

Abstract
The Wollemi pine (Wollemia nobilis) is a rare Southern conifer with striking morphological similarity to fossil pines. A small population of W. nobilis was discovered in 1994 in a remote canyon system in the Wollemi National Park (near Sydney, Australia). This population contains fewer than 100 individuals and is critically endangered. Previous genetic studies of the Wollemi pine have investigated its evolutionary relationship with other pines in the family Araucariaceae, and have suggested that the Wollemi pine genome contains little or no variation. However, these studies were performed prior to the widespread use of genome sequencing, and their conclusions were based on a limited fraction of the Wollemi pine genome. In this study, we address this problem by determining the entire sequence of the W. nobilis chloroplast genome. A detailed analysis of the structure of the genome is presented, and the evolution of the genome is inferred by comparison with the chloroplast sequences of other members of the Araucariaceae and the related family Podocarpaceae. Pairwise alignments of whole genome sequences, and the presence of unique pseudogenes, gene duplications and insertions in W. nobilis and Araucariaceae, indicate that the W. nobilis chloroplast genome is most similar to that of its sister taxon Agathis. However, the W. nobilis genome contains an unusually high number of repetitive sequences, and these could be used in future studies to investigate and conserve any remnant genetic diversity in the Wollemi pine.
Introduction
The monotypic gymnosperm Wollemia nobilis W.G. Jones, K.D. Hill & J. M. Allen (or Wollemi pine) was discovered in 1994 in the secluded warm temperate rainforests of the Wollemi National Park, New South Wales, Australia [1]. W. nobilis is similar to fossil pines from the Cretaceous period (approximately 140 million years ago) and relatives of Wollemia were once widespread [2,3], but the living population consists of fewer than 100 individuals confined to a single canyon system. This critically endangered species belongs to the Araucariaceae, a conifer family containing 30 species and three extant genera (Agathis, Araucaria, Wollemia) [4–7]. The current distributions of Araucariaceae and the closely related family Podocarpaceae are predominantly in the Southern Hemisphere [8,9]. W. nobilis can reach up to 40 m in height [1] and has the ability to form new vertical branches through coppicing [10]. Coppicing can occur in Agathis and Araucaria in response to trauma, but only Wollemia grows regularly in this manner.
Morphology alone does not resolve the position of Wollemia within the Araucariaceae [1,11], but phylogenetic studies using several chloroplast genes and ribosomal DNA data have placed Wollemia as sister to Agathis [4,5,7,12]. Molecular dating suggests that Wollemia and Agathis last shared a common ancestor between 55 and 90 million years ago [13,14]. This broad range reflects several incongruities within the literature regarding fossil calibrations and affinities with extant taxa [15–17].
It is generally accepted that the chloroplast originated from endosymbiosis of ancient cyanobacteria [18]. The consensus chloroplast genome is circular and consists of two inverted repeats (IRa and IRb), a large single-copy region (LSC), and a small single-copy region (SSC). It is estimated that on average there are 400 to 1,600 copies of the chloroplast genome in each cell [19]. The chloroplast genome is generally uniparentally inherited, typically paternally in conifers and maternally in angiosperms [20,21] but some variation in the mode chloroplast inheritance has been reported in conifers [22].
In recent years several cupressophyte chloroplast genomes including those of Agathis dammara (Lamb.) Rich. & A.Richard, Podocarpus lambertii Klotzsch ex Endl, Podocarpus totara G.Benn. ex D.Don and Nageia nagi (Thunb.) Kuntze have been published [23,24]. Comparative studies of these genomes have provided fresh insights into various aspects of conifer chloroplast genome evolution through the examination of genome size, structure, organisation and gene content [23–26].
In this study, we use a range of next generation sequencing methods to determine the complete chloroplast genome (plastome) of W. nobilis. We compare the plastome of W. nobilis with available chloroplast genomes of Araucariaceae and Podocarpaceae, and analyse genome structure and organisation to infer the steps in genome evolution. We identify repetitive sequences in W. nobilis chloroplast genes and compare these with other repetitive sequences in Araucariaceae and Podocarpaceae. The availability of new genomic datasets will deliver new tools for exploring the genetic diversity of W. nobilis, and will support future conservation management strategies [27]. Genome-level sequencing is important in W. nobilis because a previous genetic study of approximately 800 AFLP, SSR and allozyme loci did not detect any genetic diversity [28], suggesting that living W. nobilis is extensively clonal or that genetic diversity could not be detected with these markers. The chloroplast genome reported in this study could be used to perform a more extensive search for genetic diversity in the Wollemi pine.
Materials and Methods
Chloroplast DNA extraction
Foliage from Wollemia nobilis provided by the Australian Botanic Gardens, Mt Annan (Sydney, NSW, Australia) was frozen at -80°C and stored until the time of DNA extraction. W. nobilis chloroplast DNA was isolated and amplified using the method described in [29].
Genomic DNA extraction
Total DNA was extracted from young leaves using a modified cetyl trimethylammonium bromide (CTAB) method based on [30] and [28].
Chloroplast DNA sequencing and genome assembly
Two next generation sequencing (NGS) data sets were used to assemble a draft W. nobilis chloroplast genome (S1 Table). These included total genomic DNA sequenced using the Illumina GAIIx platform, and chloroplast DNA sequenced using the Roche 454 GS-FLX. To confirm the draft genome, whole genomic DNA was then sequenced in a Nextera library on the Illumina MiSeq platform. All three NGS libraries were sequenced at the Ramaciotti Centre for Genomics (University of New South Wales).
Initial Illumina and 454 reads were trimmed using clean_reads v0.2.1 [31]. Illumina sequencing data were assembled using the Velvet short read assembler (v1.1.04) [32], and the 454 chloroplast data were assembled using Mira v3.2.1 [33]. These datasets were combined using Minimus2 v3.0.1 to produce contigs with sizes greater than 10kb [34]. Scaffold confirmation, arrangement and concatenation were implemented in Burrows Wheeler Aligner (BWA) [35]. This left a single gap in the resulting chloroplast sequence.
In order to resolve this gap, the chloroplast genomes of W. nobilis and Agathis dammara (AB830884) were aligned using MAUVE 2.3.1 software [36]. We observed a 5,000 bp sequence consisting of a section of a protein-coding gene (ycf1) that was absent in the W. nobilis genome. One hundred base pairs flanking this region were extracted from A. dammara and reads from the W. nobilis Illumina MiSeq library were mapped onto this sequence. This produced continuous mapping and high coverage over the gap region.
