

Summary
AMPylation (adenylylation) has been recognized as an important post translational modification employed by pathogens to regulate host cellular proteins and their associated signaling pathways. AMPylation has potential functions in various cellular processes and is widely conserved across both prokaryotes and eukaryotes. However, despite the identification of many AMPylators, relatively few candidate substrates of AMPylation are known. This is changing with the recent development of a robust and reliable method to identify new substrates using protein microarrays, which can significantly expand the list of potential substrates. Here, we describe procedures to detect AMPylated and auto-AMPylated proteins in a sensitive, high throughput, and non-radioactive manner. The approach employs high-density protein microarrays fabricated using NAPPA (Nucleic Acid Programmable Protein Arrays) technology, which enables the highly successful display of fresh recombinant human proteins in situ. The modification of target proteins is determined via copper-catalyzed azide–alkyne cycloaddition. The assay can be accomplished within 11 hours.
Keywords: AMPylation/adenylylation, Nucleic Acid Programmable Protein Arrays (NAPPA), activity-based protein profiling (ABPP), post-translational modification, high-throughput screening, protein microarray, click chemistry, proteomics
INTRODUCTION
In the 1960s, AMPylation (adenylylation) was first found to modulate the activity of Escherichia coli glutamine synthetase through adenylylation and de-adenylylation1. However, the functions and molecular mechanism of this modification in the regulation of biological processes were not elucidated until 2009 when Orth and Dixon found AMPylation can induce cytoskeletal collapse and cytotoxicity in mammalian cells during Vibrio parahaemolyticus and Histophilus somni infections, respectively2–4. Soon after that, Müller and others’ study revealed a reversible AMPylation mechanism mediated by two Legionella pneumophila effectors, DrrA/SidM and SidD, for host vesicle transportation5–7. Similar to phosphorylation in which kinases transfer γ-phosphate in ATP, the AMPylation enzyme, or AMPylator, delivers AMP to the tyrosine or threonine residues of their respective substrates. To date, there are two AMPylation domains that have been defined, including a Fic domain (i.e., VopS from Vibrio parahaemolyticus and IbpA from Histophilus somni) and an adenylyltransferase domain (i.e., DrrA from Legionella pneumophila)4,6,7. In view of the Fic protein domain’s conservation in over 3,000 species including archea, bacteria, viruses, viroids and eukaryotes (Fig. 1)8,9, AMPylation could regulate many potential functions in a variety of cellular processes3,4,10–12. For example, Yarbrough and Woolery found that a bacterial Fic AMPylator, VopS, modifies host Rho GTPases, blocks their interaction to downstream targets and alters NFκB, Erk and JNK signaling3,11,13. Using the fly as a genetic model, Rahman revealed that Drosophia Fic (dFic) controlled visual neurotransmission and the flies became blind with the ablation of dFic by mutations12. Ham further identified a substrate, BiP, for dFic and demonstrated their participation in the unfolded protein response pathway13. However, the role of AMPylation is only beginning to be elucidated because, until recently, there have been no robust methods to identify substrates of these enzymes. The early methods developed to find AMPylation substrates, including anti-AMPylation antibodies, mass spectrometry and cell-based pull down assays, revealed only a half-dozen potential targets combined3,4,14–19.
Figure 1.
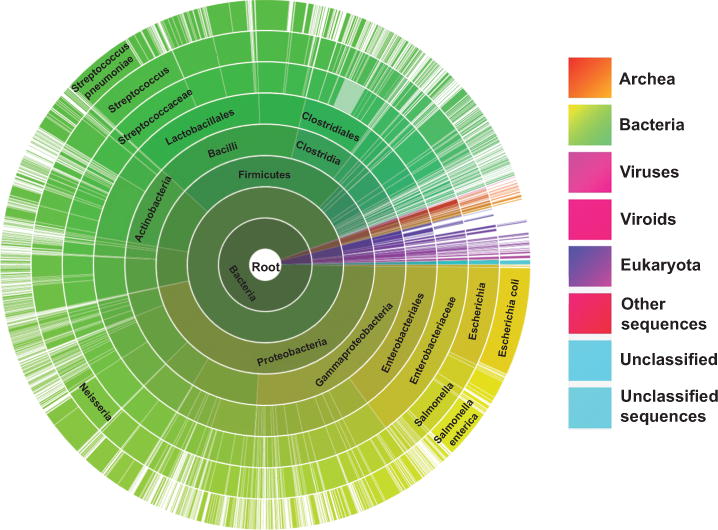
Distribution of Fic/DOC protein family sequences across a variety of 3,000+ species. The image was obtained from the pfam database (http://pfam.xfam.org/family/Fic) with minor modification9.
To address this need, we developed a high-throughput screening platform using Nucleic Acid Programmable Protein Arrays (NAPPA), which allows non-radioactive detection of AMPylated and auto-AMPylated proteins with high-sensitivity and specificity in an unbiased manner. Traditional protein arrays rely on printing purified proteins. With the NAPPA method, purified plasmid cDNA is printed on an amino-modified microscopic slide along with an anti-tag antibody, bovine serum albumin and BS3 cross-linker. This material is allowed to dry and can be stored anhydrously at ambient temperature for months without losing activity. At the time of use, the cDNA is transcribed and translated in situ into the recombinant proteins-of-interest using a mammalian cell-free expression system, and then captured to the array surface through fusion tag-anti-tag antibody with high affinity and specificity (Fig. 2)20,21. Several in vitro transcription/translation cell-free expression systems are commercially available depending on the target protein(s), for example, human HeLa cell lysate and rabbit reticulocyte lysate10,22. NAPPA makes use of our laboratory’s large >200,000 plasmid repository (DNASU Plasmid Repository, https://dnasu.org/), which includes 13,000+ plasmids that encode for unique human genes, as well as plasmids representing whole genomes for model systems and human pathogens.
Figure 2.
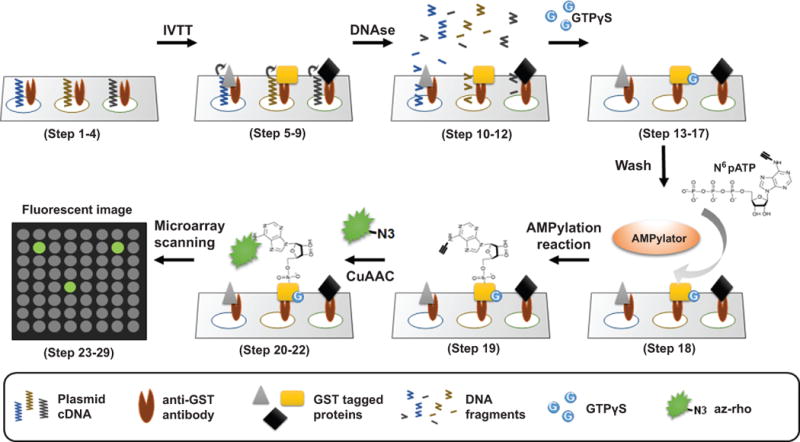
Outline of NAPPA protocol for the detection of AMPylation substrates. NAPPA arrays printed with 13,000+ human plasmid cDNAs are blocked with Tris-based SuperBlock solution to decrease nonspecific interactions (Step 1–4). The cDNA is then subjected to in vitro transcription and translation to express recombinant tagged proteins using a cell-free expression system. The proteins are captured & displayed in situ by the anti-tag antibody with high specificity and affinity (Step 5–9). DNA is removed to decrease non-specific binding using DNAse (Step 10–12). GTPγS is added in case the proteins require GTP for their activation, such as GTPases (Step 13–17). Next, the arrays are incubated with N6pATP and AMPylator enzyme to allow the transfer of AMP to the substrates (Step 18, 19). The arrays are washed and the AMPylated proteins on the array are labeled with rhodamine-labeled azide (az-rho) through copper-catalyzed azide–alkyne cycloaddition labeling (CuAAC) (Step 20–22). Finally, the slides are scanned using a microarray scanner (Steps 23–29) and the signal of substrate AMP labeling is quantified using software for microarray data analysis (Step 30–34). The AMPylators used for screening can be the enzymes with Fic and adenylyltransferase domains.
