell and is also exported from the cell, further assumptions can be made. For simplicity, assume that iron enters enterocytes at some rate a and is exported at some rate b. Then the flow of iron through enterocytes can be described by the differential equation dy/dt = a-by. This is of course a simplified illustration; in reality one would have as many equations as compartments and will have to consider shuttling between the compartments. In addition, rates might not be just simple constants, but rather complicated expressions, and would have to incorporate various regulations, for example regulation of the iron exporter ferroportin by hepcidin. From this simple example one can see that the resulting ODE model will have many unknown parameters and will require detailed information about kinetic constants or time course data in order to estimate model parameters.
Compartmental Model of Iron Homeostasis
B. J. Lao and D. T. Kamei developed a simple compartmental model of iron homeostasis calibrated to mouse data [84]. The model consists of five compartments, each denoting the amount of iron at a specific location or in a particular state: hepatocyte, FeTf (diferric transferrin), RBC (red blood cells), NTBI (non-transferrin-bound-iron), and macrophage. The system is then given by five ordinary differential equations that explicitly incorporate the roles of ferroportin, hepcidin, TfR2, and HFE. We present only one of the equations, describing the RBC compartment.
(rate of iron transfer from FeTf to RBC compartment)
kRBC;macro = 1.6 · 10−3h−1 (rate of iron transfer from RBC compartment to macrophages)
These rates are based on findings published in [85,86]. All parameters in the model were approximated using mouse data from various sources.
The resulting model was used to simulate anemia, iron overload and erythropoiesis stimulation. The following conclusions were formed.
FeTf may be involved in determining availability of iron for erythropoiesis.
To maintain proper iron absorption and intracellular iron levels, the iron-responsive elements of ferroportin are essential. Simulations performed without IRE’s resulted in normal FeTf and in iron accumulation in macrophages and hepatocytes.
Increased iron absorption by duodenal enterocytes replicated features of HFE hemochromatosis, and iron accumulation in hepatocytes was influenced by the uptake of NTBI. Moreover, increased levels of NTBI cause hepcidin decrease implying that removal of NTBI might revert hepcidin levels.
Systemic Model of Iron Homeostasis
In [87] a systemic model of iron metabolism was built based on data from normal mice (C57BL6) [88] under three diets: iron-deficient, iron-adequate, and iron-loaded. The model consists of a plasma compartment and 15 peripheral organ compartments. Each compartment (pool) is represented by its iron content and is described by a balance equation.
| Ci | iron content in ith compartment |
| vij | the rate of iron in-flux from compartment j to i |
| vji | the rate of iron out-flux from compartment i to j |
| vio | the rate of iron flux from outside into compartment i |
| voi | the rate of iron flux from compartment i out of the system. |
Time course data was obtained by administering radioactive tracer (59Fe) and then measuring at certain intervals over 28 days non-heme iron as well as hematocrit and hemoglobin content of blood. The experimental data from mice under three different diets was used to estimate parameters and calibrate the model. The authors argue that the resulting quantitative model reflects systemic properties of iron homeostasis. They conclude that this model could be used to study dietary iron perturbations and plan to use the model on genetically modified mice. Furthermore, the authors envision that this mathematical model of pools and fluxes will serve as a foundation for a whole-body model, which would ultimately include iron uptake, storage, secretion, heme synthesis, and regulatory structure.
Intracellular Model of Iron Homeostasis
A model of intracellular iron homeostasis was constructed in [89]. This model is specific to normal breast epithelial cells and represents the core control system of iron metabolism focused on iron import via TfR1, export (ferroportin), sequestration (ferritin), and regulation (iron regulatory proteins). These proteins and the labile iron pool are connected by several feedback loops that drive network dynamics. Each component in the model is defined by an ordinary differential equation that describes changes in concentration with respect to time. The resulting ODE system has five equations and 15 parameters with two parameters being external: hepcidin and the iron saturation level of extracellular transferrin. One of the assumptions made was that ferritin (Ft) is always bound to iron and undergoes natural degradation. As a result, ferritin releases iron back into the labile iron pool. It was represented by the following mechanism.