The W. nobilis chloroplast genome was then validated by mapping MiSeq reads to the final chloroplast genome. The Illumina MiSeq library was imported into CLC bio Genomics Workbench (v6.5, www.clcbio.com) using quality score settings for the Illumina Pipeline 1.8 and later. Sequences were trimmed based on a quality threshold of 0.05. Reads shorter than 150 bp and low quality reads were discarded. For the mapping, 90% of the read length was required to map with 80% similarity. A reliable reference sequence was produced since the mapping was continuous and there was consistently high coverage (average 408.54X; see S1 Table).
Raw sequence reads from the Illumina MiSeq library (total DNA) and the 454 sequencing (chloroplast DNA) have been deposited in the Sequence Read Archive (SRA) database with accession numbers SRR1927951 and SRR192612 respectively.
Genome annotation
Initial annotation of the Wollemia nobilis chloroplast genome was performed using Glimmer3 (Gene Locator and Interpolated Markov ModelER) v3.02 and Dual Organellar GenoMe Annotator (DOGMA) [37]. Genes and open reading frames (ORF) that may not have been annotated were identified with the aid of blastx (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Putative starts, stops, and intron positions were determined by comparison with homologous genes in other chloroplast genomes using MAFFT online software [38]. In addition, all tRNA genes were further verified online using tRNAscan-SE search server [39] (http://lowelab.ucsc.edu/tRNAscan-SE/). The circular W. nobilis chloroplast genome map was drawn using OGDraw v1.2 [40].
Sequence analyses and computational methods
Sequences homologous to the W. nobilis chloroplast genome were identified using Standard Nucleotide BLAST (http://blast.ncbi.nlm.nih.gov/). Whole genomes were aligned using progressive MAUVE implemented by MAUVE v2.3.1 software [36]. The AT content for the genome was calculated with Sequence Statistics on CLC Genomics Workbench v7.5 software (CLC bio). Genome annotation was performed in Geneious Pro v6.1.6 (Biomattters Ltd.), and the AT-content of protein-coding genes, tRNA genes, introns and intergenic spacers (IGSs) was determined on the basis of their annotation.
Simple sequence repeats (SSRs) were identified using Phobos Tandem Repeats Finder v3.3.12 [41]. The perfect search default settings were used and this involved a repeat unit size that ranged from one to 10 without setting a minimum satellite length constraint. A GFF file format was selected as the output option and cells were sorted based on the repeat number (with anything below three removed). Tandem repeats were identified with Tandem Repeats Finder (TRF) with default parameter settings [42]. The tandem repeat lengths were 20 bp or more with the minimum alignment score and maximum period size set as 50 and 500 (respectively), and the identity of repeats was set to ≥ 90%. REPuter [43] was used to visualize duplicated sequences in W. nobilis by forward versus reverse complement (palindromic) alignment, with the repeat size set to 200 to 5,000 bp.
Results and Discussion
General features of the W. nobilis chloroplast genome
The complete circular chloroplast genome of Wollemia nobilis (GenBank accession KP259800) is 145,630 bp. The annotated genome is shown in Fig 1 and the sequencing results are detailed in S1 Table. The genome is very similar to that of Agathis dammara (145,625 bp) and is larger than the chloroplast genomes of Podocarpus lambertii, Podocarpus totara and Nageia nagi (Podocarpaceae). However, the genome is smaller than the largest known gymnosperm chloroplast genome from Cycas taitungensis C.F.Shen, K.D.Hill, C.H.Tsou & C.J.Chen (163,403 bp; NC_009618) [44].
Fig 1. Sequence map of the Wollemia nobilis chloroplast genome.

Genes drawn outside of the circle are transcribed clockwise, while genes shown on the inside of the circle are transcribed counter-clockwise. Genes belonging to different functional groups are colour-coded. The darker gray in the inner circle indicates GC content, while the lighter gray corresponds to AT content.
The W. nobilis chloroplast genome encodes 122 genes, including 82 protein-coding genes, five ribosomal RNA genes, and 35 transfer RNA genes (Fig 1, Table 1 and Table 2). The 82 intact chloroplast protein-coding sequences are shared and are of similar length in Araucariaceae and Podocarpaceae, indicating evolutionarily conserved chloroplast gene content. A similar gene number is also shared with other cupressophytes including Cupressaceae and Cephalotaxaceae. In the gymnosperm families Cycadaceae, Ephedraceae and Ginkgoaceae, protein-coding genes are duplicated in the inverted repeat regions (IR). This increases the size of these genomes [24] but this feature is not present in the Wollemia chloroplast genome.
Table 1. Comparison of chloroplast genome characteristics in different species of Araucariaceae and Podocarpaceae.
| Araucariaceae | Podocarpaceae | ||||
|---|---|---|---|---|---|
| Characteristics | Wollemia nobilis | Agathis dammara A | Nageia nagi A | Podocarpus lambertii B | Podocarpus totara |
| GenBank Accession no. | KP259800 | AB830884 | AB830885 | NC_020361.1 | NC_020361.1 |
| Size (bp) | 145,630 | 145,625 | 133,722 | 133,734 | 133,259 |
| GC content (%) | 36.50 | 36.54 | 37.26 | 37.100 | 37.16 |
| Total number of genes | 122 | 122 (123) C | 117 (120) C | 119 (118) C | 94 (120) C |
| Total number of unique genes | 118 | (119) C | (118) C | 118 (117) C | (118) |
| Protein-coding genes | 82 | 81 (82) C | 81 (82) C | 82 | 75 (82) C |
| Ribosomal RNAs | 5 | 5 | 4 | 4 | 4 |
| Transfer RNAs | 35 | 36 | 32 (34) C | 31 (32) C | 15 (34) C |
| Protein-coding genes (bp) | 75,300 | 75,271 | 74,781 | 74,217 | 74,607 |
| Ribosomal RNAs (bp) | 4,636 | 4,638 | 4,529 | 4,504 | 4,501 |
| Transfer RNAs (bp) | 2,628 | 2,699 | 2,418 | 2,409 | 2,487 |
| Introns (bp) | 11,857 | 11,890 | 11,487 | 10,445 | 9,710 |
| Spacers (bp) | 51,189 | 51,127 | 40,507 | 42,159 | 41,821 |
| AT content (%) | |||||
| Genome | 63.51 | 63.46 | 62.74 | 62.90 | 62.84 |
| Protein-coding genes | 62.41 | 62.34 | 61.86 | 61.87 | 61.90 |
| Transfer RNA genes | 46.47 | 46.46 | 46.80 | 46.66 | 47.30 |
| Ribosomal RNA genes | 45.75 | 45.90 | 46.01 | 45.87 | 45.90 |
| Introns | 62.39 | 62.61 | 61.92 | 62.13 | 61.50 |
| Spacers | 67.88 | 67.85 | 67.44 | 67.62 | 62.84 |
Table 2. List of genes identified in the chloroplast genome of W. nobilis.