Prior to the AMPylation assay, the array printed with cDNA is incubated with a human HeLa cell-free expression system, which executes the coupled transcription and translations within two hours. Following protein expression and display, the plasmid cDNA is removed with DNAse in order to decrease non-specific binding between the DNA and AMPylation reagents. Array quality control includes assessing overall printing and DNA deposition using a fluorescent DNA stain and the analyses of protein display using an anti-tag antibody. For example, in our GST-tagged protein microarrays, the protein display correlation across arrays is R > 0.90 using a mouse anti-GST antibody and a fluorescently-labeled anti-mouse antibody, indicating a high reproducibility in the fabrication of NAPPA protein arrays (Fig. 3)10,23.
Figure 3.
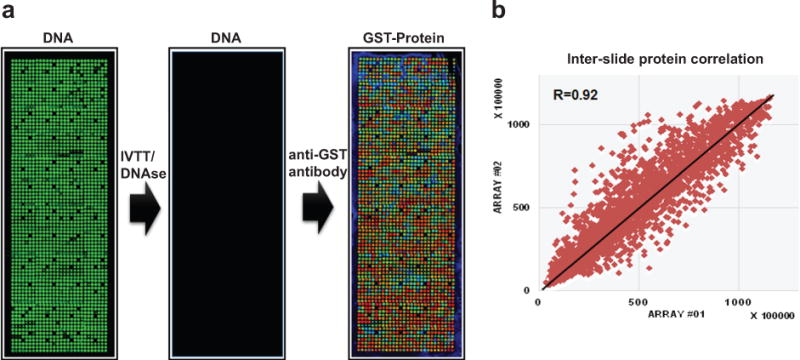
Quality control of self-assembled human protein NAPPA microarrays. (a) Expression clones (n=1,715 human ORFs) encoding the target proteins fused to a C-terminal GST tag were printed along with a polyclonal anti-GST antibody in single spot on the array surface. DNA capture was confirmed by PicoGreen staining (DNA, left), the plasmid DNA was removed using DNase after expression (DNA, middle).and the protein displayed in situ was assessed by a monoclonal anti-GST antibody (GST-Protein, right) (GST color code: red>orange>yellow>green>blue). (b) The correlation coefficient R of GST signal between two NAPPA protein arrays is 0.92.
Over the last decade, activity-based protein profiling (ABPP) using bioorthogonal chemical probes has become a powerful technology to screen for target proteins modified by active enzymes. Central to this approach is the ability to link two molecules covalently in highly specific reactions. Among the most successful chemistries is copper-catalyzed azide–alkyne cycloaddition (CuAAC) in which one participant is labeled with an alkyne group that reacts with the other participant labeled with an azide group24. The reaction is highly efficient, requires the presence of copper to trigger the reaction, and has negligible background, even in a complex mixture. The assay using bioorthogonal chemistry is also non-radioactive, robust and sensitive. The specificity and efficiency of this method, which are far superior to typical antibody-mediated interactions, and the smaller steric profiles of the alkyne and azide groups compared with common protein tags, made this the first choice for the development of a high-throughput AMPylation screening platform on NAPPA arrays10,25.
To perform AMPylation screening, the expressed NAPPA arrays are incubated with N6pATP, an alkyne containing ATP analog, and AMPylator enzyme to execute the transfer of N6-modified AMP to its substrates. After washing away the excess ATP analog, the AMPylated proteins on the array can be detected with azide-rhodamine (az-rho) based on CuAAC (Fig. 2). The slides are then scanned using a microarray scanner and the signal of substrate AMP labeling via fluorescent rhodamine is quantified using software for microarray data analysis. Finally, potential substrates are selected by comparing the slides probed with active enzyme to slides with either assay buffer alone or treated with inactive mutant enzymes (if available) as a negative control.
Applications of the method
Using this method, we have successfully confirmed known host AMPylation substrates (RhoA, Rac1 and Cdc42) of the bacterial AMPylator proteins, VopS and IbpAFic2. We also identified dozens of novel human substrates that were selectively confirmed both in vitro and in vivo. We found a novel AMPylation inhibition mechanism by phosphorylation on a novel non-GTPase substrate protein, LyGDI, in vitro. In addition, we detected the auto-AMPylation of both wild-type and mutant (E234G) mammalian AMPylator, HYPE, on NAPPA arrays in a previous study10 and the experiment described here (Fig. 4), respectively. E234G is an active HYPE mutant in which the auto-AMPylation and AMPylation activity is significantly enhanced after removing the inhibitory Glu234 that competes with binding of Arg374 to ATP γ-phosphate26,27. These results demonstrated that this method works to identify substrates of AMPylators with different folding domains from different species, as well as potential new AMPylators with auto-AMPylation activity. More in-depth characterization of AMPylation substrates and mechanisms will provide a better understanding of the functional consequences of AMPylation performed by endogenous and exogenous sources10. Moreover, the strategy described here could be adapted to other forms of PTMs. In addition, we previously demonstrated the assembly of multiprotein complexes on NAPPA arrays, indicating the potential of our method to identify the modified substrates when complexed20.
Figure 4.
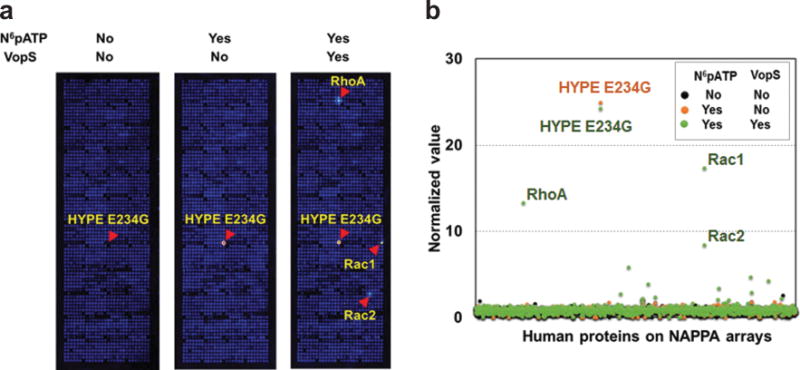
Representative results from the AMPylation screening with self-assembled human protein NAPPA microarrays. (a) Detection of auto-AMPylated and AMPylated proteins on NAPPA arrays after incubation with N6pATP and VopS, a Fic AMPylator from Vibrio parahaemolyticus. HYPE E234G is a mutant form of the human FicAMPylator that removes the wild-type inhibitory effect of αinh helix located at the front of Fic domain (i.e., active HYPE mutant). In this experiment, we employed three slides that were incubated with AMPylation buffer, N6pATP and the mix of N6pATP and VopS, respectively. Compared to the first array, we can observe strong fluorescent signals on the HYPE E234G spot on the second array, indicating the auto-AMPylation of the HYPE E234G proteins. Furthermore, with the addition of VopS, we can observe fluorescence of several additional spots, indicating Rac1, Rac2 and RhoA proteins were AMPylated by VopS enzyme. (b) Comparison of the normalized value of proteins on NAPPA arrays that detects AMPylation.
Comparison of AMPylation screening methods
A variety of methods have been developed to screen for potential AMPylation substrates using mass-spectrometry and NAPPA protein microarrays. A detailed comparison of these methods is in TABLE 1. The advantages of mass spectrometry based screening methods are their abilities to analyze natural proteins in biological samples, such as cell lysates and tissue, with high-sensitivity and flexibility. However, only a limited number of bacterial and mammalian AMPylator substrates have been identified using mass spectrometry13,14,16–19. Possible reasons for this may be due to the temporal and spatial expression or low abundance of target proteins in cells, low affinity and specificity of capture molecules, or loss of proteins during pull-down and sample preparation for mass spectrometry.