 |
| c | is a constant decay rate |
| b | is a hyperbolic rate that describes negative regulation of IRP’s on Ft, |
Here, IRP represents an inhibiting state variable, k an activation threshold, and a the maximum production rate of the regulated protein (in this case Ft). Using this information, the rate of change of the labile iron pool (LIP) can be represented by the following equation.
| a1Feex · (TfR1) | iron import (via TfR1) with the rate a1 |
| a2 · (LIP) · (Fpn) | iron export (via Fpn) with the rate a2 |
| Feex | iron saturation level of extracellular transferrin |
| b and c | as described above. |
The model was validated using data from overexpression of ferroportin. Through a combination of analytical arguments and simulations, it was shown that the model has a unique stable steady state for any choice of parameters, agreeing with experimental evidence that cellular iron is tightly controlled [90].
Including additional relevant components in the model will be the next step, with the ultimate goal of identifying basic forces and key regulators that contribute to modifications in iron homeostasis as normal breast epithelial cells transition to malignancy.
Petri Net Model of Systemic Iron Homeostasis
Petri net theory uses a different approach to simulations and analysis, and has also been applied to iron metabolism. Carl A. Petri, a German mathematician and computer scientist, introduced and formally defined the concept in the 1960s. Today, Petri nets are used in computational biology to model gene-regulatory networks, metabolic pathways, signal transduction pathways and biochemical networks.
Informally speaking, a Petri net is a directed graph that consists of two kinds of nodes, places and transitions, and arcs connecting them. Arcs only connect two nodes of different kinds; they do not join two places, or two transitions. Places, depicted by circles, represent passive elements such as proteins, protein complexes or chemical compounds, while transitions, depicted by rectangles, are active elements and represent biological interactions or chemical reactions. In the context of iron metabolism an example of a place might be Fe2+ and an example of a transition is oxidation of Fe2+ by hephaestin. Some places are marked by black dots or natural numbers, called tokens, which are dynamic elements of the net and represent the concentration of a given species in terms of moles, molecules, or even abstract concentration levels such as high, medium, and low. Tokens are distributed over places to describe a systems state, e.g., the normal body iron physiological state. The distribution of tokens over places is called a marking of the net. For a certain biological reaction to happen, its places, e.g. proteins, must contain sufficient numbers of tokens. If all places are marked (contain tokens), then a transition may fire by removing one or more tokens from each place and moving it to another appropriate place (Fig 10.6). This changes the marking of the net, i.e., the systems state. Enabled transitions do not have to fire, which makes Petri nets nondeterministic and the behavior of the system is established by all possible firing sequences. Of course, just following tokens does not represent the entire analysis. Comprehensive formal analysis must be performed to show behavioral and structural properties of the system.
Fig 10.6. A simple Petri net example.
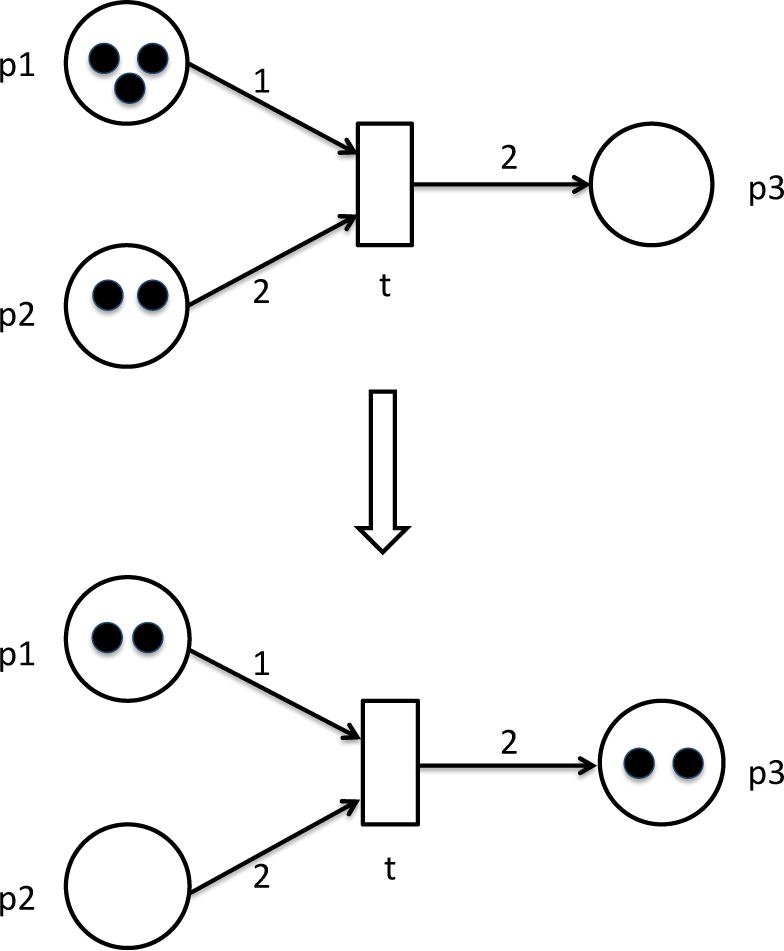
Initially input places p1 and p2 contain three and two tokens, respectively. By firing transition t one token will be removed from p1 and two tokens from p2. Transition t will consume tokens and place two of them into place p3. Transition t may fire since its preplaces p1 and p2 have sufficient number of tokens. After one firing step the marking of the net is changed: p1 has only two tokens left, p2 has no tokens, and p3 has two tokens. Transition t cannot fire anymore.