| Functional category | Group of genes | Name of genes | |||||
|---|---|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA genes | rrn16 | rrn23 | rrn4.5 | rrn5** | ||
| Transfer RNA genes | trnA-UGC* | trnC-GCA | trnD-GUC** | trnE-UUC | trnF- GAA | trnfM- CAU | |
| trnG- GCC | trnG- UCC* | trnH- GUG | trn-I CAU** | trnI- GAU* | trnK- UUU* | ||
| trnL- CAA | trnL- UAA* | trnL- UAG | trnM- CAU* | trnN- GUU | trnP- GGG | ||
| trnP- UGG | trnQ- UUG | trnR- ACG | trnR- CCG | trnR-UCU** | trnS- UGA | ||
| trnS-GCU | trnS-GGA | trnT- GGU | trnT- UGU | trnV- GAC | trnV- UAC* | ||
| trnW- CCA | trnY-GUA | ||||||
| Small subunit of ribosome | rps11 | rps12* | rps14 | rps15 | rps18 | rps19 | |
| rps2* | rps3 | rps4 | rps7 | rps8 | |||
| Large subunit of ribosome | rpl14 | rpl16* | rpl2 | rpl20 | rpl22 | rpl23 | |
| rpl32 | rpl33 | rpl36 | |||||
| DNA-dependent RNA polymerase | rpoA | rpoB | rpoC1* | rpoC2 | |||
| Translational initiation factor | infA | ||||||
| Genes for photosynthesis | Subunits of photosystem I | psaA | psaB | psaC | psaI | psaJ | psaM |
| ycf3* | ycf4 | ||||||
| Subunits of photosystem II | psbA | psbB | psbC | psbD | psbE | psbF | |
| psbH | psbI | psbJ | psbK | psbL | psbM | ||
| psbN | psbT | psbZ | |||||
| Subunits of cytochrome | petA | petB* | petD* | petG | petL | petN | |
| Subunits of ATP synthase | atpA | atpB | atpE | atpF* | atpH | atpI | |
| Large subunit of Rubisco | rbcL | ||||||
| Chlorophyll biosynthesis | chlB | chlL | chlN | ||||
| Subunits of NADH dehydrogenase | ndhA* | ndhB* | ndhC | ndhD | ndhE | ndhF | |
| ndhG | ndhH | ndhI | ndhJ | ndhK | |||
| Other genes | Maturase | matK | |||||
| Envelope membrane protein | cemA | ||||||
| Subunit of acetyl-CoA | accD | ||||||
| C-type cytochrome synthesis gene | ccsA | ||||||
| Protease | clpP | ||||||
| Component of TIC complex | ycf1 | ||||||
| Genes of unknown function | Conserved open reading frames | ycf2 | |||||
*genes with introns
**duplicated genes
Table 1 details the results of a comparative analysis of the W. nobilis, A. dammara, P. lambertii, P. totara and N. nagi chloroplast genomes. The gene content of these genomes was determined using both the annotation methods described in this study, and by reference to the previously published annotations on NCBI Genome (http://www.ncbi.nlm.nih.gov/genome). Differences between these annotations (probably due to differences in annotation methodology) have been noted in Table 1, with the values observed in our annotation shown in parentheses. The major differences between our annotations and the previously published annotations were: (1) the published annotation for P. totara (NC_020361.1) was very incomplete and had many missing protein-coding genes and tRNAs; (2) the matK gene was absent in both A. dammara and N. nagi even though high similarity was observed when aligned to a similar sequence in W. nobilis; (3) the two tRNAs (trnC and trnQ) were not annotated in the previously published N. nagi annotation; and (4) the gene number in the published P. lambertii annotation included a pseudogene, but we have omitted this from the total number of genes shown for the P. lambertii plastome in Table 1.
Base composition
GC base pairs are more thermodynamically stable than AT base pairs, and so GC content influences chloroplast genome stability. The GC content of the Wollemia chloroplast genome (36.5%) is very similar to A. dammara but slightly lower than the GC content of Podocarpaceae chloroplast genomes (which range from 37.1 to 37.26%; Table 1). The GC content of the W. nobilis chloroplast genome is also higher than members of Cupressaceae such as Taiwania cryptomerioides Hayata (34.63%), Calocedrus formosana (Florin) W.C.Cheng & L.K.Fu (35.38%) and Cryptomeria japonica (Thunberg ex Linnaeus f.) D.Don (34.83%) [25].
Previous studies have found that the AT content in genomic regions may be associated with the dynamics of repeats (e.g. [45,46]) and may also be associated with the codon bias of chloroplast protein-coding genes and hence the regulation of gene expression (e.g. [45,46]). AT-rich regions in the Wollemia chloroplast genome include intergenic (67.88%), protein-coding (62.41%) and intronic (62.39%) regions, while rRNAs (45.75%) and tRNAs (46.47%) have a much lower AT content. These patterns are similar across all species listed in Table 1, as well as in the plastomes of many other plants (e.g. [25,47]).