TABLE 1.
Comparison of current AMPylation screening methods.
| No | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Platform | AP-MS | AP-MS | MS | AP-MS | NAPPA |
| Method | Click chemistry+MS |
Fluorescent ATP+MS |
Isotope labeling+MS |
Anti-AMPylation antibody+MS |
Click chemistry+NAPPA |
| Type of ATP | N6pATP | Fl-ATP | Isotope-labeled ATP | ATP | N6pATP |
| Source of substrate proteins | Cell lysate | Cell lysate | Cell lysate | Cell lysate | cell-free produced recombinant human proteins |
| Isolation of AMPylated proteins | Pull-down | Pull-down | No | Pull-down | No |
| Identification | MS | MS | MS | MS | Fluorescence |
| Potential target identified | 1 known substrate for VopS (Cdc42) | 6 for VopS, including 2 known substrates (Rac1 and Cdc42) | 1 for Bep2 | None reported | 27 for VopS and 29 for IbpA, including 3 known substrates for each AMPylator (RhoA, Rac1 and Cdc42) |
| Advantages | Natural proteins, with PTMs | Natural proteins, with PTMs | Natural proteins, with PTMs | Natural proteins, with PTMs | No protein abundance issue |
| Possible reason of failed target identification | Low abundance in cell lysate; quality of capture molecules; loss of proteins during pull-down and sample preparation | Low abundance in cell lysate; quality of capture molecules; loss of proteins during pull-down and sample preparation | Isotopic toxicity; Low abundance in cell lysate; loss of proteins during pull-down and sample preparation | Low abundance in cell lysate; quality of anti-AMPylation antibodies; loss of proteins during pull-down and sample preparation | Protein may not be well-folded or with PTMs |
| Reference | Grammel M, et al.19 | Lewallen DM, et al.16 | Pieles K, et al.14 | Hao Y, et al.17 | Yu X, et al.10 |
AP-MS, affinity-purification mass spectrometry; NAPPA, Nucleic Acid Programmable Protein Arrays.
The use of protein microarrays appears to eliminate the challenge of studying low abundance proteins as the proteins are displayed in an unbiased manner on the array23,28–32. By combining click chemistry with NAPPA, we identified 27 potential substrates of VopS and 29 potential substrates of IbpAFic2, thus increasing the total number of reported targets for those enzymes about ten-fold14–19. Moreover, two new GTPase substrates (Rac2 and Rac3) were selectively validated in HEK293T cells using a Vibrio parahaemolyticus stain containing VopS only10. It will be important to perform in vivo confirmation of other substrates that were initially identified in vitro.
Advantages of the method
There are several distinct advantages using a NAPPA-based approach over other published methods to identify potential AMPylator substrates: (1) Higher sensitivity. This method has identified many times more candidate substrates compared with all previous methods10,14,15,17–19; (2) High throughput. Thousands of unique proteins can be tested simultaneously. There are currently 13,000 unique human proteins available on the platform with new clones added regularly; (3) Highly consistent display levels. About 95% of human proteins can be displayed on NAPPA, with very consistent display levels among the proteins. We have found that 93% of proteins have levels within two fold of the mean, which avoids abundance biases and thus tests every protein21,23,33. (4) Improved translation using human cell-free expression system. The use of human ribosomes and chaperones increases the likelihood of complete protein translation and natural protein folding10,34; (5) Fast. The experiment can be performed in only a few days; (6) Cost-effective. The ability to test so many proteins so quickly significantly lowers the cost of this approach. The NAPPA arrays themselves are cost effective to produce because preparing and printing DNA is much easier and more reliable than expressing, purifying and printing proteins.; (7) Non-radioactive. N6pATP based click detection provides an alternative safe approach compared with radioactive assays; and (8) Flexibility. This may be the most important advantage of this approach. The use of programmable arrays allows for great flexibility in selecting candidate substrates – as long as cDNAs are available, the corresponding proteins can be included on the arrays. Thus, researchers who wish to test the proteomes of other organisms, or different variations of proteins, such as splice variants or mutants, can be accommodated. With ability to synthesize genes de novo now, nearly any protein can be included.
Limitations of the method
First, the success of AMPylation substrate screening relies upon the availability of an active purified AMPylator enzyme, which can bind and transfer N6pATP, to probe the arrays. The previous detection of dozens of AMPylation substrates of VopS and lbpAFic, as well as the auto-AMPylation of HYPE on NAPPA, demonstrated the great potential of our method to find substrates of uncharacterized enzymes10. Preparing well-characterized and active AMPylator enzyme is not trivial and requires significant biochemistry effort. It should be noted that this is a limitation of all in vitro AMPylation screening methods. However, now that a high-throughput method exists for identifying targets, more laboratories will invest in producing purified active enzyme because of the opportunity for greater return. Second, this method relies on having access to NAPPA based protein microarrays. We have widely published detailed protocols for producing NAPPA arrays, as well as published on recent improvements in the method21,35–37. In addition, we have published a video tutorial for how to build NAPPA arrays (https://www.youtube.com/watch?v=Ur1fg9jQv04)36. Furthermore, researchers who do not have direct access to resources to build, print, screen, scan, or analyze their own arrays can make use of our central plasmid repository (https://dnasu.org/) and NAPPA core service and facility (http://nappaproteinarray.org/), which distribute plasmid cDNAs and NAPPA arrays at academic prices, respectively28,38. Third, this method can only test targets for which there are available cDNAs. Currently, there are ~13,000 unique genes available for testing; however, efforts are underway to expand this number to obtain one representative clone for every human gene. Fourth, the substrate proteins expressed and displayed on NAPPA may not be folded correctly, may not have appropriate PTM, or may require a cofactor of some kind. For studies on human targets, the use of human lysate including chaperone proteins to produce the proteins is favorable compared to most other protein production methods, but comes with no guarantees. Moreover, the ATP modified with an alkyne group might interfere with the modification of potential substrates. However we did not observe these issues in our experiments for known AMPylation substrates, which were confirmed with this NAPPA platform10. Fifth, as with any in vitro method, candidate substrates may be detected that represent false positives and may not occur naturally during the course of a true infection. Additional steps will be needed to confirm all detected candidate substrates. And last, NAPPA platform adaptation and optimization will be needed for other PTMs, such as phosphorylation, phosphocholination or UMPylation39. Preliminary studies show that phosphorylation can be monitored using anti-phosphorylation antibodies on NAPPA40. The development of phosphocholination and UMPylation assays on NAPPA might be executed using anti-phosphocholine antibody41 and C8-Alkyne-dUTP42, respectively.
Experimental design
Preparation of AMPylators
Not surprisingly, recombinant bacterial AMPylators used in AMPylation studies have been primarily expressed in bacteria. However, many proteins do not express well in bacteria giving low or no yield, possibly due to the size, cytotoxicity and insolubility of target proteins. Bacteria may also not possess the appropriate chaperones and co-factors for folding the recombinant proteins into their native conformations. To circumvent this issue, AMPylator truncations containing only the Fic or adenylyltransferase domain can be constructed, which can be expressed more easily from the bacteria system by reducing the size or hydrophobic part of the protein sequence3,4. Theoretically the truncations could alter substrate specificity, though these modified forms have yielded a number of verified targets43. Cell-free expression is an alternative choice that avoids cytotoxicity and has been shown to express many more proteins successfully compared to bacterial expression systems, especially for large sized proteins or membrane proteins44,45. However, it is much more expensive than bacteria, and would be a second choice if bacteria can produce active protein.
