

Summary
Resistance to cytotoxic chemotherapy drugs, including doxorubicin, is a significant obstacle to the effective treatment of breast cancer. Here, we have identified a mechanism by which the PI3K/Akt pathway mediates resistance to doxorubicin. In addition to inducing DNA damage, doxorubicin triggers sustained activation of Akt signaling in breast cancer cells. We show that Akt contributes to chemotherapy resistance such that PI3K or Akt inhibitors sensitize cells to doxorubicin. We identify MERIT40, a component of the BRCA1-A DNA damage repair complex, as an Akt substrate that is phosphorylated following doxorubicin treatment. MERIT40 phosphorylation facilitates assembly of the BRCA1-A complex in response to DNA damage and contributes to DNA repair and cell survival following doxorubicin treatment. Finally, MERIT40 phosphorylation in human breast cancers is associated with estrogen receptor positivity. Our findings suggest that combination therapy with PI3K or Akt inhibitors and doxorubicin may constitute a successful strategy to overcome chemotherapy resistance.
Keywords: DNA damage, chemotherapy, PI3K, Akt, MERIT40
Introduction
As a component of both monotherapy and combination therapy regimens, the anthracycline antibiotic doxorubicin is the primary route of treatment in a wide range of cancers, including breast cancer. The cytotoxicity of doxorubicin is primarily mediated by inhibition of DNA topoisomerase II and subsequent generation of DNA double strand breaks (Tewey et al., 1984). The extent of DNA damage induced by doxorubicin exceeds the DNA repair capacity of tumor cells leading to cell cycle arrest and cell death. Although breast tumors are often initially responsive, the clinical efficacy of doxorubicin is severely limited by intrinsic and acquired resistance. Characterization of mechanisms that contribute to drug resistance and the identification of novel strategies to circumvent doxorubicin resistance would provide new and more effective therapies in the management of breast cancer.
The phosphoinositide 3-kinase (PI3K) pathway plays a critical role in virtually all aspects of tumor biology by regulating fundamental cellular functions including cell proliferation and survival. The PI3K pathway is frequently hyperactivated in breast cancer and numerous small molecule inhibitors have been developed to specifically inactivate this pathway for cancer therapy (Baselga, 2011). Elevated PI3K pathway activity has been associated with diminished sensitivity to conventional chemotherapy agents and the class I PI3K inhibitor GDC-0941 enhances the anti-tumor activity of doxorubicin in breast and ovarian cancer cells that depend on PI3K for survival (Isakoff et al., 2005; Wallin et al., 2010). A major effector of the PI3K pathway is the serine-threonine kinase Akt. Phosphorylation at two sites, Thr308 and Ser473, increases the enzymatic activity of Akt and leads to the phosphorylation of numerous substrates containing the consensus RxRxxS/T motif. Akt activation in response to oncogenic PI3K pathway mutations and in response to growth factor signaling has been well documented. However, Akt is also regulated by the PI3K-related kinase family member DNA-Dependent Protein Kinase (DNA-PK), which phosphorylates Akt at Ser473 in response to DNA damage (Bozulic et al., 2008). The mechanisms by which Akt influences cell survival following DNA damage are poorly understood.
Here, we demonstrate that doxorubicin induces Akt activation in breast cancer cell lines and show that PI3K and Akt inhibitors dramatically enhance the cytotoxicity of doxorubicin. Mechanistically, we identify MERIT40 as an Akt substrate and demonstrate that MERIT40 phosphorylation contributes to the resolution of DNA damage following doxorubicin exposure. We propose that inhibition of MERIT40 phosphorylation and disruption of DNA damage repair contributes to the efficacy of combination therapy with PI3K/Akt inhibitors and doxorubicin.
Results
PI3K and Akt inhibitors sensitize cells to doxorubicin-induced death
We first examined the ability of doxorubicin to induce Akt activation. Exposure of MCF10A breast epithelial cells to doxorubicin results in elevated Akt phosphorylation at both Ser473 and Thr308 in a time- and concentration-dependent manner (Fig. 1A and 1B). Akt phosphorylation is accompanied by an increase in phosphorylation of the Akt substrate PRAS40 and is also coincident with phosphorylation of histone H2A.X, a marker of DNA damage and genomic instability. Doxorubicin also enhances Akt phosphorylation in T47D and SUM-159 breast cancer cells (Fig. 1C), despite the fact that both cell lines harbor activating mutations in the PIK3CA gene that promote constitutive PI3K pathway activity. The ability of doxorubicin to induce Akt activation is blocked by the DNA-PK inhibitor Nu7441 consistent with the notion that DNA-PK is directly involved in activating Akt downstream of DNA damage (Fig. 1D). Interestingly, Akt phosphorylation is also largely disrupted by the class I PI3K inhibitor BKM120, consistent with a recent study demonstrating that doxorubicin induces activation of receptor tyrosine kinase (RTK)/PI3K/Akt signaling (Bezler et al., 2012).