Structure of rps16
Ribosomal protein S16 (Rps16) is essential for the translation of chloroplast genes in tobacco [48] and can be found in some cupressophytes (e.g. Cephalotaxus oliveri Mast. and Cryptomeria japonica; [26,49]). The rps16 gene is situated between the chlB gene and the trnK-UUU gene in a conserved part of the genome. The coding sequence is 268 bp in length and has an 853 bp intron in C. oliveri. When the chlB/trnK region of Wollemia nobilis is aligned with that of C. oliveri, only remnants of the rps16 gene are evident due to the absence of an initiation codon. A similar rps16 remnant region is also present in Agathis dammara and has ~95% similarity to the corresponding W. nobilis sequence. In comparison, the chlB/trnK intergenic regions in P. lambertii, P. totara and N. nagi do not resemble the rps16 gene at all. A possible slower mutation rate in Araucariaceae compared to Podocarpaceae could explain the complete absence of rps16 in the latter group. The absence of a functional rps16 gene in this location could indicate that this gene is not essential for translation in Agathis and Wollemia chloroplasts. Alternatively, its function could be replaced by another ribosomal protein or by an intact nuclear copy of rps16, as in some legumes [50].
Within the Cupressaceae the rps16 gene is present in Calocedrus formosana and C. japonica, but is absent from Juniperus scopulorum Sarg. Similarly in the Taxaceae it is present in Taxus mairei (Lemée & Lév.) S.Y.Hu ex T.S.Liu but absent in Amentotaxus formosana H.L.Li. This suggests that there have been multiple independent losses of the rps16 gene within the gymnosperms. Further study to trace rps16 gene loss through the conifer lineages could aid in understanding the process of chloroplast genome evolution in gymnosperms.
Comparative analysis of introns and intergenic regions
There are 16 intron-containing chloroplast genes in W. nobilis, including six tRNA genes and 10 protein-coding genes. Similar intronic features were observed in A. dammara. Nearly all of these genes contain a single intron except for the two introns in ycf3 and rps12. The trnK-UUU gene has an unusual intron that encodes a matK ORF. This trnK intron is observed in many plants and has been extensively used as a phylogenetic marker (e.g. [51,52]). Additionally, we observed a 31 bp overlap between ndhC and ndhK, and a 53 bp overlap between psbC and psbD.
W. nobilis and A. dammara have 120 highly similar intergenic regions. However, the intergenic region between rbcL and trnR-CCG in W. nobilis contains a trnD-GUC in A. dammara, producing the intergenic regions rbcL/trnD and trnD/trnR. Four intergenic regions (rpoC1/rpoC2, psaB/psaA, psbF/psbE and ndhH/ndhA) are identical in sequence between these two species.
The psbA/trnH intergenic region is the most widely used plastid barcode for species differentiation in land plants including Araucaria [53,54]. It is highly variable in sequence and in length [27,54,55], with a non-coding region flanked by two conserved coding regions, psbA (which encodes photosystem II protein D1) and trnH-GUG. We observed 646 bp of additional sequence in the 847 bp psbA/trnH intergenic region in W. nobilis. This sequence was absent in A. dammara where the psbA/trnH intergenic region was 201 bp in length. BLAST analyses of the W. nobilis psbA/trnH intergenic region indicate that this indel is present in all 19 Araucaria species. The length of psbA/trnH in Podocarpaceae ranges from 600 to 626 bp. This suggests that a deletion may have occurred in this region in Agathis after the divergence of Agathis and Wollemia.
Comparative analysis of tRNAs
Wollemia nobilis and A. dammara have the same 32 unique tRNAs, but have different numbers of tRNA coding sequences due to gene duplication events (Table 1). W. nobilis has 35 tRNAs because it only has two of the three copies of trnD-GUC observed in A. dammara. The trnD-GUC gene in W. nobilis is associated with a 760 bp direct repeat, and the trnR-UCU is also duplicated and is associated with a direct repeat of 310 bp. These tRNA-containing repeats are not present in A. dammara and the impact of these repeats on chloroplast genome function is unclear.
Analyses of plastid tRNAs could support a better understanding of the divergence among conifers [23]. The trnR-CCG gene is entirely absent in Cupressaceae, Taxaceae and Cephalotaxaceae, but is found in both Pinaceae and Podocarpaceae. It is present in W. nobilis and Agathis, and may be generally present in Araucariaceae. This provides further evidence for a major loss of the trnR-CCG gene in the Taxaceae/Taxodiaceae/Cupressaceae group [23]. The trnR-CCG gene may have been readily lost because it is not essential for translation in land plants [56].
Remnant inverted repeats in W. nobilis and Araucariaceae
A large inverted repeat (IR) is found in many land plants and typically includes a pair of ycf2 and ribosomal operons. However, in several gymnosperms (including Pinaceae, Cupressaceae, Cephalotaxaceae and Podocarpaceae) only short remnants of the IR have been observed [24,26]. We identified two short IRs in W. nobilis: a 602 bp IR region that includes the rrn5 gene, and another region of 73 bp that includes the trnI-CAU gene. Both of these IRs are also found in A. dammara. The rrn5-containing short IR was not found in any of the cupressophytes (Cupressaceae, Cephalotaxaceae, Podocarpaceae or Taxaceae).
Duplicated and inverted tRNAs were observed in W. nobilis as well as in A. dammara. The duplicated tRNA, trnI-CAU, is inverted in W. nobilis as well as in Taiwania cryptomerioides and Pinus thunbergii Parlatore [49,57]. Other tRNAs including trnN-GUU in Podocarpaceae [23,25] and trnQ-UUG in Cephalotaxaceae [49] have also been identified.
Whole genome comparative analysis
The W. nobilis chloroplast genome was aligned with the chloroplast genomes of other closely related gymnosperms to compare the organisation of these genomes. Fig 2 shows two locally collinear blocks (LCBs) between the W. nobilis and A. dammara chloroplast genomes. These blocks suggest a high level of similarity in genome organisation between these two species, although they are inverted relative to each other (Fig 2). More comparisons of W. nobilis to members of the Podocarpaceae produced chloroplast genome alignments with several inversions and translocations. There are seven LCBs between W. nobilis vs. P. lambertii, nine LCBs between W. nobilis vs. N. nagi and 10 LCBs between W. nobilis vs. P. totara (S1 Fig). These comparisons show that the chloroplast genomes of P. lambertii and P. totara are both very different in structure as previously reported [23]. Examination of local pairwise alignments between the chloroplast genomes of W. nobilis and A. dammara also shows a high level of sequence similarity (96.6%). Collectively these alignments confirm the close evolutionary relationship between Wollemia and Agathis species [14,58,59], and the more distant relationship between Wollemia and Podocarpus or Nageia.