Full-length proteins may also interfere with AMPylation screening as Engel et al.26 found an α-helix (αinh) with a conserved (S/T) XXXE (G/N) motif within Fic AMPylators that prevents ATP γ-phosphate binding and inhibits the enzyme activity. The mutation of this inhibitory motif could significantly enhance the auto-AMPylation and AMPylation of enzymes, e.g., N. meningitids Fic (NmFic) and Homo sapiens Fic (HYPE)26.
Either inactive enzyme or assay buffer can be used as a negative control for high-throughput AMPylation substrate screening. Most Fic AMPylators have a conserved motif (HPFXXGNG)3 and the adenylyltransferase AMPylators have a conserved motif (G-X11-D-X-D)6. Thus, inactive enzyme can sometimes be made by mutating key residues in AMPylators’ motifs. For example, the inactive VopS, IbpA and HYPE enzymes were prepared by changing the histidine of Fic motif to alanine3,4,26. The inactive SidM enzyme was prepared by changing two aspartates of adenylyltransferase motif to alanines41.
Preparation of NAPPA protein microarrays
13,000+ sequenced-verified human ORF sequences were transferred into the T7-based mammalian expression vector, pANT7_cGST, containing a C-terminal GST tag. An advantage of placing the GST tag at the protein C-terminus is that the presence of the tag confirms full-length translation of the target protein. We use the antibody to the GST tag to assess protein levels because the tag is common to all proteins on the array and because the stoichiometry is only one GST tag per protein. Thus, the signal for binding to GST can be compared from protein to protein as an indirect measure of moles of protein per feature33.
All these plasmids are available in the non-profit DNASU plasmid repository (https://dnasu.org/). Highly purified plasmid DNA is prepared by our automated DNA factory robot following the same basic chemistry published previously35. NAPPA arrays containing ~2,000 human ORFs per slide were printed by the non-profit NAPPA Protein Array Core (http://nappaproteinarray.org/) according to previously described methods21,35, and can be obtained at cost upon request (see further information below regarding access to NAPPA arrays in limitations section). Printing and protein display were assessed with PicoGreen and anti-GST antibody, respectively (Fig. 3).
Most mammalian cell-free expression systems can be used with NAPPA, including the commercialized rabbit reticulocyte lysate and human HeLa lysate kits. We found that the overall signal intensity and signal-to-noise ratio of protein expression & display and AMPylation on NAPPA were significantly enhanced using human HeLa lysate-based cell-free expression system instead of reticulocyte lysate-based cell-free expression system10,34. It is possible that expression and folding may be improved by the human ribosomal machinery and chaperones in HeLa cell lysates compared with the rabbit translational machinery10,34.
Removal of plasmid DNA from NAPPA with DNAse
After the expression of NAPPA arrays, the plasmid DNA and their corresponding mRNA molecules on the slide surface might non-specifically absorb the N6pATP, enzyme or fluorescent molecules during incubation and increase the background noise. In our previous experiment, we systematically compared the performance of AMPylation assay using NAPPA with DNAse and RNAse treatment10. The results showed that the background signal decreased after the removal of plasmid DNA from NAPPA using DNAse, and the signal to noise improved correspondingly. However, we did not observe any additional improvement with the addition of RNAse. The results indicated that most mRNA molecules were washed away after protein expression and they did not affect AMPylation screening. This approach can also be applied to different assays on the NAPPA platform such as protein-protein and protein-DNA interactions.
Loading GTPγS to NAPPA protein arrays
Thus far, most of the confirmed substrates of bacterial AMPylators are GTPases3,4,10,13,14. For example, VopS (Vibrio parahaemolyticus) employs AMPylation on activated Rho GTPases thereby inhibiting downstream signaling pathways (e.g., NFκB, Erk and JNK) and affecting actin rearrangements. Furthermore, it prevents the degradation of GTPases by E3 ubiquitin ligases3,4,10,11. Whereas SidM and SidD (Legionella pneumophila) reversibly modify activated Rab1 to regulate host vesicle trafficking using AMPylation and De-AMPylation, respectively6,7. Thus, we prefer to activate GTPase proteins on NAPPA prior to AMPylation screening by incubating the NAPPA with GTPγS, a non-hydrolyzable GTP analog in which an oxygen of γ-phosphate of GTP is replaced with a sulfur group. The nucleotides that exist on GTPase proteins can be exchanged to GTPγS using 7.5 mM EDTA in Tris-HCl buffer based solution, and the reaction is then stopped with addition of 50 mM MgCl23. However, this step might not be necessary if there is no evidence of AMPylator’s targets or activity associated with GTPase.
Screening for AMPylation substrates using NAPPA protein microarrays and click chemistry
To perform AMPylation screening using NAPPA, the expressed and DNAse-treated NAPPA protein arrays are incubated with N6pATP and AMPylator in AMPylation buffer at 30°C for 1h. The optimal assay conditions might differ depending on the specific AMPylator used. After the AMPylation reaction, the modified substrates are detected using az-rho through CuAAC. This method of detection is more sensitive than using antibodies directed at AMPylated peptides10. The criteria of target selection are described in detail below (Step 34). To obtain a reliable data set, the screening experiment should be done at least three times on independent days. Only the targets reproducibly detected in all experiments should be selected as potential substrates to decrease the number of false positives, which can be a concern particularly in large screenings (e.g., high throughput arrays).
All standard fluorescent microarray scanners compatible with 75×25mm slides can be used for NAPPA arrays. The procedure is basically the same for different scanners. Here we employ the Tecan PowerScanner in our lab as an example and show the critical parameters for the array scanning and obtaining of fluorescent images, including laser, emission filter, PMT, gain and resolution (step 23 – 29). The wavelengths of the laser for excitation and emission filter need to match to that of fluorescent rhodamine (586, 601nm). A slight difference of excitation/emission wavelengths between scanner and fluorescent dye will result in the decrease of quantified signals. Different fluorescent dyes (i.e., Cy3 and Cy5) conjugated with azide, which are commercially available, can be used as an alternative. PMT and gain are used for the linear adjustment of signal levels from 0.01% to 1000%. The PMT and gain should be tested prior to formal scanning to optimize the dynamic range, such that the background signals are low while ensuring that the strong signals are maximized but not saturated. Increasing resolution will enhance the pixel number for each microarray spot, but will slow the scanning speed. Normally we use 10 μm resolution for the NAPPA scanning, which takes about 8 min for each slide. The produced fluorescent images are sufficient for the spot examination and subsequent signal quantification.
Validation of selected targets in vitro and in vivo
Confirmation of selected targets can be performed using an independent rapid bead-based AMPylation assay in vitro10. The principle of bead-based AMPylation assay employs GST-tagged substrate proteins that are immobilized on magnetic beads displaying polyclonal anti-GST antibody. The AMPylation reaction is performed by incubating the substrate protein-coated magnetic beads with N6pATP and AMPylator. The beads are then washed with PBST and the azide-fluorescein is added so that the substrates AMPylated with alkyne will be labeled through CuAAC. Labeled substrate proteins are then released from the beads under heating (95°C, 5min) in 1× SDS-loading buffer and then loaded on a SDS-PAGE gel, where the AMPylated substrates are detected through in-gel fluorescence. Comparisons to total substrate protein are accomplished by using the same gel for a western blot with monoclonal anti-GST antibody. The assay can be easily adapted to be high-throughput using a 96-well microplate and magnet10.
Promising substrates eventually require validation in vivo. This is often accomplished in cell-based assays; however, a detailed description of how to validate substrates in vivo is beyond the scope of this article. Such assays require careful optimization and must be individualized based on the specific candidates tested. In previous experiments, we have observed success with the stable expression of both AMPylator and substrate in cells followed by pull down and confirmation by either antibodies or mass spectrometry10. It should be noted that there are technical reasons that a bona fide substrate may fail to validate in vivo, such as competing PTMs or specific cellular growth requirements.