Fig. 1.

Influence of doxorubicin on Akt activity and contribution of PI3K/Akt signaling toward cell survival following doxorubicin exposure.
MCF10A cells were serum-starved and treated with (A) 2 μM doxorubicin over a 10 hour time course or (B) increasing concentrations of doxorubicin for 10 hours. (C) T47D and SUM159 cells were serum-starved and treated with 0.5 μM doxorubicin for 24 hours. (D) MCF10A cells were serum-starved and pre-treated with 2 μM BKM120, 2 μM MK2206 or 2 μM Nu7441 for 30 minutes before exposure to 2 μM doxorubicin for 10 hours. (E) MCF10A cells were pre-treated with 1 μM MK2206, 1 μM BKM120 or 1 μM Nu7441 for 24 hours before exposure to increasing concentrations of doxorubicin for an additional 48 hours. Cell viability is expressed as a percentage of viability observed in untreated cells (t test; * P < 0.05, ** P < 0.01, *** P < 0.001). (F) MCF10A cells were pre-treated with 1 μM MK2206, 1 μM BKM120 or 1 μM Nu7441 for 24 hours before addition of 0.5 μM doxorubicin for an additional 24 hours. (G) T47D, SUM159 and MCF7 cells were pre-treated with 1 μM MK2206 or 1 μM BKM120 for 24 hours before exposure to 0.5 μM doxorubicin for an additional 48 hours. Cell viability is expressed as a percentage of viability observed in untreated cells (t test; * P < 0.05, ** P < 0.01, *** P < 0.001).
We next determined the contribution of Akt activity to cell survival following doxorubicin exposure. Single-agent doxorubicin limits the viability of MCF10A cells in a concentration-dependent manner (Fig. 1E). However, the cytotoxicity of doxorubicin is significantly enhanced when MCF10A cells are pre-treated with either Nu7441, BKM120 or MK2206, an allosteric pan-Akt inhibitor, suggesting that DNA-PK/PI3K/Akt signaling contributes to cell survival following DNA damage. Strikingly, Akt inhibition with MK2206 is as effective at sensitizing cells to doxorubicin as DNA-PK or PI3K inhibition, indicating that Akt plays a critical role in regulating cell survival after DNA damage (Fig. 1E). It should be noted that the effect of single-agent Nu7441, BKM120 or MK2206 on the viability of MCF10A cells is minimal. Doxorubicin-induced cell death by apoptosis is exacerbated in cells pre-treated with Nu7441, BKM120 or MK2206 as demonstrated by enhanced cleavage of poly (ADP-ribose) polymerase (PARP), a signature marker of apoptosis (Fig. 1F). Inhibition of DNA-PK, PI3K or Akt also increases phosphorylation of histone H2A.X following doxorubicin exposure, suggesting that Akt directly contributes to DNA damage repair. Moreover, inhibition of PI3K or Akt sensitizes PI3K-mutant breast cancer cell lines T47D, SUM159 and MCF7 to doxorubicin, and in all cases the combination of Akt inhibitor and doxorubicin is as effective as combination PI3K inhibitor plus doxorubicin (Fig. 1G). Taken together, these data demonstrate that Akt drives a survival pathway that promotes DNA repair and thereby desensitizes cells to the toxicity of doxorubicin.