Fig 2. MAUVE alignment of W. nobilis and A. dammara chloroplast genomes.
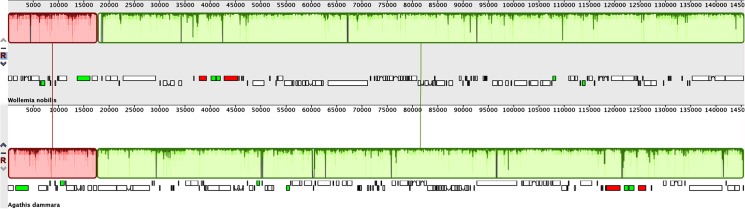
The W. nobilis genome is shown at top as the reference genome. Within each of the alignments, local collinear blocks are represented by blocks of the same colour connected by lines. Note that the two LCBs in the A. dammara genome are both inverted relative to the W. nobilis genome.
Repetitive sequences in the chloroplast genome of W. nobilis
Although large numbers of tandem repeats have been reported in conifers [23,49], the mechanisms underlying the origin of these tandem repeats remain unclear. Nonetheless, they are known to be associated with gene duplication [60], gene expansion [23,49] and chloroplast DNA rearrangement [61]. We identified 28 tandem repeats of more than 20 bp in length in the W. nobilis chloroplast genome (Table 3), of which 12 are in intergenic regions, 14 in coding regions, and two extend from an intergenic region into a coding region. The length of the repeat units in these regions varied between 11 and 60 bp, and up to 11 repeat units were present.
Table 3. Distribution of tandem repeats in the W. nobilis chloroplast genome.
| Serial No. | Indices | Repeat length | Size of repeat unit X Copy number | Location |
|---|---|---|---|---|
| 1 | 4445–4492 | 54 | 18x3 | atpF/atpA (IGS) |
| 2 | 18715–18751 | 36 | 12x3 | trnH/chlL (IGS) |
| 3 | 25945–25971 | 24 | 12x2 | ycf2 (CDS) |
| 4 | 26323–26359 | 36 | 18x2 | ycf2 (CDS) |
| 5 | 34174–34345 | 171 | 57x3 | rps7 (CDS) |
| 6 | 36587–36660 | 72 | 36x2 | rps12/trnV (IGS) |
| 7 | 37477–37502 | 24 | 12x2 | trnV/rrn16 (IGS) |
| 8 | 37734–37762 | 28 | 14x2 | trnV/rrn16 (IGS) |
| 9 | 47091–47126 | 32 | 16x2 | trnR/trnN (IGS) |
| 10 | 64129–64185 | 60 | 15x4 | ycf1 (CDS) |
| 11 | 65803–65865 | 66 | 33x2 | ycf1 (CDS) |
| 12 | 65853–65894 | 45 | 15x3 | ycf1 (CDS) |
| 13 | 66632–66672 | 42 | 21x2 | ycf1 (CDS) |
| 14 | 67290–67332 | 42 | 21x2 | ycf1 (CDS) |
| 15 | 69017–69356 | 330 | 30x11 | ycf1 (CDS) |
| 16 | 72719–72756 | 38 | 19x2 | rpl23 (CDS) |
| 17 | 74646–74676 | 32 | 16x2 | rpl2/rps19 (IGS) |
| 18 | 84689–84713 | 26 | 13x2 | psbT/psbB (IGS) |
| 19 | 91442–91485 | 44 | 22x2 | trnP/psaJ (IGS) |
| 20 | 92695–92802 | 108 | 54x2 | rps18 (CDS) and rps18/psbF (IGS) |
| 21 | 100704–101063 | 360 | 60x6 | accD (CDS) |
| 22 | 101250–101295 | 45 | 45 | accD (CDS) |
| 23 | 101313–101337 | 24 | 12x2 | accD (CDS) |
| 24 | 113049–113073 | 24 | 12x2 | ndhJ/trnF (IGS) |
| 25 | 115787–115820 | 32 | 16x2 | rps4/trnS (IGS) |
| 26 | 123920–124039 | 120 | 60x2 | psaB/rps14 (IGS) and rps14 (CDS) |
| 27 | 125626–125659 | 39 | 13x3 | trnS/psbC (IGS) |
| 28 | 140203–140242 | 42 | 21x2 | rpoC1 (CDS) |
The accD gene encodes the acetyl-CoA carboxylase beta subunit. The ORF of this gene is variable among land plants, with cupressophytes having the largest expansions of the accD ORF (ranging from 700 to 1,056 codons; [49]). The large size of the accD ORF in cupressophytes has been attributed to the accumulation of tandem repeat sequences within the gene [26,49]. The accD reading frame of W. nobilis is 800 codons in length, and hence is shorter than that of A. dammara (820 codons) but longer than that of P. lambertii (684 codons). Large insertions are usually found in the middle of the accD ORF [49], and this was also the case for W. nobilis in which several tandem repeats were observed in accD. The longest tandem repeat was 360 bp in length (as shown in Table 3), and consisted of six copies of 20 imperfect amino acid sequences starting with an LDREEK motif. The other tandem repeats are located downstream of this repeat, such that there are three copies of the motif, PEEEV and then two copies of the motif QWVN. Nine similar repeats were found in the accD gene in C. oliveri, and this gene remained functional [49]. Hence, the Wollemia accD gene is also expected to retain its normal function.
The protein-coding region ycf1 contains higher numbers of tandem repeats and SSRs than any other gene within the W. nobilis chloroplast genome. This includes 17 poly-A repeats, and six different tandem repeats (Tables 3 and 4). The ycf1 gene is often the largest protein-coding gene in plastomes (e.g. 7,830 bp in W. nobilis, 7,914 bp in A. dammara) and encodes a chloroplast envelope protein translocase (part of the TIC complex; [62]). High numbers of tandem repeats and SSRs (11 tandem repeats and 148 SSRs) were also reported in the ycf1 gene in P. lambertii [23]. Internal stop codons are absent in both W. nobilis and P. lambertii ycf1, suggesting that the ycf1 gene in these species encodes a functional protein.