Bioinformatics is very helpful for prioritizing candidates based on the annotation of their protein classes, functions and subcellular locations, as well as searching for motifs that are conserved in previously validated substrate candidates. This approach can provide information about network interactions and biological processes10,46. In previous work, we identified a YxPVTF motif in six GTPases and one non-GTPase protein. We ectopically expressed candidate substrates in HEK293T cells, which were then challenged with a Vibrio parahaemolyticus strain expressing VopS. Target AMPylation was identified by immunoprecipitation using an anti-AMPylation antibody followed by mass spectrometry10. Among the five targets tested (Rac2, Rac3, LyGDI, LC3 and LENG1), Rac2 and Rac3, which contained the YxPVTF motif, were confirmed for their AMP modification by VopS in vivo.
MATERIALS
REAGENTS
SuperBlock (TBS) Blocking Buffer (Thermo Scientific, cat. no. NC0276612)
Sodium chloride (NaCl, Sigma-Aldrich, cat. no. S3014)
Sodium phosphate dibasic (Na2HPO4, Sigma-Aldrich, cat. no. S3264)
Potassium chloride (KCl, Sigma-Aldrich, cat. no. P9541)
Potassium phosphate monobasic (KH2PO4, Sigma-Aldrich, cat. no. P5655)
-
Human HeLa lysate based cell-free expression system (1-Step Human Coupled IVT Kit, Thermo Scientific, cat. no. 88882)
! CAUTION The human cell-free expression system should be stored at − 80°C and limited to three freeze/thaw cycles.
DNase I (Sigma-Aldrich, cat. no. AMPD1)
Bovine serum albumin (BSA, Sigma-Aldrich, cat. no. A7906-1KG)
Tris-[hydroxymethyl]aminomethane (Tris, MP Biomedicals, cat. no. 04819638 – 5 kg)
-
Hydrogen chloride (HCl, Sigma-Aldrich, cat. no. 295426-227G)
! CAUTION HCl is a strong corrosive. Wear a safety face shield, gloves and a lab coat. Prepare the solution in a chemical fume hood.
-
Sodium hydroxide (NaOH, Sigma-Aldrich, cat. no. S8045-1KG)
! CAUTION NaOH is a strong corrosive. Wear a safety face shield, gloves and a lab coat. Prepare the solution in a chemical fume hood.
Ethylenediaminetetraacetic acid (EDTA, G-Biosciences, cat. no. RC-048)
Guanosine- 5′- O- (3- thiotriphosphate) (GTPγS, BIOLOG, cat. no. G019-05)
Magnesium chloride (MgCl2, EMD, cat. no. MX0045)
4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES, Sigma-Aldrich, cat. no. H4034-25G).
-
Potassium hydroxide (KOH, Sigma-Aldrich, cat. no. P5958-250G)
! CAUTION KOH is corrosive. Wear a safety face shield, gloves and a lab coat. Prepare the solution in a chemical fume hood.
DL-Dithiothreitol (DTT, Sigma-Aldrich, cat. no. 43819-5G)
N6-Propargyl-ATP (N6pATP, Jena Bioscience, cat. no. CLK-NU-001)
Triethanolamine (Sigma-Aldrich, cat. no. 90278)
Tris(2-carboxyethyl)phosphine (TCEP, Sigma-Aldrich, cat. no.646547-10X1ML)
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, Sigma-Aldrich, cat. no. 678937-50MG)
Copper(II) sulfate, anhydrous (CuSO4, Sigma-Aldrich, cat. no. 451657-10G)
Sulforhodamine 101 Azide (az-rho) (Click Chemistry Tools, cat. no. AZ110-5)
-
NAPPA microarrays printed with target ORF sequences in a mammalian expression vector with a T7 promoter and a carboxyl-terminal GST tag (e.g., pANT7-cGST) are available from NAPPA Protein Array Core (http://nappaproteinarray.org/). The NAPPA microarrays can also be fabricated by the users according to the published protocol21,35–37 and tutorial video (https://www.youtube.com/watch?v=Ur1fg9jQv04)36.
! CAUTION The NAPPA arrays can be stored under anhydrous conditions up to six months at room temperature.
The individual human ORF clones, DNASU (http://dnasu.asu.edu/DNASU/).
EQUIPMENT
ELGA LabWater Purification system (ELGA, cat. no. PURELAB Option S-R 7-15)
Make type II water, up to 15 MΩ-cm, for the preparation of general solutions in lab
CELLSTAR four-well Plate (Greiner bio-one, cat. no. 96077307)
HybriWellSealing System (Grace BIO-LABS, cat. no. 440904)
VWR Rocking platform (VWR, cat. no. 4000-304)
Centrifuge (Beckman Coulter, cat. no. Allegr X-12R)
SX4750A ARIES™ Rotor “Swinging Bucket” Rotor “Assembly”(Beckman Coulter, cat. no. 369704)
SX4750 Rotor Multiwell-Plate Carriers (Beckman Coulter, cat. no. 392806)
Programmable Chilling/Heating Incubator (EchoTherm, cat. no. IN30)
Tecan’s PowerScanner (Tecan)
Array-Pro Analyzer, version 6.3 (Media Cybernetics)
Excel2013 software (Microsoft)
REAGENTS SETUP
10 × PBS Mix 80 g NaCl, 14.4 g Na2HPO4, 2 g KCl and 2.4 g KH2PO4 with 800 ml H2O. Adjust the pH to 7.4 with HCl and add H2O to 1 liter. The final concentration of each component is 1.37 M NaCl, 100 mM Na2HPO4, 27 mM KCl and 18 mM KH2PO4. Store the solution at 4°C for up to one year.
1 × PBST Mix 100 ml 10× PBS with 2 ml Tween 20, and add H2O to 1-liter. The final concentration of Tween 20 is 0.05 % (vol/vol). Store the solution at room temperature for less than one week.
1 × DNAse Mix 100 ul of 10× DNAse, 100ul of 10× DNAse buffer and 800ul H2O to 1 ml. Always use fresh solution for the experiments.
- Prepare human HeLa lysate-based cell free expression system as below (Step 6).
Name Volume [μl]
HeLa lysate 75
Accessory proteins 15
Reaction mix 30
Nuclease-free Water 30
Total/Slide 150
1 % BSA Dissolve 1 g BSA in 100 ml of PBST. Store at 4°C for less than 2 days.
1 M Tris Dissolve 60.57 g Tris with 0.4 liter of H2O, adjust the pH to 7.5 with HCl, and add H2O to 0.5 liter. Store the solution at room temperature for up to one year.
0.5 M EDTA Dissolve 94.5 g with 0.4 liter of H2O, adjust the pH to 8.0 with NaOH, and add H2O to 0.5 liter. Store the solution at room temperature for up to one year.
GTPγS buffer Mix 50 ml of 1 M Tris-HCl, 15 ml of 0.5 M EDTA and H2O to the final volume of 1 liter. Store the solution at room temperature for up to one month.
1 M MgCl2 Dissolve 20.3 g MgCl2 in 100 ml of H2O. Store the solution at room temperature for up to one year.
1 M HEPES Dissolve 23.83 g HEPES in 80 ml H2O, adjust the pH to 7.5 with NaOH, and add H2O to 100 ml. Store the solution at room temperature for up to one year.
GTP washing buffer Mix 5 ml of 1M HEPES, 2.5 ml of 1M MgCl2 and H2O to the final volume of 500 ml. Store the solution at room temperature for up to one year.
1 M DTT Dissolve 0.15 g in 1 ml H2O. Always use fresh solution for the experiments.
2.5 M NaCl Dissolve 73.05 g NaCl in H2O of 500ml. Store the solution at room temperature for up to one year.
AMPylation buffer Mix 10 ml of 1M HEPES, 20 ml of 2.5 M NaCl, 2.5 ml of 1M MgCl2, 50 mg BSA, 0.5ml of 1M DTT and H2O to 500 ml. Store the solution at 4°C for less than a week.