Akt phosphorylates MERIT40 in response to doxorubicin exposure
As part of three distinct protein complexes, the tumor suppressor breast cancer susceptibility gene 1 (BRCA1) plays a critical role in regulating the cellular response to DNA damage. The BRCA1-A complex, containing BRCA1, Abraxas, Rap80, BRE, BRCC36 and MERIT40, forms at sites of DNA double strand breaks and contributes to the resolution of DNA damage (Feng et al., 2009; Shao et al., 2009; Wang et al., 2009). A global phosphoproteomic screen identified that MERIT40 is phosphorylated in a sequence that conforms to the optimal Akt consensus motif, RxRxxS/T (Fig. 2A) (Moritz et al., 2010). Using a phospho-MERIT40 Ser29-specific antibody, we show that purified recombinant Akt1, Akt2 or Akt3 can directly phosphorylate MERIT40 at Ser29 (Fig. 2B). Moreover, IGF-1 stimulation promotes phosphorylation of MERIT40 and this is blocked in cells pretreated with BKM120, MK2206, the mTORC1/2 inhibitor Torin1, but not the mTORC1 inhibitor rapamycin, thereby implicating Akt as the kinase responsible for MERIT40 phosphorylation at Ser29 (Fig. 2C). Specific knockdown of Akt isoforms also prevents MERIT40 phosphorylation in response to IGF-1 stimulation (Fig. 2D). Expression of constitutively active PIK3CA alleles into MCF10A cells promotes hyperactivation of Akt and enhances MERIT40 phosphorylation in the absence of growth factors (Fig. 2E). Constitutively active, myristoylated Akt1, Akt2 and Akt3 constructs are also able to induce phosphorylation of MERIT40 in the absence of growth factor (Fig. 2F). Strikingly, doxorubicin also induces rapid and sustained phosphorylation of MERIT40 at Ser29 (Fig. 2G and S1) and this is blocked by Nu7441, Torin1 and MK2206, but not rapamycin (Fig. 2H). Consistent with the notion that doxorubicin induces activation of RTK/PI3K/Akt signaling, the EGFR inhibitor gefitinib and BKM120 also block doxorubicin-induced MERIT40 phosphorylation. In addition, specific knockdown of Akt isoforms completely inhibits MERIT40 phosphorylation and exacerbates histone H2A.X phosphorylation in response to doxorubicin exposure (Fig. 2I). These data show that MERIT40 is an Akt substrate phosphorylated in response to growth factor-induced Akt activation, hyperactivation of Akt via PI3K oncogenic mutations, and downstream of DNA damage-induced activation of Akt.
Fig. 2.

MERIT40 is an Akt substrate.
(A) Domain structure of MERIT40 highlighting the Akt consensus phosphorylation motif (24RPRTRS29), two TNKS binding motifs (28RSNPEGAE35 and 48RSEGEGE54) and a von Willebrand A domain (VWA). (B) HA-Flag-MERIT40 and MERIT40 Ser29Ala were used as substrates in an in vitro kinase assay with recombinant active Akt1, Akt2 or Akt3. (C) MCF10A cells were serum-starved and pre-treated with the indicated inhibitors for 30 minutes before stimulation with IGF-1 for 30 minutes. (D) MCF10A cells were infected with empty vector or Akt shRNA constructs. Cells were serum-starved before stimulation with IGF-1 for 30 minutes. (E) MCF10A cells were infected with vector control, PIK3CA wild-type (WT), PIK3CA E545K (EK) or PIK3CA H1047R (HR) constructs and serum-starved. (F) MCF10A cells were transfected with vector control or myristoylated Akt alleles (myrAkt1/2/3) and serum-starved. (G) MCF10A cells were serum-starved and exposed to 2 μM doxorubicin over a 10 hour time course. See also Fig. S1. (H) MCF10A cells were serum-starved and pre-treated with the indicated inhibitors for 30 minutes before exposure to 2 μM doxorubicin for 10 hours. (I) MCF10A cells were infected with empty vector or Akt shRNA constructs. Cells were serum-starved before exposure to 2 μM doxorubicin for 10 hours.
MERIT40 phosphorylation promotes assembly of the BRCA1-A complex
MERIT40 is an integral component of the nuclear BRCA1-A complex and is required for BRCA1-A complex stability and DNA damage resistance (Feng et al., 2009; Shao et al., 2009; Wang et al., 2009). MERIT40 harbors two binding motifs for the PARP family member tankyrase (TNKS) (Fig. 2A) (Guettler et al., 2011). Interestingly, one TNKS binding motif includes the Ser29 residue. We therefore determined if TNKS is a component of the BRCA1-A complex and also examined the consequence of phosphorylation on the ability of MERIT40 to interact with TNKS. Previous studies have shown that MERIT40 is required to maintain the stability of components of the BRCA1-A complex (Hu et al., 2011). In addition to destabilization of