Table 4. Characteristics of simple sequence repeats identified in the chloroplast genomes of W. nobilis, A. dammara and P. lambertii.
| Mono | Di | Tri | Tetra | Penta | Hexa | Total | |
|---|---|---|---|---|---|---|---|
| W. nobilis | |||||||
| Total counts | 239 | 69 | 62 | 15 | 1 | 1 | 387 |
| Total Repeat Length (repeat unit X number of repeat) (bp) | 1991 | 744 | 621 | 184 | 15 | 18 | 3573 |
| Density (Total repeat length/genome size) [bp/kb] | 13.67 | 5.11 | 4.26 | 1.26 | 0.10 | 0.12 | 24.53 |
| Proportion among other SSR (%) | 55.72 | 20.82 | 17.38 | 5.15 | 0.42 | 0.50 | 100 |
| Mean Length | 8.33 | 10.78 | 10.02 | 12.27 | 15 | 18 | 9.23 |
| Standard Deviation | 1.84 | 4.45 | 3.64 | 1.03 | 0 | 0 | |
| A. dammara | |||||||
| Total counts | 250 | 68 | 64 | 12 | 2 | 1 | 397 |
| Total Repeat Length (repeat unit X number of repeat) (bp) | 2168 | 720 | 615 | 152 | 30 | 18 | 3703 |
| Density (Total repeat length/genome size) [bp/kb] | 14.89 | 4.94 | 4.22 | 1.04 | 0.21 | 0.12 | 25.43 |
| Proportion among other SSR (%) | 58.55 | 19.44 | 16.61 | 4.10 | 0.81 | 0.49 | 100 |
| Mean Length | 8.67 | 10.59 | 9.61 | 12.67 | 15 | 18 | 9.33 |
| Standard Deviation | 2.38 | 3.88 | 1.53 | 1.56 | 0 | 0 | |
| P. lambertii | |||||||
| Total counts | 198 | 63 | 52 | 9 | 1 | 1 | 324 |
| Total Repeat Length (repeat unit X number of repeat) (bp) | 1558 | 586 | 498 | 112 | 15 | 18 | 2787 |
| Density (Total repeat length/genome size) [bp/kb] | 11.65 | 4.38 | 3.72 | 0.84 | 0.11 | 0.13 | 20.84 |
| Proportion among other SSR (%) | 55.90 | 21.03 | 17.87 | 4.02 | 0.54 | 0.65 | 100 |
| Mean Length | 7.87 | 9.30 | 9.58 | 12.44 | 15 | 18 | 8.60 |
| Standard Deviation | 1.56 | 2.81 | 1.33 | 1.33 | 0 | 0 |
Short simple repeats in the W. nobilis chloroplast genome
Simple sequence repeats (SSRs) usually have a higher rate of mutation compared with other neutral regions of DNA due to slipped strand mispairing. Chloroplast SSRs are often used as molecular markers in genetic studies analysing population structure as these short repeats are haploid and uniparentally inherited [63,64]. Here, we compared the perfect SSRs between the three species W. nobilis, A. dammara and P. lambertii (Table 4) using summarised data collected from Phobos (S2 Table). The largest number of SSRs was found in A. dammara, followed by W. nobilis and P. lambertii. Given the varying genome sizes, we observed the overall SSR density and found W. nobilis (24.53 bp every 1000 bp) and A. dammara (25.43 bp every 1000 bp) were more similar to each other than to P. lambertii (20.84 bp every 1000 bp). The average repeat lengths of the mono-, di- and tri- nucleotides for W. nobilis (9.71 bp) and A. dammara (9.62 bp) were similar whereas in P. lambertii the average repeat length was 8.91 bp. Mononucleotide repeats were found to be the most common type of SSR in all three species (Table 4), and poly-A repeats were more abundant than poly-C repeats (S2 Table). The function of these repeats (if any) could be investigated by further characterisation of SSRs at specific genomic regions such as coding sequences, introns or intergenic spacers. The SSRs in W. nobilis could also be used to investigate its genetic diversity.
It is important to note that previous studies have used varied algorithms for SSR detection [64,65]. Hence, any further comparisons between W. nobilis and other species would have to be made using the SSR criteria described in this study or another common set of SSR criteria.
Conclusion
We used a combination of de novo assembly and reference to the A. dammara chloroplast genome to obtain the complete chloroplast genome sequence for Wollemia nobilis, a critically endangered Southern conifer with a very small extant population. Although Wollemia is a monotypic genus, we observe a close similarity between the chloroplast genomes of A. dammara and W. nobilis in terms of genome size, organisation and sequence. The shared genomic features include rrn5 and trnI IR remnants, a syntenic rps16 pseudogene and an insertion/deletion hotspot in the psbA/trnH intergenic region. Our data provide an insight into the evolution of the Araucariaceae plastid genome in the wider context of plastid evolution in conifers. A striking feature of the W. nobilis chloroplast genome is its large number of repetitive sequences, notably within the accD gene and including a large number of SSRs. These sequences could be used as molecular markers in future studies aimed at identifying and conserving genetic diversity in the Wollemi pine.
Supporting Information
a. W. nobilis vs. P. lambertii, b. W. nobilis vs. P. totara, c. W.nobilis vs. N. nagi.
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
The authors would like to acknowledge and thank Amanda Rollason, Cathy Offord and other staff at the Australian Botanic Garden, Mt. Annan (Sydney) for maintaining the W. nobilis collection and providing access to fresh material. The authors would also like to acknowledge: Oliver Deusch, Peter Lockhart and Patrick Biggs (Massey University, New Zealand) for reading the manuscript and advice on genome assembly and annotation; Carolyn Connelly and other staff at the Royal Botanic Gardens (Sydney) for laboratory support; UNSW students involved in the initial DNA sequencing; Sven Warris (Hanze University) and Nandan Deshpande (UNSW) for advice on genome assembly; and Professor Ian Dawes (UNSW) for his strong support for the project. The authors would also like to dedicate this paper to the memory of Alan Wilton (1953–2011), who initiated the project as part of his long commitment to the innovative teaching of genetics at the University of New South Wales.
Data Availability
The annotated Wollemi pine chloroplast genome sequence is available from GenBank (accession number KP259800). Raw sequence reads have been deposited in the Sequence Read Archive (SRA) database (accession numbers SRR1927951 and SRR192612).