4ST Mix 100 ml SDS, 30ml of 2.5 M NaCl, 25 ml of 1 M Triethanolamine and H2O to 500 ml. Store the solution at room temperature for up to one year.
50 mM TCEP Mix 1 ml 0.5 M TCEP with 9 ml H2O, and aliquot to 1 ml for each. Store the solution at − 80°C for up to one year.
2 mM TBTA Dissolve 50 mg in 47 ml H2O, and aliquot to 1 ml for each. Store the solution at − 80°C for up to two years. Store the solution at − 80°C for up to one year.
50 mM CuSO4 Dissolve 0.4 g CuSO4 in H2O to 50 ml. Store the solution at room temperature for up to one year.
-
Prepare az-rho solution as below (Step 20).
Name Stock [mM] Volume [μl]
4ST 52.5
az-rho 10 1.75
TCEP 50 3.5
TBTA 2.5 8.75
CuSO4 50 3.5
H2O 105
Total/Slide 175
EQUIPMENT SETUP
Programmable Chilling/Heating Incubator setup
Set up a program with the temperature of 30°C for 1.5 h and 15°C for 0.5 h, which is used for the protein expression on NAPPA arrays.
VWR Rocking platform setup
Set up the rocking platform to the setting of “2”, which is approximately 50–70 tilts per minute, and use for washing NAPPA slides after incubation.
Centrifuge setup
To remove excess liquid from the slides, set the centrifuge to 1000 × g for 2 min at 4 °C.
Tecan’s PowerScanner setup
Turn on the scanner and computer to allow connection. Open the microarray scanning software and choose the green laser with the emission filter of 575 ± 30 nm for the scanning of NAPPA probed with fluorescent rhodamine.
PROCEDURE
Blocking and in vitro transcription and translation of NAPPA protein arrays • TIMING ~ 3.5 h
Obtain or prepare the NAPPA arrays21,35–37. Here we use three slides from one array (n=1,715 human genes) as an example to show the processing of AMPylation assay using NAPPA and click chemistry (Fig. 5a).
Transfer three slides into a four-well plate, add 3 ml superblock solution to each well and incubate at room temperature for 1 h on rocking platform with the setting of “2”.
Wash the slides briefly with H2O
Remove the liquid on slides surface by centrifugation with 1,000 × g, 2 min at 4°C.
-
Carefully align the HybridWell hybridization chamber to the slides surface and seal the chamber to the slides by rubbing around the seal using a plastic stick (Fig. 5b).
▲CRITICAL STEP Avoid touching the HybridWell hybridization chamber to the microarray spots on glass slide.
?TROUBLESHOOTING
-
Inject 140 μl of Human cell-free expression system (see Reagent Setup) into the chamber through the hole at the right side of slide, and seal the holes using a circle sticker (Fig. 5c–d).
▲CRITICAL STEP Remove the air bubbles generated during the injection by gently tapping the HybridWell while holding the slide at an angle. The presence of bubbles may affect the expression & display of some proteins on NAPPA.
■ PAUSE POINT Any remaining human cell-free expression system solution can be stored at − 80 °C. However, the solution should be used within three freeze/thaw cycles to avoid significant loss in activity.
-
Start the in vitro transcription and translation of NAPPA arrays by incubating the slides for 1.5 h at 30°C and following with 0.5 h at 15°C in the programmable incubator.
?TROUBLESHOOTING
Remove the slide chamber and briefly wash the slides with PBST.
Tap the slide on its edge on a paper towel to remove excess buffer, and immediately go to the next step.
Figure 5.

The main steps for the in vitro transcription and translation of plasmid DNAs that are printed on NAPPA arrays. (a) Obtain NAPPA arrays printed with plasmid cDNAs encoding ~1, 715 human full-length ORFs (Step 1); (d) Seal HybridWell chamber surrounding the printed array on the slide using a plastic stick (Step 5); (c) Inject human cell-free expression system (Step 6); (d) Seal holes on chamber using a circle sticker (Step 6);
Remove plasmid DNA from NAPPA protein arrays • TIMING ~ 2 h
-
Transfer 500 μl 1× DNase to the microarray surface and cover the microscopic slide with a full-length coverslip, and then incubate for 20 min at room temperature.
▲CRITICAL STEP Avoid the generation of air bubbles during operation.
Remove the digested DNA fragments and DNAse by washing the slides three times with PBST, 5 min for each washing.
Transfer the slides into a four-well plate, add 3 ml 1% BSA solution to each well and incubate at room temperature for 1 h on a rocking platform with the setting of “2” (see Equipment Setup).
Load GTPγS to NAPPA protein arrays • TIMING ~ 1 h
Discard the BSA solution and briefly rinse slides using PBST.
Add 2.5 ml 50 μM GTPγS to each well and incubate for 15 min at 30°C in the programmable incubator.
Add 125 μl 1 M MgCl2 to each well and incubate the slides for additional 5 min at 30°C to stop the reaction.
Wash the slides with GTP washing buffer two times, 5 min for each washing.
Centrifuge the slides (placed on its side in a metal rack such that the protein spots are not in contact with any surface) at 1,000 × g for 2 min at 4 °C.
NAPPA AMPylation assay • TIMING ~ 3 h
-
18 | Apply a HybridWell hybridization chamber to the slide surface, and incubate the array with 160 μl AMPylation solution containing 40 μg/ml VopS and 250 μM N6pATP for 1 h at 30 °C. In parallel, we incubate the other two slides with the assay buffer, with and without N6pATP, which are used as controls to detect the proteins with AMPylation and auto-AMPylation respectively (see Experimental Design and Anticipated Results).
▲CRITICAL STEP The concentration of different AMPylators might differ, which depends on the enzymes’ AMPylation activity. The concentration range from 40 μg/ml to 200 μg/ml is recommended.
?TROUBLESHOOTING
-
After the AMPylation reaction, wash the three slides three times with PBST, and remove the liquid from the slides surface by centrifuging the slides with 1,000 × g for 2 min at 4°C.
?TROUBLESHOOTING
-
Apply a HybridWell hybridization chamber, add 165 μl az-rho solution (see Reagent Setup) and incubate for 1 h at room temperature.
▲CRITICAL STEP Thaw the reagents for 30 min at room temperature before preparing the az-rho solution.
-
Remove the chamber and wash the slides three times with PBST on the rocking platform with setting of “2”, 5 min for each wash (see Equipment Setup).
▲CRITICAL STEP The signal-to-noise ratio could be improved by increasing the washing time to overnight at 4°C.
?TROUBLESHOOTING
Remove the liquid from the slides surface by centrifuging the slides at 1,000 × g for 2 min at 4 °C.
Scan NAPPA protein arrays • TIMING ~ 1h for six slides with one setting
-
23 | Load NAPPA slides into the microarray scanner.
?TROUBLESHOOTING
-
24 | Choose the green laser with a 575/30 nm emission filter for the scanning of fluorescent dyes such as rhodamine, Cy3, DyLight™549 and Alexa Fluor® 555, etc.
▲CRITICAL STEP The user should choose the red laser with a 676/37 nm filter if Cy5, DyLight™649 and Alexa Fluor® 647 dyes are used for detection.
?TROUBLESHOOTING
-
25 | Remove the “Autogain”.
▲CRITICAL STEP With “autogain” an automatic gain will be assigned for each individual slide and the scanning parameters might be changed from slide to slide. Herein the “autogain” is not recommended.
-
26 | For the AMPylation detection with az-rho, we normally choose the PMT from 200 % to 400 % and a Gain of 100 % at the resolution of 10 μm. The user can choose to set up multi settings for each individual slide.
▲CRITICAL STEP The scanning setting might be different for different fluorescent dyes. Optimize the PMT and Gain within the range from 0.01% to 1000%, and then choose the optimal one with clear non-saturated fluorescent signals and low background for the image examination and signal quantification.