Rap80, Abraxas, BRE and BRCC36, MERIT40 depletion causes a dramatic reduction in TNKS expression (Fig. 3A). TNKS expression, as well as expression of additional components of the BRCA1-A complex, is fully rescued by re-expression of either wild-type MERIT40 or a phosphorylation-deficient MERIT40 Ser29Ala mutant (Fig. 3A). This implies that MERIT40 phosphorylation does not impact the ability of MERIT40 to influence the stability of BRCA1-A complex components or TNKS. These data do however suggest that TNKS could represent a previously uncharacterized component of the BRCA1-A complex. Consistent with this model, co-immunoprecipitation experiments reveal that endogenous TNKS interacts with endogenous BRCA1 and this interaction is enhanced following doxorubicin exposure (Fig. 3B). Co-immunoprecipitation experiments also show that MERIT40 interacts with TNKS (Fig. 3C). However, whereas wild-type MERIT40 binds TNKS with high affinity, MERIT40 Ser29Ala shows dramatically reduced binding to TNKS (Fig. 3C). In cells exposed to doxorubicin, an increase in the interaction of wild-type MERIT40 with endogenous Rap80 and TNKS is observed (Fig. 3D). By contrast, doxorubicin does not stimulate the association of either Rap80 or TNKS with MERIT40 Ser29Ala, indicating that phosphorylation of MERIT40 promotes the association of BRCA1-A complex components in response to DNA damage. This conclusion is also supported by the observation that doxorubicin triggers an increase in the association of endogenous TNKS and Rap80 with endogenous MERIT40 (Fig. 3E). Strikingly, the interaction of MERIT40 with TNKS and Rap80 in response to DNA damage is disrupted by the Akt inhibitor MK2206, indicating that MERIT40 phosphorylation is required to enhance the association of BRCA1-A complex components following DNA damage (Fig. 3E). Importantly, a phosphorylation-dependent increase in the association of BRCA1-A complex components is not observed in cells stimulated with IGF-1, indicating that BRCA1-A complex formation specifically requires a DNA damage signal in addition to Akt-dependent phosphorylation of MERIT40 (Fig. 3F). Consistent with previous studies, MERIT40 depletion compromises Rap80 focus formation upon DNA damage (Fig. 3E) (Feng et al. 2009). Rap80 focus formation is completely rescued by re-expression of wild-type MERIT40 but not by phosphorylation-deficient MERIT40 Ser29Ala. These data demonstrate that MERIT40 phosphorylation plays an integral role in assembly of the BRCA1-A complex following doxorubicin exposure.
Fig. 3.

Effect of MERIT40 phosphorylation on stability and formation of the BRCA1-A complex.
(A) MCF10A cells were infected with empty vector or MERIT40 shRNA constructs and empty vector, wild-type MERIT40 or MERIT40 S29A mutant expression constructs. (B) MCF10A cells were serum-starved before exposure to 2 μM doxorubicin for 1 hour. Endogenous BRCA1 was immunoprecipitated from nuclear extracts and co-immunoprecipitation with endogenous TNKS and Rap80 was monitored. (C) HEK-293T cells were transfected with wild-type or mutant MERIT40 constructs and a wild-type TNKS expression plasmid. HA-tagged MERIT40 was immunoprecipitated from cells and co-immunoprecipitation of myc-tagged TNKS was monitored. Alternatively, myc-tagged TNKS was immunoprecipitated from cells and co-immunoprecipitation of HA-tagged TNKS was monitored. (D) MCF10A cells were infected with wild-type or mutant MERIT40 constructs. Cells were serum-starved and exposed to 2 μM doxorubicin for 1 hour. HA-tagged MERIT40 was immunoprecipitated from cells and co-immunoprecipitation with endogenous TNKS and Rap80 was monitored. (E) MCF10A cells were serum-starved and pre-treated with 2 μM MK2206 for 30 minutes before exposure to 2 μM doxorubicin for 1 hour. Endogenous MERIT40 was immunoprecipitated from cells and co-immunoprecipitation with endogenous TNKS and Rap80 was monitored. (F) MCF10A cells were serum-starved and treated with 2 μM doxorubicin for 1 hour or IGF-1 for 30 minutes. Endogenous MERIT40 was immunoprecipitated from cells and co-immunoprecipitation with endogenous TNKS and Rap80 was monitored. (G) T47D cells were infected with empty vector or a MERIT40 shRNA construct and empty vector, wild-type MERIT40 or MERIT40 S29A mutant expression constructs. Cells were exposed to 0.5 μM doxorubicin for 1 hour, washed with PBS and incubated in fresh media for an additional hour. Rap80 focus formation was monitored by immunofluorescence.
