

Abstract
Duchenne muscular dystrophy (DMD) affects 1in 5,000 newborn males. These boys appear healthy as infants and young children, but then experience a heartbreaking decline, as muscle tissue gradually wastes away, leaving patients nonambulant by their late teens. The heart and respiratory muscles are similarly weakened, compromising life span. DMD has served as a test bed for population genetics in the 1950’s, gene identification methods (reverse genetics) in the 1980’s, and the integration of molecular diagnostics into patient diagnosis and family counseling. Additional landmarks are held by the dystrophin gene: It is the largest in the human genome—2.3 million base pairs—and has the highest known spontaneous mutation rate of all human genes. Although we have made robust progress in understanding the molecular basis of DMD, translation of these advances to improvements in patient care has been painfully slow. Early hints of success with stem cell transplantation and replacement gene therapy hit technical roadblocks of delivery and immunological barriers that have remained persistently impassable.
With this backdrop, the innovative exon-skipping therapy for DMD, which rescues dystrophin production, caught the rapt attention of scientists, physicians, and patients. This approach treats a DMD patient systemically with antisense oligonucleotides directed against the dystrophin mRNA transcript. These oligos are designed to rescue the target mRNA, restoring the proper reading frame and thus protein production; in contrast, antisense therapy for other disorders focus on destroying or inhibiting the target mRNA. Dystrophin rescue in DMD is accomplished by excluding an exon that neighbors a deletion mutation, effectively transforming an out-of-frame (nonsense, loss-of-function) transcript to an in-frame transcript capable of de novo dystrophin production in patient muscle. The newly produced dystrophin protein lacks amino acids but retains some of its cytoskeletal function. The concept that internally truncated dystrophin could be functional derived from observations that similar mutations underlie the milder Becker muscular dystrophy patients.
Since the first report of exon skipping occurring spontaneously in patient cells in 1991 (1), more than 350 publications have validated this corrective strategy. Impressive systemic rescue of dystrophin has been seen in the mdx mouse model of DMD in many independent laboratories (2–5) and was further supported by preclinical efficacy in a large animal model of DMD (6). Early DMD patient trials first used local intramuscular injection of antisense oligos and have been followed by intravenous and subcutaneous injections focused on systemic dystrophin rescue (7–9).
TRIALS AND TRIBULATIONS
In late 2013, a series of public reports caused a pregnant pause in the progress. A large phase 3 study of 186 DMD patients (125 on drug and 61 placebo) carried out by GlaxoSmithKline (GSK) in partnership with Prosensa failed to show significant improvement of the primary outcome measure—the six-minute walk test (6 MWT). GSK has since terminated its relationship with Prosensa on the exon-skipping program in DMD. Simultaneous with this late-stage failure, an exon-skipping oligo generated by an alternative oligonucleotide chemistry (morpholino; Sarepta) showed impressive safety profiles and a wide therapeutic index in phase 2b trials in DMD patients. However, when Sarepta sought accelerated approval, the U.S. Food and Drug Administration (FDA) did not find the data compelling enough to grant conditional approval.
What happened? Is this yet another case in which impressive preclinical data and an enticing mechanism of action failed to translate into human patient efficacy? The scenario actually makes scientific sense, but also serves to underscore the shifting sands and blind alleys that disorient the modern orphan drug developer, as well as the families anxious for a drug that can slow the tragic progression of DMD. The problems faced by the two programs (GSK/Prosensa, Sarepta) are quite different, and these differences are instructive both for other drugs in the orphan pipeline and for the path forward in exon skipping therapeutics.