27 | Define the name of each image and the location where the image needs to be saved.
28 | Start the scanning. It takes 10 min to prewarm the laser before array scanning.
29 | The images will be saved as 16-bit TIFF format.
Data quantification and candidate selection • TIMING ~ 12 h
-
30 | Extract the average fluorescent signal intensity from microarray spots in a TIFF image and produce a data file (.csv) using Array-Pro Analyzer microarray software.
▲CRITICAL STEP Most commercial microarray analyzing software can be used for this work, such as ScanArray Express (Perkin Elmer) and GenePix (Molecular Devices). There also are several free software applications available, such as P-SCAN and ProteinScan, which can be downloaded from htt://mttab.cancer.gov47.
31 | Open the data file (.csv) using Microsoft Excel2013 software. Older Excel versions can also be used for this purpose.
-
32 | Normalizethe raw signal intensity using the equation below.
Where Xi.j and,Yi.j are the raw signal intensity and normalized value of each individual spot, respectively.
Bkg is the signal value at the first quartile of microarray spots printed with buffer-only as negative control, which is assumed to be the background noise induced by the non-specific binding of ATP analog, AMPylation enzyme and fluorescent molecules, and M is the median of fluorescent intensity of all protein spots.
▲CRITICAL STEP The aim of data normalization is to decrease the background variation between individual arrays. The equation was generated based on the hypothesis that only a minority of targets exist on high-density NAPPA protein arrays for unbiased screenings, so that the median of all protein spots can be used as a measure of background10,28,48.
33 | Examine the array images and remove any obvious false positive values (i.e., caused by the spot shape, dust and non-specific binding, etc.) according to their position in image and data file.
34 | Select the candidate targets using the following criteria: 1.Select proteins with a normalized value of 20 % percent above the median as positives; 2. If a parallel screen was performed using an inactive control enzyme as a negative control, calculate the ratio of substrate signal with AMPylator to its buffer or inactive mutant control, and select the potential substrates with a ratio higher than 1.2; 3. Repeat each AMPylation screening experiment three times on independent days. Positive targets in all experiments, as explained in criteria (1) and (2), should be considered substrate candidates.
TIMING
Steps 1 – 9, Blocking and in vitro transcription and translation of NAPPA protein arrays: ~ 3.5 h
Steps 10 – 12, Remove plasmid DNA from NAPPA protein arrays: ~ 2 h
Steps 13 – 17, Load GTPγS to NAPPA protein arrays: ~ 1 h
Steps 18 – 22, NAPPA AMPylation assay: ~ 3 h
Steps 23 – 29, Scan NAPPA protein arrays: ~1 h
Steps 30 – 34, Data quantification and candidate selection: ~ 12 h
TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 33 | No signals | Array slides loaded upside down in Hybridization chamber resulting in no target hybridization of probes (Step 5, 18, 20) | Repeat experiment and ensure that the hybridization chamber is covered on the slide surface with microarray spots |
| 33 | Low expression of human proteins on NAPPA (Step 6) | Check the human cell-free expression system for the protein production in vitro, which may lose its activity during shipment and storage. Check the protein expression on NAPPA or western blot using monoclonal mouse anti-GST antibody35. |
|
| 33 | The AMPylator may have no AMPylation enzyme activity (Step 18) | Check other PTMs that are mediated by the proteins with Fic domain39,49,50 | |
| 33 | N6pATP may have no activity (Step 18) | Check the activity of N6pATP using in-gel fluorescence with VopS and Rho GTPase substrates10,19 | |
| 33 | Array slides are loaded upside down in the Tecan microarray scanner (Step 23) | Load slide again in Tecan microarray scanner with the microarray spots facing up | |
| 33 | Wrong laser is chosen for array scanning (Step 24) | Chose the laser with appropriate excision and emission wavelengths | |
| 33 | Low signals | There may have an inhibitory α-helix which inhibits the AMPylation enzyme activity (Step 18) | To remove the inhibitory α-helix upstream of the Fic domain26 |
| 33 | The enzyme might lose its activity during storage (Step 18) | To prepare new enzyme proteins, aliquot and store at − 80 °C | |
| 33 | Low concentration of AMPylators or click reagents (Step 18, 20) | To increase the concentrations of AMPylators or click reagents | |
| 33 | High background | The concentration of AMPylators or click reagents might be too high (Step 18, 20) | To decrease the concentrations of AMPylators or click reagents To increase the washing time using PBST |
| 33 | Nonspecific binding of AMPylators with strong auto-AMPylation fluorescence (Step 18) | To remove the non-specific bound AMPylators from the array surface by increasing the washing time with PBST |
ANTICIPATED RESULTS
The detection of proteins with auto-AMPylation and AMPylation on NAPPA are shown in Fig. 4. In this example, we employed a NAPPA array containing 1,715 human full-length proteins and then performed the auto-AMPylation and AMPylation assays as described above (Step 18–20). The first array was incubated with AMPylation buffer to serve as the negative control. The second array was incubated with N6pATP to observe the proteins with auto-AMPylation. The third array was incubated with N6pATP and VopS to detect the AMPylation of VopS substrates. By comparing the second array to the first negative control array, HYPE E234G proteins had a strong fluorescent signal, indicating that HYPE proteins were auto-AMPylated (Fig. 4a). Moreover, with the addition of VopS, we can see that several additional proteins fluoresced when compared to the second array, indicating that these proteins were specifically modified with AMP by the VopS enzyme. We normalized the fluorescent signal intensity of all human proteins on NAPPA and drew a plot graph (Fig. 4b). The results indicate that HYPE E234G and several RhoGTPase proteins (e.g., Rac1, Rac2 and RhoA) had significant normalized values above the control array. These results confirm the ability of our NAPPA arrays to detect proteins with auto-AMPylation and AMPylation.
Acknowledgments
We thank Early Detection Research Network (5U01CA117374). We thank Kim Orth lab (Department of Molecular Biology, UT Southwestern Medical Center) and Howard C. Hang lab (The Laboratory of Chemical Biology and Microbial Pathogenesis, The Rockefeller University) for providing the purified AMPylator proteins and click reagents, respectively. We thank Brianne Petritis and Kristi Barker for the critical reading of manuscript.
Footnotes
AUTHOR CONTRIBUTIONS X.Y. designed and performed the experiments, and wrote the manuscript; J.L. designed the study and wrote the manuscript.
COMPETING FINANTIAL INTERESTS The authors declare no competing financial interests.
Link to The Virginia G. Piper Center for Personalized Diagnostics, Biodesign, ASU: http://cpdlab.biodesign.asu.edu/
TWEET: Detecting AMPylation using NAPPA arrays
Contributor Information
Xiaobo Yu, Email: xiaobo.yu@asu.edu.
Joshua LaBaer, Email: jlabaer@asu.edu.