MERIT40 phosphorylation contributes to the resolution of DNA damage
To further evaluate the functional significance of MERIT40 phosphorylation by Akt, we investigated the ability of MERIT40 to influence DNA damage repair. Depletion of MERIT40 from T47D cells dramatically enhances doxorubicin-induced DNA damage, revealed by an increase in histone H2A.X phosphorylation (Fig. 4A) and phospho-H2A.X focus formation (Fig. 4B and 4C). This response is rescued by re-expression of wild-type MERIT40, but not MERIT40 Ser29Ala. These data demonstrate that MERIT40 phosphorylation at Ser29 is required for the resolution of doxorubicin-induced DNA damage. In addition, MERIT40 depletion dramatically enhances spontaneous DNA damage in MCF10A cells, and this can be rescued by re-expression of wild-type MERIT40, but again not by MERIT40 Ser29Ala (Fig. S2). Importantly, MERIT40 depletion sensitizes cells to doxorubicin and this response is rescued by re-expression of wild-type MERIT40 but not MERIT40 Ser29Ala (Fig. 4D and 4E). Taken together, these data demonstrate that Akt-dependent phosphorylation of MERIT40 promotes DNA repair and MERIT40 phosphorylation contributes to cell survival following doxorubicin exposure.
Fig. 4.
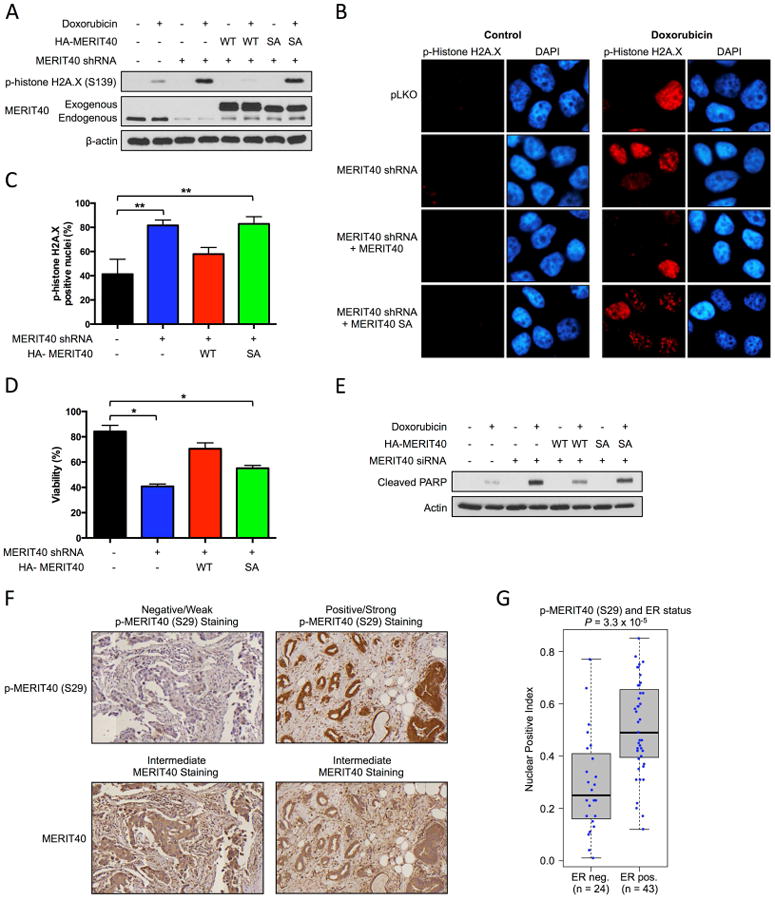
Contribution of MERIT40 phosphorylation toward the resolution of DNA damage and cell survival following doxorubicin exposure and detection of phospho-MERIT40 in human breast tumors.
(A) T47D cells were infected with empty vector or a MERIT40 shRNA construct and empty vector, wild-type MERIT40 or MERIT40 S29A mutant expression constructs. Cells were exposed to 0.5 μM doxorubicin for 1 hour, washed with PBS and incubated in fresh media for an additional 6 hours. See also Fig. S2. (B) T47D cells were infected with empty vector or a MERIT40 shRNA construct and empty vector, wild-type MERIT40 or MERIT40 S29A mutant expression constructs. Cells were exposed to 0.5 μM doxorubicin for 1 hour, washed with PBS and incubated in fresh media for an additional 6 hours. Phosphorylation of histone H2A.X was monitored by immunofluorescence. (C) The percentage of cells with nuclear p-Histone H2A.X foci in Figure 4C was quantified by counting 200 cells in each treatment condition. (t test, ** P < 0.01). (D) MCF10A cells were transfected with control or MERIT40 siRNA constructs and empty vector, wild-type MERIT40 or MERIT40 S29A mutant expression constructs. Cells were exposed to 0.2 μM doxorubicin for 48 hours. Cell viability is expressed as a percentage of viability observed in untreated cells (t test, * P < 0.05). (E) MCF10A cells were transfected with control or MERIT40 siRNA constructs and empty vector, wild-type MERIT40 or MERIT40 S29A mutant expression constructs. Cells were exposed to 0.2 μM doxorubicin for 24 hours. (F) Detection of p-MERIT40 and MERIT40 in invasive breast tumor tissue samples by immunohistochemistry (IHC). (G) Relationship between p-MERIT40 IHC staining in human breast tumor samples and ER status.