Funding Statement
Funding for this work was provided by a grant from Bioplatforms Australia (www.bioplatforms.com.au) to AW and SKD, the Ramaciotti Centre for Genomics (University of New South Wales)(www.ramaciotti.unsw.edu.au), the Royal Botanic Gardens and Domain Trust (Sydney) (www.rbgsyd.nsw.gov.au) and an Early Career Research Grant from the Faculty of Science, University of New South Wales to SKD. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Jones W, Hill K, Allen J. Wollemia nobilis, a new living Australian genus and species in the Araucariaceae. Telopea (Syd). 1995;6: 173–176. [Google Scholar]
- 2. Chambers TC, Drinnan AN, McLoughlin S. Some morphological features of Wollemi pine (Wollemia nobilis: Araucariaceae) and their comparison to Cretaceous plant fossils. Int J Plant Sci. 1998: 160–171. [Google Scholar]
- 3. Macphail M, Hill K, Partridge A, Truswell E, Foster C. Wollemi Pine-old pollen records for a newly discovered genus of gymnosperm. Geology Today. 1995;11: 48–50. [Google Scholar]
- 4. Gilmore S, Hill K. Relationships of the Wollemi Pine (Wollemia nobilis) and a molecular phylogeny of the Araucariaceae. Telopea (Syd). 1997;7: 275–291. [Google Scholar]
- 5. Stefanoviac S, Jager M, Deutsch J, Broutin J, Masselot M. Phylogenetic relationships of conifers inferred from partial 28S rRNA gene sequences. Am J Bot. 1998;85: 688–688. [PubMed] [Google Scholar]
- 6. Conran JG, Wood GM, Martin PG, Dowd JM, Quinn CJ, Gadek AP, et al. Generic relationships within and between the gymnosperm families Podocarpaceae and Phyllocladaceae based on an analysis of the chloroplast gene rbcL . Aust J Bot. 2000;48: 715–724. [Google Scholar]
- 7. Quinn C, Price R, Gadek P. Familial concepts and relationships in the conifer based on rbcL and matK sequence comparisons. Kew Bulletin. 2002: 513–531. [Google Scholar]
- 8. Kershaw P, Wagstaff W. The Southern conifer family Araucariaceae: history, status and value for palaeoenvironmental reconstruction. Annu Rev Ecol Syst. 2001;32: 397–414. [Google Scholar]
- 9. Enright N, Hill R. Ecology of the Southern Conifers Melbourne: Melbourne University Press; 1995. [Google Scholar]
- 10. Burrows G, Offord C, Meagher P, Ashton K. Axillary meristems and the development of epicormic buds in Wollemi pine (Wollemia nobilis). Ann Bot. 2003;92: 835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burrows G, Meagher P, Heady R. An anatomical assessment of branch abscission and branch-base hydraulic architecture in the endangered Wollemia nobilis . Ann Bot. 2007;99: 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zonneveld B. Genome sizes of all 19 Araucaria species are correlated with their geographical distribution. Plant Syst Evol. 2012;298: 1249–1255. [Google Scholar]
- 13. Kranitz ML, Biffin E, Clark A, Hollingsworth ML, Ruhsam M, Gardner MF, et al. Evolutionary diversification of New Caledonian Araucaria . PloS One. 2014;9: e110308 10.1371/journal.pone.0110308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biffin E, Hill RS, Lowe AJ. Did kauri (Agathis: Araucariaceae) really survive the Oligocene drowning of New Zealand? Systematic Biology. 2010;59: 594–602. 10.1093/sysbio/syq030 [DOI] [PubMed] [Google Scholar]
- 15. Hill RS, Lewis T, Carpenter RJ, Whang SS. Agathis (Araucariaceae) macrofossils from Cainozoic sediments in south-eastern Australia. Aust J Bot. 2008;21: 162–177. [Google Scholar]
- 16. Knapp M, Mudaliar R, Havell D, Wagstaff SJ, Lockhart PJ. The drowning of New Zealand and the problem of Agathis . Syst Biol. 2007;56: 862–870. [DOI] [PubMed] [Google Scholar]
- 17. Lee DE, Bannister JM, Lindqvist JK. Late Oligocene-early Miocene leaf macrofossils confirm a long history of Agathis in New Zealand. New Zealand J Bot. 2007;45: 565–578. [Google Scholar]
- 18. Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet. 2004;5: 123–135. [DOI] [PubMed] [Google Scholar]
- 19. Pyke KA. Plastid division and development. Plant Cell. 1999;11: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reboud X, Zeyl C. Organelle inheritance in plants. Heredity. 1994;72: 132–140. [Google Scholar]
- 21. Mogensen HL. The hows and whys of cytoplasmic inheritance in seed plants. Am J Bot. 1996;83: 383–404. [Google Scholar]
- 22. Jansen RK, Ruhlman TA. Plastid genomes of seed plants Genomics of chloroplasts and mitochondria: Springer; 2012. pp. 103–126. [Google Scholar]
- 23. do Nascimento Vieira L, Faoro H, Rogalski M, de Freitas Fraga HP, Cardoso RLA, de Souza EM, et al. The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS One. 2014;9: e90618 10.1371/journal.pone.0090618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu C-S, Wang Y-N, Hsu C-Y, Lin C-P, Chaw S-M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol Evol. 2011;3: 1284–1295. 10.1093/gbe/evr095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu CS, Chaw SM. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): evolution towards shorter intergenic spacers. Plant Biotechnol J. 2014;12: 344–353. 10.1111/pbi.12141 [DOI] [PubMed] [Google Scholar]
- 26. Hirao T, Watanabe A, Kurita M, Kondo T, Takata K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: diversified genomic structure of coniferous species. BMC Plant Biol. 2008;8: 70 10.1186/1471-2229-8-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S. Plant DNA barcoding: from gene to genome. Biol Rev. 2015;90: 157–166. 10.1111/brv.12104 [DOI] [PubMed] [Google Scholar]
- 28. Peakall R, Ebert D, Scott LJ, Meagher PF, Offord CA. Comparative genetic study confirms exceptionally low genetic variation in the ancient and endangered relictual conifer, Wollemia nobilis (Araucariaceae). Mol Ecol. 2003;12: 2331–2343. [DOI] [PubMed] [Google Scholar]
- 29. McPherson H, van der Merwe M, Delaney SK, Edwards MA, Henry RJ, McIntosh E, et al. Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 2013;13: 8 10.1186/1472-6785-13-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12: 13–15. [Google Scholar]