References
- 1.Stadtman ER, et al. Multiple molecular forms of glutamine synthetase produced by enzyme catalyzed adenylation and deadenylylation reactions. Advances in enzyme regulation. 1970;8:99–118. doi: 10.1016/0065-2571(70)90011-7. [DOI] [PubMed] [Google Scholar]
- 2.Itzen A, Blankenfeldt W, Goody RS. Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem Sci. 2011;36:221–228. doi: 10.1016/j.tibs.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Yarbrough ML, et al. AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science. 2009;323:269–272. doi: 10.1126/science.1166382. [DOI] [PubMed] [Google Scholar]
- 4.Worby CA, et al. The fic domain: regulation of cell signaling by adenylylation. Mol Cell. 2009;34:93–103. doi: 10.1016/j.molcel.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan Y, Luo ZQ. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature. 2011;475:506–509. doi: 10.1038/nature10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neunuebel MR, et al. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science. 2011;333:453–456. doi: 10.1126/science.1207193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller MP, et al. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science. 2010;329:946–949. doi: 10.1126/science.1192276. [DOI] [PubMed] [Google Scholar]
- 8.Kinch LN, Yarbrough ML, Orth K, Grishin NV. Fido, a novel AMPylation domain common to fic, doc, and AvrB. PLoS One. 2009;4:e5818. doi: 10.1371/journal.pone.0005818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Research. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, et al. Copper-catalyzed azide-alkyne cycloaddition (click chemistry)-based detection of global pathogen-host AMPylation on self-assembled human protein microarrays. Mol Cell Proteomics. 2014;13:3164–3176. doi: 10.1074/mcp.M114.041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woolery AR, Yu X, LaBaer J, Orth K. AMPylation of Rho GTPases Subverts Multiple Host Signaling Processes. J Biol Chem. 2014;289:32977–32988. doi: 10.1074/jbc.M114.601310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahman M, et al. Visual neurotransmission in Drosophila requires expression of Fic in glial capitate projections. Nat Neurosci. 2012 doi: 10.1038/nn.3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ham H, et al. Unfolded protein response-regulated dFic reversibly AMPylates BiP during endoplasmic reticulum homeostasis. J Biol Chem. 2014 doi: 10.1074/jbc.M114.612515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pieles K, Glatter T, Harms A, Schmidt A, Dehio C. An experimental strategy for the identification of AMPylation targets from complex protein samples. Proteomics. 2014;14:1048–1052. doi: 10.1002/pmic.201300470. [DOI] [PubMed] [Google Scholar]
- 15.Lewallen DM, et al. Inhibiting AMPylation: a novel screen to identify the first small molecule inhibitors of protein AMPylation. ACS chemical biology. 2014;9:433–442. doi: 10.1021/cb4006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewallen DM, Steckler CJ, Knuckley B, Chalmers MJ, Thompson PR. Probing adenylation: using a fluorescently labelled ATP probe to directly label and immunoprecipitate VopS substrates. Mol Biosyst. 2012;8:1701–1706. doi: 10.1039/c2mb25053e. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Al-Eryani R, Yarbrough ML, Orth K, Ball HL. Characterization of AMPylation on threonine, serine, and tyrosine using mass spectrometry. J Am Soc Mass Spectrom. 2011;22:752–761. doi: 10.1007/s13361-011-0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao YH, et al. Characterization of a rabbit polyclonal antibody against threonine-AMPylation. J Biotechnol. 2011;151:251–254. doi: 10.1016/j.jbiotec.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grammel M, Luong P, Orth K, Hang HC. A chemical reporter for protein AMPylation. J Am Chem Soc. 2011;133:17103–17105. doi: 10.1021/ja205137d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramachandran N, et al. Self-assembling protein microarrays. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran N, et al. Next-generation high-density self-assembling functional protein arrays. Nat Methods. 2008;5:535–538. doi: 10.1038/nmeth.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miersch S, et al. Serological autoantibody profiling of type 1 diabetes by protein arrays. J Proteomics. 2013 doi: 10.1016/j.jprot.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 23.Prados-Rosales R, et al. Mycobacterial membrane vesicles administered systemically in mice induce a protective immune response to surface compartments of Mycobacterium tuberculosis. mBio. 2014;5:e01921–01914. doi: 10.1128/mBio.01921-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grammel M, Hang HC. Chemical reporters for biological discovery. Nat Chem Biol. 2013;9:475–484. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westcott NP, Hang HC. Chemical reporters for exploring ADP-ribosylation and AMPylation at the host-pathogen interface. Curr Opin Chem Biol. 2014;23C:56–62. doi: 10.1016/j.cbpa.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engel P, et al. Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature. 2012;482:107–110. doi: 10.1038/nature10729. [DOI] [PubMed] [Google Scholar]
- 27.Bunney TD, et al. Crystal Structure of the Human, FIC-Domain Containing Protein HYPE and Implications for Its Functions. Structure. 2014 doi: 10.1016/j.str.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu X, et al. Exploration of panviral proteome: high-throughput cloning and functional implications in virus-host interactions. Theranostics. 2014;4:808–822. doi: 10.7150/thno.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin YY, et al. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ptacek J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, et al. Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nature communications. 2014;5:4961. doi: 10.1038/ncomms5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, Schneiderhan-Marra N, Joos TO. Protein microarrays for personalized medicine. Clin Chem. 2010;56:376–387. doi: 10.1373/clinchem.2009.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X, et al. Quantifying antibody binding on protein microarrays using microarray nonlinear calibration. Biotechniques. 2013;54:257–264. doi: 10.2144/000114028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Festa F, et al. Robust microarray production of freshly expressed proteins in a human milieu. Proteomics Clin Appl. 2013;7:372–377. doi: 10.1002/prca.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu J, LaBaer J. Nucleic acid programmable protein array a just-in-time multiplexed protein expression and purification platform. Methods Enzymol. 2011;500:151–163. doi: 10.1016/B978-0-12-385118-5.00009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miersch S, LaBaer J. Nucleic Acid programmable protein arrays: versatile tools for array-based functional protein studies. Current protocols in protein science/editorial board, John E. Coligan … [et al.] 2011;Chapter 27:22. doi: 10.1002/0471140864.ps2702s64. Unit27. [DOI] [PubMed] [Google Scholar]
- 37.Sibani S, LaBaer J. Immunoprofiling using NAPPA protein microarrays. Methods Mol Biol. 2011;723:149–161. doi: 10.1007/978-1-61779-043-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seiler CY, et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014;42:D1253–1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Pino A, Zenkin N, Loris R. The many faces of Fic: structural and functional aspects of Fic enzymes. Trends Biochem Sci. 2014;39:121–129. doi: 10.1016/j.tibs.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Festa F, Mendoza A, Vatten K, LaBaer J. Study of the kinase activity using NAPPA protein microarray expressed with Human IVTT system; AACR 103rd Annual Meeting 2012; April 15, 2012; p. 2072. LB-2414 (Cancer Research, 2012) [Google Scholar]
- 41.Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. Legionella pneumophila LidA Affects Nucleotide Binding and Activity of the Host GTPase Rab1. Journal of Bacteriology. 2012;194:1389–1400. doi: 10.1128/JB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kovacic S, et al. Construction and Characterization of Kilobasepair Densely Labeled Peptide-DNA. Biomacromolecules. 2014;15:4065–4072. doi: 10.1021/bm501109p. [DOI] [PubMed] [Google Scholar]
- 43.Kim SY, Kim IG, Chung SI, Steinert PM. The structure of the transglutaminase 1 enzyme. Deletion cloning reveals domains that regulate its specific activity and substrate specificity. J Biol Chem. 1994;269:27979–27986. [PubMed] [Google Scholar]
- 44.Saul J, et al. Development of a full-length human protein production pipeline. Protein Sci. 2014;23:1123–1135. doi: 10.1002/pro.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlson ED, Gan R, Hodgman CE, Jewett MC. Cell-free protein synthesis: applications come of age. Biotechnology advances. 2012;30:1185–1194. doi: 10.1016/j.biotechadv.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joshi P, et al. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spurrier B, Ramalingam S, Nishizuka S. Reverse-phase protein lysate microarrays for cell signaling analysis. Nat Protoc. 2008;3:1796–1808. doi: 10.1038/nprot.2008.179. [DOI] [PubMed] [Google Scholar]
- 48.Anderson KS, et al. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J Proteome Res. 2011;10:85–96. doi: 10.1021/pr100686b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cruz JW, et al. Doc toxin is a kinase that inactivates elongation factor Tu (vol 289, pg 7788, 2014) Journal of Biological Chemistry. 2014;289:19276–19276. doi: 10.1074/jbc.M113.544429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng F, et al. A Xanthomonas uridine 5′-monophosphate transferase inhibits plant immune kinases. Nature. 2012;485:114–U149. doi: 10.1038/nature10962. [DOI] [PubMed] [Google Scholar]