Elevated MERIT40 expression has been identified in epithelial ovarian cancer and SNPS within the MERIT40 gene are associated with breast cancer risk (Antoniou et al., 2010; Bolton et al., 2010). However, the expression of MERIT40 and phospho-MERIT40 in human breast tumors has not been examined. Tissue microarrays containing cores of invasive breast cancer tissue obtained from archival pathology specimens were used to examine expression of MERIT40 and phospho-MERIT40 by immunohistochemistry. This analysis reveals strong phospho-MERIT40 staining in a subset of cases that is present in both the nuclei and cytoplasm (Fig. 4F, positive/strong). A subset of cases showed no phospho-MERIT40 staining (Fig. 4F, negative/weak). Overall, the total MERIT40 staining shows more intermediate staining with fewer strongly positive cases (Fig. 4F, intermediate). We also used computational analysis to assess the association of phospho-MERIT40 nuclear staining with breast cancer subtypes. This analysis reveals a strong, statistically-significant association of nuclear phospho-MERIT40 with ER status (P=3.3×10-5) (Fig. 4G).
Discussion
Resistance to cytotoxic chemotherapy agents is a common phenomenon in breast cancer that has a drastic impact on patient survival. This underscores the need to identify strategies to overcome drug resistance. One approach that has been favored in recent years is the development of drug regimens combining cytotoxic chemotherapy with molecularly targeted drugs that inhibit signaling cascades critical to breast cancer survival and progression. The PI3K and Akt pathway is hyperactive in more than 70% of breast tumors and is critical for tumor progression and resistance to anti-cancer drugs (Courtney et al., 2010). In addition, as highlighted in this study, an undesirable response to chemotherapy exposure is activation of PI3K/Akt signaling. Rationally-designed small molecule inhibitors that target either PI3K or Akt have recently been developed and are currently in phase I or II clinical trials (Dienstmann et al., 2014). Unfortunately, modest anti-tumor responses have been reported following PI3K and Akt inhibitor monotherapy and substantial tumor regression is rarely observed (Rodon et al., 2013). In an attempt to improve response rates to PI3K and Akt inhibitors, clinical trials that incorporate these inhibitors and traditional chemotherapeutic drugs are now in progress (Paplomata and O'Regan, 2014). Our study provides evidence that PI3K and Akt inhibitors dramatically sensitize breast cancer cells to the DNA-damaging chemotherapy agent doxorubicin. The frequency of PI3K pathway alterations in breast cancer combined with the ability of doxorubicin to induce PI3K/Akt activity provides rationale to assess combination therapy with doxorubicin and PI3K inhibitors in the clinic.
In chemotherapy-sensitive cancer cells, DNA damage resulting from doxorubicin exposure leads to cell death via induction of apoptosis. The ability of the PI3K and Akt pathway to promote cell survival has been widely reported and a number of substrates that contribute to inhibition of apoptosis have been identified (Manning and Cantley, 2007). Although it is well-established that Akt activity promotes resistance to DNA damage, the substrates that directly impact DNA damage repair are poorly characterized (Xu et al., 2012). Our study identifies MERIT40, a component of the BRCA1-A DNA damage repair complex, as a unique Akt substrate that directly influences the cellular response to DNA damage. Specifically, we show that MERIT40 phosphorylation at Ser29 facilitates the association of MERIT40 with the ubiquitin-binding protein Rap80. Furthermore, we show that MERIT40 phosphorylation promotes nuclear Rap80 focus formation. Rap80 is a critical scaffold protein in the BRCA1-A complex required to relocate and target the remaining subunits of the BRCA1-A complex to sites of DNA damage (Bian et al., 2012; Kim et al., 2007; Sobhian et al., 2007; Wang et al., 2007; Wu et al., 2012). Future studies to investigate the effect of MERIT40 phosphorylation in facilitating the association of MERIT40 with additional components of the BRCA1-A complex are warranted. Importantly, we also demonstrate that MERIT40 phosphorylation promotes DNA damage repair and contributes to cell survival following doxorubicin exposure. We therefore propose that inhibition of MERIT40 phosphorylation and disruption of DNA damage repair contributes to the efficacy of combination therapy with PI3K/Akt inhibitors and doxorubicin.