In a series of papers a Petri net model of systemic iron homeostasis was constructed [91–93]. The model consists of 47 places and 57 transitions and has been verified through extensive analysis. In the latest article in the series [92] the authors focused on some aspects of the anemia of chronic disorders and, based on their analysis of the net, they have made some observations, listed below. The predictions made by the model were validated using data from patients with chronic anemia that were treated with recombinant human erythropoietin (rHuEPO). The conclusions based on the model and laboratory tests are:
Transferrin receptor levels are not influenced by inflammation.
There is a strong positive correlation between the dose of recombinant human erythropoietin and soluble transferrin receptor.
There is a strong negative correlation between the dose of rHuEPO and hepcidin, suggesting a reverse relationship.
The TfR1 serum level was confirmed to be a suitable indicator of erythropoietic activity.
It is worth commenting on some differences among the mathematical models presented here. The majority of the models consist of a system of ordinary differential equations. These capture the continuous rate of change of the concentrations of the different molecular quantities over (continuous) time. The last model consists of a Petri net, that is, a graph structure of a certain type, together with rules that govern the state of the different nodes in this graph. In the model presented, the states are integer values, specifying how many tokens are placed at a particular node at a given time. Thus, the states are discrete, rather than continuous concentrations.
Furthermore, time progresses in discrete steps also. There are also other types of time- and/or state-discrete models in use in systems biology [83]. The different modeling methods each have their pros and cons. What particular modeling framework is best for a particular type of system depends on several factors, such as availability of experimental data, kinetic parameters, the type of question to be answered, and others.
Conclusion
Iron metabolism and its relationship to a variety of disorders and diseases is difficult or impossible to fully understand without a systems approach. Regulatory mechanisms in different parts of the organism, operating at different time and spatial scales, are connected to each other and interact through complex feedback loops. Without an understanding of how these interdependencies affect dynamic changes in iron homeostasis, systematic therapeutic approaches will remain elusive. Systems biology and mathematical modeling promise such a rigorous understanding. As detailed in this chapter, much has been discovered about the mechanistic foundations of iron regulation. However, key parts of the system remain poorly understood.
So-called reductionist biology has an important role to play in uncovering additional features of iron metabolism. These can then be integrated into system level models, such as the case studies presented here. Systems biology brings another valuable approach to the problem through the generation and analysis of high-throughput “-omics” data, which have not yet been used extensively to study iron metabolism. Large-scale gene expression studies using DNA microarrays or high-throughput sequencing can help in discovering new genes involved in iron regulation and their connections to the known control network. Proteomics analyses using mass spectrometry based methods can shed light on posttranscriptional regulation and further help identify important players in the network. The application of one or more of a variety of network inference algorithms can be used to build up a more complete regulatory network structure that can be used to generate experimental hypotheses, to be validated in the laboratory.