- 31. Blanca JM, Pascual L, Ziarsolo P, Nuez F, Cañizares J. ngs_backbone: a pipeline for read cleaning, mapping and SNP calling using Next Generation Sequence. BMC Genomics. 2011;12: 285 10.1186/1471-2164-12-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18: 821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chevreux B. MIRA: an automated genome and EST assembler. PhD Thesis, Ruprecht-Karls University. 2005. Available: http://www.chevreux.org/uploads/media/chevreux_thesis_MIRA.pdf
- 34. Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8: 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinform. 2009;25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5: e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wyman SK, Jansen RK, Boore JL Automatic annotation of organellar genomes with DOGMA. Bioinform. 2004;20: 3252–3255. [DOI] [PubMed] [Google Scholar]
- 38. Katoh K, Kuma K-i, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schattner P, A.F. B, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33: W686–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52: 267–274. [DOI] [PubMed] [Google Scholar]
- 41. Mayer C, Leese F, Tollrian R. Genome-wide analysis of tandem repeats in Daphnia pulex—a comparative approach. BMC Genomics. 2010;11: 277 10.1186/1471-2164-11-277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27: 573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29: 4633–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu C-S, Wang Y-N, Liu S-M, Chaw S-M. Chloroplast genome (cpDNA) of Cycas taitungensis and 56 cp protein-coding genes of Gnetum parvifolium: insights into cpDNA evolution and phylogeny of extant seed plants. Mol Biol Evol. 2007;24: 1366–1379. [DOI] [PubMed] [Google Scholar]
- 45. Morton BR. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J Mol Evol. 2003;56: 616–629. [DOI] [PubMed] [Google Scholar]
- 46. Rouwendal GJ, Mendes O, Wolbert EJ, De Boer AD. Enhanced expression in tobacco of the gene encoding green fluorescent protein by modification of its codon usage. Plant Mol Biol. 1997;33: 989–999. [DOI] [PubMed] [Google Scholar]
- 47. Steane DA. Complete nucleotide sequence of the chloroplast genome from the Tasmanian blue gum, Eucalyptus globulus (Myrtaceae). DNA Res. 2005;12: 215–220. [DOI] [PubMed] [Google Scholar]
- 48. Fleischmann TT, Scharff LB, Alkatib S, Hasdorf S, Schöttler MA, Bock R. Nonessential plastid-encoded ribosomal proteins in tobacco: a developmental role for plastid translation and implications for reductive genome evolution. Plant Cell. 2011;23: 3137–3155. 10.1105/tpc.111.088906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yi X, Gao L, Wang B, Su Y-J, Wang T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol. 2013;5: 688–698. 10.1093/gbe/evt042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doyle JJ, Doyle JL, Palmer JD. Multiple independent losses of two genes and one intron from legume chloroplast genomes. Syst Bot. 1995: 272–294. [Google Scholar]
- 51. Chaw S-M, Walters TW, Chang C-C, Hu S-H, Chen S-H. A phylogeny of cycads (Cycadales) inferred from chloroplast matK gene, trnK intron, and nuclear rDNA ITS region. Mol Phylogenet Evol. 2005;37: 214–234. [DOI] [PubMed] [Google Scholar]
- 52. Hausner G, Olson R, Simon D, Johnson I, Sanders ER, Karol KG, et al. Origin and evolution of the chloroplast trnK (matK) intron: a model for evolution of group II intron RNA structures. Mol Biol Evol. 2006;23: 380–391. [DOI] [PubMed] [Google Scholar]
- 53. Kress WJ, Erickson DL. A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One. 2007;2: e508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94: 275–288. 10.3732/ajb.94.3.275 [DOI] [PubMed] [Google Scholar]
- 55. Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, et al. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot. 2005;92: 142–166. 10.3732/ajb.92.1.142 [DOI] [PubMed] [Google Scholar]
- 56. Sugiura C, Sugita M. Plastid transformation reveals that moss tRNAArg-CCG is not essential for plastid function. Plant J. 2004;40: 314–321. [DOI] [PubMed] [Google Scholar]
- 57. Wakasugi T, Tsudzuki J, Ito S, Nakashima K, Tsudzuki T, Sugiura M. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii . Proc Natl Acad Sci. 1994;91: 9794–9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Escapa IH, Catalano SA. Phylogenetic analysis of Araucariaceae: Integrating molecules, morphology, and fossils. Int J Plant Sci. 2013;174: 1153–1170. [Google Scholar]
- 59.Stöckler K, Daniel IL, Lockhart PJ. New Zealand kauri (Agathis australis (D. Don) Lindl., Araucariaceae) survives Oligocene drowning. Syst Biol. 2002: 827–832. [DOI] [PubMed]
- 60. Do HDK, Kim JS, Kim J-H. A trnI_CAU Triplication Event in the Complete Chloroplast Genome of Paris verticillata M.Bieb. (Melanthiaceae, Liliales). Genome Biol Evol. 2014;6: 1699–1706. 10.1093/gbe/evu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cosner ME, Jansen RK, Palmer JD, Downie SR. The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr Genet. 1997;31: 419–429. [DOI] [PubMed] [Google Scholar]
- 62. Kikuchi S, Bédard J, Hirano M, Hirabayashi Y, Oishi M, Imai M, et al. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science. 2013;339: 571–574. 10.1126/science.1229262 [DOI] [PubMed] [Google Scholar]
- 63. Echt CS, DeVerno L, Anzidei M, Vendramin G. Chloroplast microsatellites reveal population genetic diversity in red pine, Pinus resinosa Ait. Mol Ecol. 1998;7: 307–316. [Google Scholar]
- 64. Leclercq S, Rivals E, Jarne P. Detecting microsatellites within genomes: significant variation among algorithms. BMC Bioinformatics. 2007;8: 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Merkel A, Gemmell N. Detecting microsatellites in genome data: variance in definitions and bioinformatic approaches cause systematic bias. Evol Bioinform 2008;4: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a. W. nobilis vs. P. lambertii, b. W. nobilis vs. P. totara, c. W.nobilis vs. N. nagi.
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
The annotated Wollemi pine chloroplast genome sequence is available from GenBank (accession number KP259800). Raw sequence reads have been deposited in the Sequence Read Archive (SRA) database (accession numbers SRR1927951 and SRR192612).