

Abstract
Genetic and environmental factors contribute to the onset and progression of lupus. CD4+ T cells from patients with active lupus show a decreased ERK signaling pathway, which causes changes in gene expression. The defect points to its upstream regulator, PKCδ, which exhibits a deficient activity due to oxidative stress. Our aim was to investigate the effect of a defective PKCδ in the development of lupus.
We generated a double transgenic C57BL6 × SJL mouse that expresses a doxycycline-induced dominant negative PKCδ (dnPKCδ) in T cells. The transgenic mice displayed decreased T cell ERK signaling, decreased DNMT1 expression and overexpression of methylation sensitive genes involved in the exaggerated immune response in the pathogenesis of lupus. The mice developed anti-dsDNA autoantibodies and glomerulonephritis with IgG deposition.
The study indicates common pathogenic mechanisms with human lupus, suggesting that environmentally-mediated T cell PKCδ inactivation plays a causative role in lupus.
Keywords: Lupus, T cells, PKCδ, transgenic mouse model, extracellular signal-regulated kinase (ERK), autoimmunity
1. INTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic relapsing autoimmune disease characterized by the development of autoantibodies to nuclear components and immune complex deposition in tissues including the kidney, lung and others, causing end-organ damage. Environmental factors and predisposing genetic variants interact to cause the development and flares of this disease [1]. While autoantibody formation is a prominent immunologic abnormality in lupus, a growing body of evidence indicates that epigenetic deregulation of T cell gene expression, caused by impaired T cell DNA methylation, underlies the autoimmune response and autoantibody formation in idiopathic, as well as some forms of drug induced human lupus [2]. A causative role for T cell DNA demethylation in lupus was first suggested by reports that CD4+ T cells treated with 5-azacytidine, a DNA methylation inhibitor, were sufficient to cause lupus-like autoimmunity when injected into syngeneic mice [3], and that patients with active lupus had similar decreases in T cell DNA methylation [4]. Procainamide and hydralazine, which cause antinuclear antibody production in a majority of people and drug-induced lupus in a genetically predisposed subset [5], were subsequently shown to inhibit T cell DNA methylation [6], and murine T cells treated with these drugs also caused lupus-like autoimmunity when injected into syngeneic mice [7]. Procainamide was found to be a competitive inhibitor of Dnmt1 enzymatic activity [8; 9], while hydralazine blocks ERK pathway signaling, preventing upregulation of Dnmt1 as cells enter mitosis [10].
The methylation defect in T cells from patients with active lupus was traced to impaired ERK pathway signaling [11; 12], similar to that caused by hydralazine [10], and T cells treated with ERK pathway signaling inhibitors also caused a lupus-like disease when injected into syngeneic mice [6]. A causative role for impaired ERK pathway signaling in lupus was demonstrated by creating a double transgenic mouse strain in which expression of a dominant negative MEK (dnMEK) could be selectively induced in T cells by adding doxycycline to their drinking water. Importantly, doxycycline only induced anti-DNA antibodies and an “interferon signature” in C57BL6 mice [13], but caused an immune complex glomerulonephritis when the C57BL6 double transgenic were crossed with SJL mice, which are genetically more susceptible to autoimmunity [14]. This is consistent with extensive evidence indicating a genetic requirement for lupus to develop [15; 16]. This is analogous to drug-induced lupus, where hydralazine and procainamide cause antinuclear antibodies and drug-induced lupus in genetically predisposed people, but only antinuclear antibodies in people without genetic susceptibility to lupus [5]. More recent studies traced the ERK pathway signaling defect to PKCδ [17] . PMA directly activates PKCδ by inducing phosphorylation on its activation loop. However, PMA-stimulated PKCδ phosphorylation is impaired in both hydralazine treated CD4+ T cells and CD4+ T cells from patients with active lupus [17], suggesting an intrinsic PKCδ defect. Lupus is characterized by an environmentally-induced oxidative state [18; 19], and we subsequently reported that the lupus T cell PKCδ activation defect is due to oxidative damage, causing impaired ERK pathway signaling in lupus T cells. The same PKCδ signaling defect was found in T cells treated with oxidizing agents in vitro [20].