Since iron regulation is a highly dynamic process, we require dynamic computational models for its study. This chapter describes some recent examples of such models. Given the complexity of the entire process, substantially more sophisticated models will be required. Their construction needs to be based on comprehensive time-resolved data at different scales and in different cell types. The confluence of new and improved mathematical and computational techniques, together with sophisticated new measurement techniques, brings such models into the realm of the possible. Thus, the promise of systems biology is yet to be fully realized in the study of iron metabolism and its relation to human health.
Acknowledgments
This work was supported in part by Grants NIH R21 CA156133-01A1 (R.L., S.V.T.) and NIH Cancer Biology Training Grant T32-CA079448 (J.C.).
References
- 1.Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics. 2009;2:2. doi: 10.1186/1755-8794-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hower V, et al. A general map of iron metabolism and tissue-specific subnetworks. Mol Biosyst. 2009;5(5):422–43. doi: 10.1039/b816714c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Josephs HW. Absorption of iron as a problem in human physiology; a critical review. Blood. 1958;13(1):1–54. [PubMed] [Google Scholar]
- 4.Laufberger V. Sur la cristallisation de la ferritine. Soc Chim Biol. 1937;19:1575–1582. [Google Scholar]
- 5.Feder JN, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 6.Trowbridge IS, Omary MB. Human cell surface glycoprotein related to cell proliferation is the receptor for transferrin. Proc Natl Acad Sci USA. 1981;78(5):3039–3043. doi: 10.1073/pnas.78.5.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hentze MW, Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci U S A. 1996;93(16):8175–82. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275(26):19906–12. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 9.Donovan A, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403(6771):776–81. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 10.Krause A, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480(2–3):147–50. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 11.McKie AT, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5(2):299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 12.Park CH, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–10. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 13.Pigeon C, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276(11):7811–9. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 14.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112(2):219–30. doi: 10.1182/blood-2007-12-077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 16.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–33. doi: 10.1182/blood-2011-01-258467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hentze MW, et al. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 18.Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 19.De Domenico I, Ward DM, Kaplan J. Hepcidin regulation: ironing out the details. J Clin Invest. 2007;117(7):1755–8. doi: 10.1172/JCI32701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunshin H, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388(6641):482–8. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 21.Fleming MD, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16(4):383–6. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 22.Canonne-Hergaux F, et al. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood. 1999;93(12):4406–17. [PubMed] [Google Scholar]
- 23.Oakhill JS, et al. Functional characterization of human duodenal cytochrome b (Cybrd1): Redox properties in relation to iron and ascorbate metabolism. Biochim Biophys Acta. 2008;1777(3):260–8. doi: 10.1016/j.bbabio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Turi JL, et al. Duodenal cytochrome b: a novel ferrireductase in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(2):L272–80. doi: 10.1152/ajplung.00342.2005. [DOI] [PubMed] [Google Scholar]
- 25.Ferris CD, et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol. 1999;1(3):152–7. doi: 10.1038/11072. [DOI] [PubMed] [Google Scholar]
- 26.Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta. 2010;1800(8):783–92. doi: 10.1016/j.bbagen.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Theil EC. Ferritin: at the crossroads of iron and oxygen metabolism. J Nutr. 2003;133(5 Suppl 1):1549S–53S. doi: 10.1093/jn/133.5.1549S. [DOI] [PubMed] [Google Scholar]
- 28.Vulpe CD, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21(2):195–9. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 29.Schade AL, Caroline L. An Iron-binding Component in Human Blood Plasma. Science. 1946;104(2702):340–1. doi: 10.1126/science.104.2702.340. [DOI] [PubMed] [Google Scholar]