GSK/Prosensa used a traditional chemistry for their oligonucleotide drug 2′-O-methyl phosphorothioate (2OMePS). Similar chemistry has been used in about 60 clinical trials for many different disorders, and nearly all studies showed challenges with a sufficient therapeutic window, namely, difficulty in delivering enough of the oligo inside the cells to achieve potent modulation of RNA metabolism without hitting a toxicity ceiling. The toxicity limit on 2OMePS chemistry is about 20 mg/kg before activation of the complement and innate immune systems directed against the DNA-like drug. It was never clear that the 20 mg/kg/wk subcutaneous dose could produce sufficient truncated dystrophin to anticipate clinical benefit. A subset of patients in the phase 2 trials showed an impressive stabilization in the 6 MWT, and this was used as a rationale for moving ahead to phase 3 studies. But the 6 MWT, especially in children, is far from perfect as an outcome measure. Recent public meetings between the FDA and DMD stakeholder groups suggested that alternatives to the 6 MWT (such as acute measures of strength) may be a better primary outcome for future trials. Ideally, this program could have been de-risked at phase 2, by altering dose and chemistry until better de novo dystrophin production was observed. Prosensa continues to test additional exon-skipping drugs using the 2OMePS chemistry, likely with this intent.
Morpholinos, such as that used by Sarepta in their DMD trials, are chemically less similar to DNA than 2OMePS but retain strong sequence-specific base pairing to the mRNA target. Morpholinos appear capable of evading many or all of the toxicities plaguing the 2OMePS chemistry. Whereas 2OMePS hit toxicity ceiling at 20 mg/kg, morpholinos have been taken to 960 mg/kg in mice, 320 mg/kg in monkeys, and 50 mg/kg in human trials (9).
Preclinical studies in multiple laboratories have shown a clear dose-dependence of morpholino oligo rescue of dystrophin, and more appears to be better. Furthermore, morpholinos are quickly cleared by the kidney and are excreted intact. However, morpholinos are costly; dosed at 50 mg/kg/wk, raw drug production costs may exceed $100,000 per patient per year, making morpholinos potentially cost prohibitive.
Still, if morpholino chemistry is safer and capable of yielding higher dystrophin production in preclinical studies, why has the FDA balked at accelerated approval? First, the reliability of dystrophin as a biomarker has been challenged. Determining what constitutes an “adequate” dystrophin level to predict later clinical benefit is further complicated by difficulties in obtaining representative muscle biopsies. Fundamentally, dystrophin rescue levels are likely quite variable across a DMD patient’s muscle (given dog and mouse exon-skipping data). Also, as a DMD boy ages, his myofibers are replaced in a patchy fashion by fibrofatty connective tissue, leaving less and less muscle to rescue by oligos or any other approach. Many exon-skipping trials have focused on older boys, because these patients, with their low six-minute walk distances, could show the greatest benefit. However, older patients may be hard to treat than younger patients, because older boys have less intact muscle. On the other hand, younger patients display greater six-minute walk performance, which may remain stable over the course of a clinical trial—a natural history potentially seen in untreated patients. Thus, the six-minute walk test might not be an appropriate primary endpoint in young patients. Underlying all of these problems is an inherent variability in biochemical and clinical outcomes- even neighboring cells show different extents of rescue in exon skipping and Becker Muscular Dystrophy muscle. Understanding this vari- ability is necessary to make the therapeutic approach more robust and dystrophin production data more compelling.
STEERING AROUND THE POTHOLE
Understandably, DMD parents who have seen impressive stabilization in six-minute walk distance in a subset of morpholino-treated patients have pressed FDA for accelerated approval. independent of increases in dystrophin content, because this biomarker is an imperfect proxy for clinical benefit. FDA has been very attentive, holding many meetings with stakeholders. But on what data can an accelerated approval be based—clinical (six-minute walk test) or surrogate biochemical (de novo dystrophin in patient muscle)? Dystrophin is the primary site of the biochemical defect in DMD, and restoration of dystrophin by an experimental treatment is a logical basis for accelerated approval. However, the Sarepta trial results have shown dystrophin levels to be highly variable among patients and significantly increased in too few patients to build a compelling case for approval. Although stabilization of the six-minute walk in a subset of patients is indeed impressive, it is a very small number of patients (N=10) in an unblinded phase 2b trial. Stabilization of walk distance was used by GSK/Prosensa to justify the resource-intensive phase 3 trial that then failed, and control groups in other trials have also shown impressive stabilization (likely the result of a training effect). Thus FDA may be hesitant to run that same play again.