Our studies also identify the PARP family member TNKS as a component of the BRCA1 A complex. We demonstrate that MERIT40 interacts with TNKS and furthermore that MERIT40 phosphorylation promotes the association of MERIT40 with TNKS. TNKS has previously been implicated in the regulation of DNA damage repair and has been shown to influence stability of the catalytic subunit of DNA-PK via poly-ADP-ribosylation (PARsylation) (Dregalla et al., 2010). In addition, PARP1 has been shown to PARsylate BRCA1 and thereby maintain stability of the BRCA1-A complex (Hu et al., 2014). The ability of TNKS to influence DNA damage repair via PARsylation of DNA-PK, BRCA1, or additional BRCA1-A complex components, clearly merits further investigation. In addition, it will be interesting to evaluate the contribution of MERIT40 phosphorylation to DNA damage-induced protein PARsylation.
Molecular markers that might predict the efficacy of combination therapy with PI3K/Akt pathway inhibitors and cytotoxic agents, like doxorubicin, are lacking. Another interesting observation arising in this study is that phospho-MERIT40 staining is associated with ER status in invasive breast tumor tissue samples. It should be noted that activating mutations in the PIK3CA gene occur in a significant proportion of ER-positive breast cancers (Network, 2012). Our results suggest that MERIT40 phosphorylation could be used as a biomarker in ER-positive breast cancer patients to predict sensitivity to traditional cytotoxic chemotherapy agents and also indicates that patients with high levels of phospho-MERIT40 would likely benefit from combination therapy with Akt inhibitors and doxorubicin.
In summary, our study identifies a mechanism by which the PI3K and Akt pathway mediates DNA repair in response to chemotherapy exposure and indicates that combining PI3K/Akt inhibitors with doxorubicin may constitute a successful strategy to overcome chemotherapy resistance in breast cancer, at least in part by disrupting the phosphorylation of MERIT40.
Experimental Procedures
Immunoblotting
Cells were washed with PBS and lysed in RIPA buffer. Lysates were resolved by SDS-PAGE and transferred electrophorectically to nitrocellulose membrane (Bio-Rad) followed by immunoblotting.
Sulforhodamine B assay
Cell viability was monitored using the Sulforhodamine B (SRB) assay. Adherent cells were fixed by addition of 12.5% (w/v) trichloroacetic acid and incubation at 4 °C for 1 hour. Wells were washed with water and cells were stained by addition of SRB solution (0.5% (w/v) SRB, 1% acetic acid). Wells were washed twice with 1% acetic acid and allowed to dry at room temperature. SRB was solubilized with 10 mmol/L Tris, pH 10.5 and absorbance at 510 nm was measured.
In vitro kinase assay
MSCV-HA-Flag-MERIT40 or MSCV-HA-Flag-MERIT40 Ser29Ala MERIT40 was immunoprecipitated from cell extracts and incubated with 500 ng of recombinant Akt1, Akt2 or Akt3 (Sigma-Aldrich) in a kinase buffer containing 250 μmol/L cold ATP for 1 hour at 30°C. Eluates were resolved by SDS-PAGE.
Immunoprecipitation
Nuclear extracts were prepared by high salt extraction. For whole cell lysate preparation cells were washed with PBS and lysed in EBC lysis buffer. Lysates were incubated with 1-2 μg of antibody overnight at 4 °C followed by inc ubation with protein A/G Sepharose beads (Amersham Biosciences). Immune complexes were washed with NETN buffer and eluted by incubation for 5 minutes at 95°C in S DS-PAGE sample buffer. Eluates were resolved by SDS-PAGE.
Immunofluorescence
Cells plated on coverslips were fixed with 2% paraformaldehyde for 10 minutes, permeabilized with 0.5% Triton X-100, and blocked with 1% BSA in 20 mmol/L Tris-HCl, pH 7.5, for 20 minutes. Coverslips were then incubated with the appropriate antibodies. After washing twice with PBS, coverslips were mounted with Prolong Gold antifade reagent containing DAPI (Life Technologies). Images of cells were acquired using a fluorescence microscope (Nikon Eclipse Ti) and digital image analysis software (NIS-Elements, Nikon).
Tissue microarrays and image analysis of immunohistochemistry
Two tissue microarrays containing breast tissue specimens from the archives of the Department of Pathology at the Beth Israel Deaconess Medical Center, were constructed as previously described (Elloul et al., 2014). Immunohistochemistry staining was done for total MERIT40 and p-MERIT40 Ser29. Computational image analysis of protein expression was performed using Definiens TissueStudio 3.6.1 (Munich, Germany) to yield the intensity of nuclear expression of p-MERIT40 Ser 29 and MERIT 40 in the cancer epithelium. The proportion of positively staining epithelial nuclei was recorded for each core and reported as the Nuclear Positive Index.