- 30.Ohgami RS, et al. The Steap proteins are metalloreductases. Blood. 2006;108(4):1388–94. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw GC, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440(7080):96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 32.Krishnamurthy P, Xie T, Schuetz JD. The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther. 2007;114(3):345–58. doi: 10.1016/j.pharmthera.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Keel SB, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319(5864):825–8. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 34.Donovan A, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood. 2004;104(13):4287–93. doi: 10.1182/blood-2004-06-2477. [DOI] [PubMed] [Google Scholar]
- 36.Kawabata H, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274(30):20826–32. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 37.Robb A, Wessling-Resnick M. Regulation of transferrin receptor 2 protein levels by transferrin. Blood. 2004;104(13):4294–9. doi: 10.1182/blood-2004-06-2481. [DOI] [PubMed] [Google Scholar]
- 38.Liuzzi JP, et al. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. 2006;103(37):13612–7. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 40.De Domenico I, et al. Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with Jak2. Proc Natl Acad Sci U S A. 2009;106(10):3800–5. doi: 10.1073/pnas.0900453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao B, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012;15(6):918–24. doi: 10.1016/j.cmet.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross SL, et al. Molecular Mechanism of Hepcidin-Mediated Ferroportin Internalization Requires Ferroportin Lysines, Not Tyrosines or JAK-STAT. Cell Metab. 2012;15(6):905–17. doi: 10.1016/j.cmet.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Babitt JL, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–9. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 44.Andriopoulos B, Jr, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41(4):482–7. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meynard D, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41(4):478–81. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 46.Wang RH, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2(6):399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 47.Lee DH, et al. Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis. Blood. 2010;115(15):3136–45. doi: 10.1182/blood-2009-11-251199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang AS, et al. Hemojuvelin-neogenin interaction is required for bone morphogenic protein-4-induced hepcidin expression. J Biol Chem. 2009;284(34):22580–9. doi: 10.1074/jbc.M109.027318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silvestri L, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502–11. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao J, et al. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9(3):217–27. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, et al. Hepatocyte-targeted HFE and TFR2 control hepcidin expression in mice. Blood. 2010;115(16):3374–81. doi: 10.1182/blood-2009-09-245209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallace DF, et al. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology. 2009;50(6):1992–2000. doi: 10.1002/hep.23198. [DOI] [PubMed] [Google Scholar]
- 53.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–9. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pietrangelo A, et al. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology. 2007;132(1):294–300. doi: 10.1053/j.gastro.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 55.Verga Falzacappa MV, et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–8. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 56.Andrews NC. Anemia of inflammation: the cytokine-hepcidin link. J Clin Invest. 2004;113(9):1251–3. doi: 10.1172/JCI21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shayeghi M, et al. Identification of an intestinal heme transporter. Cell. 2005;122(5):789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 58.Qiu A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127(5):917–28. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 59.Andrews NC. When is a heme transporter not a heme transporter? When it’s a folate transporter. Cell Metab. 2007;5(1):5–6. doi: 10.1016/j.cmet.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 60.Rajagopal A, et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature. 2008;453(7198):1127–31. doi: 10.1038/nature06934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferreira GC, Gong J. 5-Aminolevulinate synthase and the first step of heme biosynthesis. J Bioenerg Biomembr. 1995;27(2):151–9. doi: 10.1007/BF02110030. [DOI] [PubMed] [Google Scholar]
- 62.Yoshida T, et al. Human heme oxygenase cDNA and induction of its mRNA by hemin. Eur J Biochem. 1988;171(3):457–61. doi: 10.1111/j.1432-1033.1988.tb13811.x. [DOI] [PubMed] [Google Scholar]