So, has exon skipping hit a roadblock, detour, or bump in the road? Perhaps the best analogy is a very large pothole—one that gives a driver pause. Increased potency without toxicity and at an acceptable cost will be needed to traverse the pothole. There are many promising ways to boost potency, including with the use of adjuvants or modified oligos, but these tweaks will take time to develop and implement. A quicker pothole patch would be to make modifications, to drug-development strategies, that more fully de-risk clinical studies earlier in the translational path and pave the way to a more compelling accelerated package (for example, dose optimization leading to greater dystrophin detection, using robust detection methods). With sufficient de-risking, it may be possible to develop a more compelling case for dystrophin as a prognostic biomarker in short-term trials, perhaps in younger patients. There are emerging international consensus standards for dystrophin measurements in human muscle as well as new applications of mass spectrometry that can be applied to very small amounts of muscle. These manipulations may help scientists steer around the pothole.
Exon skipping remains one of the most promising approaches for dystrophin restoration in the near term, especially for the subset of DMD patients with “skip friendly” gene mutations. The road to drug approvals for orphan diseases is rarely smooth, but emphasis on de-risking and regulatory changes that encourage accelerated approvals has everyone working to improve driving conditions.
Fig. 1. Repairing disrupted dystrophin.
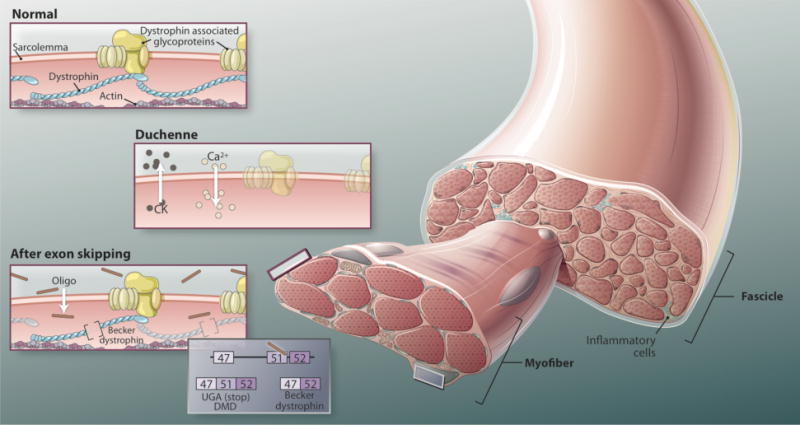
In exon-skipping therapy, for DMD patients are treated systemically with antisense oligonucleotides di- rected against the dystrophin mRNA transcript. These oligos rescue the target mRNA, restoring the proper reading frame and thus protein production. Dystrophin rescue is accomplished by excluding an exon that neighbors a deletion mutation, effectively transforming an out-of-frame (nonsense, loss-of-function) transcript to an in-frame transcript capable of de novo dystrophin production in patient muscle This dystrophin protein lacks some amino acids but retains cytoskeletal function.
REFERENCES AND NOTES
- 1.Matsuo M, Masumura T, Nishio H, Nakajima T, Kitoh Y, Takumi T, Koga J, Nakamura H. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy kobe. J Clin Invest. 1991;87:2127–2131. doi: 10.1172/JCI115244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, Partridge TA, Lu QL. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 3.Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Wilton SD. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med. 2006;8:207–216. doi: 10.1002/jgm.838. [DOI] [PubMed] [Google Scholar]
- 4.Malerba A, Thorogood FC, Dickson G, Graham IR. Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum Gene Ther. 2009;20:955–965. doi: 10.1089/hum.2008.157. [DOI] [PubMed] [Google Scholar]
- 5.Aoki Y, Yokota T, Nagata T, Nakamura A, Tanihata J, Saito T, Duguez SM, Nagaraju K, Hoffman EP, Partridge T, Takeda S. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc Natl Acad Sci USA. 2012;109:13763–13768. doi: 10.1073/pnas.1204638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ SJ, van Deutekom JC. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 9.Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J, Malik V, Shontz K, Walker CM, Flanigan KM, Corridore M, Kean JR, Allen HD, Shilling C, Melia KR, Sazani P, Saoud JB, Kaye EM. Eteplirsen Study Group. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]