Supplementary Material
Highlights.
Doxorubicin triggers activation of Akt signaling in breast cancer cells
MERIT40 is phosphorylated by Akt in response to doxorubicin exposure.
MERIT40 phosphorylation contributes to DNA repair and cell survival.
PI3K and Akt inhibitors sensitize breast cancer cells to doxorubicin.
Acknowledgments
The authors thank the histology core at BIDMC for their technical support for immunohistochemistry; Steven Elledge for providing MSCV-HA-Flag-MERIT40; Frank Sicheri for providing pLP-dmyc-TNKS2; and members of the Toker laboratory for discussions. This work was supported in part by grants from the Department of Defense Breast Cancer Research Program (BC110900, A.T.), a John Gavin Post-doctoral Fellowship from the Genesis Oncology Trust of New Zealand (K.K.B.), and the National Library of Medicine of the National Institutes of Health (K22LM011931, A.H.B).
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures and two figures.
Author Contributions: K.K.B. and A.T. designed the experiments and wrote the paper. K.K.B. performed all of the experiments. All authors contributed to analysis and interpretation of the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antoniou AC, Wang X, Fredericksen ZS, McGuffog L, Tarrell R, Sinilnikova OM, Healey S, Morrison J, Kartsonaki C, Lesnick T, et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet. 2010;42:885–892. doi: 10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16:12–19. doi: 10.1634/theoncologist.2011-S1-12. [DOI] [PubMed] [Google Scholar]
- Bezler M, Hengstler JG, Ullrich A. Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances apoptosis of ovarian cancer cells. Mol Oncol. 2012;6:516–529. doi: 10.1016/j.molonc.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian C, Wu R, Cho K, Yu X. Loss of BRCA1-A complex function in RAP80 null tumor cells. PLoS One. 2012;7:e40406. doi: 10.1371/journal.pone.0040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton KL, Tyrer J, Song H, Ramus SJ, Notaridou M, Jones C, Sher T, Gentry-Maharaj A, Wozniak E, Tsai YY, et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet. 2010;42:880–884. doi: 10.1038/ng.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–213. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- Dregalla RC, Zhou J, Idate RR, Battaglia CL, Liber HL, Bailey SM. Regulatory roles of tankyrase 1 at telomeres and in DNA repair: suppression of T-SCE and stabilization of DNA-PKcs. Aging. 2010;2:691–708. doi: 10.18632/aging.100210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elloul S, Kedrin D, Knoblauch NW, Beck AH, Toker A. The adherens junction protein afadin is an AKT substrate that regulates breast cancer cell migration. Mol Cancer Res. 2014;12:464–476. doi: 10.1158/1541-7786.MCR-13-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Huang J, Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009;23:719–728. doi: 10.1101/gad.1770609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guettler S, LaRose J, Petsalaki E, Gish G, Scotter A, Pawson T, Rottapel R, Sicheri F. Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell. 2011;147:1340–1354. doi: 10.1016/j.cell.2011.10.046. [DOI] [PubMed] [Google Scholar]
- Hu X, Kim JA, Castillo A, Huang M, Liu J, Wang B. NBA1/MERIT40 and BRE interaction is required for the integrity of two distinct deubiquitinating enzyme BRCC36-containing complexes. J Biol Chem. 2011;286:11734–11745. doi: 10.1074/jbc.M110.200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014;4:1430–1447. doi: 10.1158/2159-8290.CD-13-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paplomata E, O'Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6:154–166. doi: 10.1177/1758834014530023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- Shao G, Patterson-Fortin J, Messick TE, Feng D, Shanbhag N, Wang Y, Greenberg RA. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23:740–754. doi: 10.1101/gad.1739609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- Wallin JJ, Guan J, Prior WW, Edgar KA, Kassees R, Sampath D, Belvin M, Friedman LS. Nuclear phospho-Akt increase predicts synergy of PI3K inhibition and doxorubicin in breast and ovarian cancer. Sci Transl Med. 2010;2:48ra66. doi: 10.1126/scitranslmed.3000630. [DOI] [PubMed] [Google Scholar]
- Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23:729–739. doi: 10.1101/gad.1770309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Liu C, Chen J, Yu X. RAP80 protein is important for genomic stability and is required for stabilizing BRCA1-A complex at DNA damage sites in vivo. J Biol Chem. 2012;287:22919–22926. doi: 10.1074/jbc.M112.351007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.