- 63.Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341(26):1986–95. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 64.Allen KJ, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221–30. doi: 10.1056/NEJMoa073286. [DOI] [PubMed] [Google Scholar]
- 65.Roetto A, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–2. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 66.Papanikolaou G, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36(1):77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 67.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115(8):2180–6. doi: 10.1172/JCI25683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nemeth E, et al. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105(4):1803–6. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 69.Sham RL, et al. Hereditary hemochromatosis due to resistance to hepcidin: high hepcidin concentrations in a family with C326S ferroportin mutation. Blood. 2009;114(2):493–4. doi: 10.1182/blood-2009-04-216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris ZL, et al. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci U S A. 1995;92(7):2539–43. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshida K, et al. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat Genet. 1995;9(3):267–72. doi: 10.1038/ng0395-267. [DOI] [PubMed] [Google Scholar]
- 72.Beutler E, et al. Molecular characterization of a case of atransferrinemia. Blood. 2000;96(13):4071–4. [PubMed] [Google Scholar]
- 73.Nemeth E, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–3. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 74.Nemeth E, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torti SV, Torti FM. Ironing out cancer. Cancer Res. 2011;71(5):1511–4. doi: 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kotsopoulos J, et al. Plasma micronutrients, trace elements, and breast cancer in BRCA1 mutation carriers: an exploratory study. Cancer Causes Control. 2012;23(7):1065–74. doi: 10.1007/s10552-012-9975-0. [DOI] [PubMed] [Google Scholar]
- 77.Hann HW, Stahlhut MW, Blumberg BS. Iron nutrition and tumor growth: decreased tumor growth in iron-deficient mice. Cancer Res. 1988;48(15):4168–70. [PubMed] [Google Scholar]
- 78.Richardson DR. Iron chelators as therapeutic agents for the treatment of cancer. Crit Rev Oncol Hematol. 2002;42(3):267–81. doi: 10.1016/s1040-8428(01)00218-9. [DOI] [PubMed] [Google Scholar]
- 79.Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet. 1980;2(8191):390–2. doi: 10.1016/s0140-6736(80)90440-7. [DOI] [PubMed] [Google Scholar]
- 80.Shan L, et al. Visualizing head and neck tumors in vivo using near-infrared fluorescent transferrin conjugate. Mol Imaging. 2008;7(1):42–9. [PubMed] [Google Scholar]
- 81.Pinnix ZK, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2(43):43–56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miller LD, et al. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011;71(21):6728–37. doi: 10.1158/0008-5472.CAN-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laubenbacher R, et al. A systems biology view of cancer. Biochim Biophys Acta. 2009;1796(2):129–39. doi: 10.1016/j.bbcan.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lao BJ, Kamei DT. A compartmental model of iron regulation in the mouse. J Theor Biol. 2006;243(4):542–54. doi: 10.1016/j.jtbi.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 85.Rivera S, et al. Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood. 2005;106(6):2196–9. doi: 10.1182/blood-2005-04-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117(3):285–97. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 87.Lopes TJ, et al. Systems analysis of iron metabolism: the network of iron pools and fluxes. BMC Syst Biol. 2010;4:112. doi: 10.1186/1752-0509-4-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schumann K, et al. A method to assess 59Fe in residual tissue blood content in mice and its use to correct 59Fe-distribution kinetics accordingly. Toxicology. 2007;241(1–2):19–32. doi: 10.1016/j.tox.2007.08.082. [DOI] [PubMed] [Google Scholar]
- 89.Chifman J, et al. The core control system of intracellular iron homeostasis: A mathematical model. J Theor Biol. 2012;300:91–9. doi: 10.1016/j.jtbi.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Epsztejn S, et al. Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem. 1997;248(1):31–40. doi: 10.1006/abio.1997.2126. [DOI] [PubMed] [Google Scholar]
- 91.Formanowicz D, et al. Petri net based model of the body iron homeostasis. J Biomed Inform. 2007;40(5):476–85. doi: 10.1016/j.jbi.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 92.Formanowicz D, et al. Some aspects of the anemia of chronic disorders modeled and analyzed by petri net based approach. Bioprocess Biosyst Eng. 2011;34(5):581–95. doi: 10.1007/s00449-010-0507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sackmann A, et al. An analysis of the Petri net based model of the human body iron homeostasis process. Comput Biol Chem. 2007;31(1):1–10. doi: 10.1016/j.compbiolchem.2006.09.005. [DOI] [PubMed] [Google Scholar]