Based on these observations, we hypothesized that environmentally-induced T cell PKCδ inactivation may cause a lupus-like disease. We therefore generated a double transgenic, C57BL6 × SJL mouse in which doxycycline induces expression of a dominant negative PKCδ (dnPKCδ) selectively in T cells, reproducing the environmentally induced PKCδ inactivation found in lupus T cells [17; 20]. Inducing expression of the T cell specific dnPKCδ in these mice decreases ERK pathway signaling and Dnmt1 levels, causing overexpression of genes normally suppressed by DNA methylation, and the mice develop anti-dsDNA antibodies and an immune-complex glomerulonephritis resembling human lupus. These results thus support the hypothesis that environmentally-induced T cell PKCδ inactivation contributes to the development of human lupus.
2. MATERIALS AND METHODS
2.1 Generation of a dnPKCδ/PCR2.1 construct
A dnPKCδ cDNA was PCR amplified from a plasmid encoding a dominant negative form of mouse PKC-δK376R-pEGFP-N1 fusion protein, generously donated by Dr. Stuart H. Yuspa [21], using primers with an EcoR1 restriction site at the 5’ end and a BamH1 site at the 3’ end. A stop codon was added to the 3’ end, using High Fidelity Taq polymerase (Roche). “A” overhangs were added using Taq polymerase (Invitrogen), and then the construct was subcloned into the PCR 2.1 vector using TA cloning method. The entire sequence was verified by sequencing, and confirmed the K376R mutation and the absence of any other PCR induced base changes.
2.2 DnPKCδ/pTRE-Tight construct and transgene
The dnPKCδ cDNA was excised from the dnPKCδ/PCR 2.1 construct using EcoR1 and BamH1 then ligated into pTRE-Tight to provide a tightly controlled expression system. Subcloning was confirmed by sequencing, performed by the DNA Sequencing Core at the University of Michigan. The dnPKCδ/pTRE-Tight construct was then digested with Xho1 to excise the dnPKCδ along with the tet-on promoter and the poly A tail for microinjection.
2.3 Tet-on dnPKCδ transgenic mice
DnPKCδ/CD2rtTA double transgenic mice were developed by the Transgenic Animal Model Core of the University of Michigan's Biomedical Research Core Facilities. Double transgenic mice were generated by crossing dnPKCδ-TRE transgenic mice with CD2-rtTA mice kindly donated by Dr. R. Zamoyska [22]. Briefly, the dnPKCδ transgene generated was injected into fertilized eggs from C57BL/6 × SJL mice and implanted into pseudopregnant females. Mice with the transgene were backcrossed onto an SJL background and bred with SJL transgenic strain containing the reverse tetracycline transactivator (rtTA) under the control of a CD2 promoter (CD2-rtTA). Mice were backcrossed onto SJL background for at least 10 generations. Animals were maintained in a specific pathogen-free environment. All protocols were approved by the University of Michigan Committee on the Use and Care of Animals (UCUCA). Pups were weaned at 20 days of age and genotyped for the presence of the dnPKCδ and CD2rtTA transgenes confirmed by PCR using genomic DNA isolated from tail-snips (Qiagen Blood & Tissue Kit). PCR primers specific to each gene were obtained from Integrated DNA Technologies (IDT, Coralville, IA); the sequences were: dnPKCδ Fw: 5’-TATCAGTG ATAGAGAACGTATG-3’ and Rv; 5’-CAGCACAGAAAGGCTGGCTTGCTTC-3’. Primer sequences used for the CD2rtTA were previously described [13]. Transgene expression was induced by giving 2 mg/ml of doxycycline (doxy) in the drinking water and supplemented with 5% of sucrose for palatability as previously described by our group [13]. Double transgenic control animals were given 5% sucrose alone. Urinary protein was measured using Chemstrip 6 dipsticks (Roche, Madison, WI).
Doxycycline hydrochloride (doxy) (Clontech Lab.Inc, Mountainview, CA) was dissolved in water and prepared fresh before use. The bottles were protected from light and changed every 4 days.
2.4 RNA isolation
Mouse tissues were homogenized in Trizol (Invitrogen, Carlsbad CA) using an Ultraturrax (IKA, Staufen, Germany) disperser. The aqueous layer was mixed with an equal volume of 70% ethanol, then RNA purified using an RNeasy kit (Qiagen, Valencia CA) according to the manufacturer's instructions. DNA digestion was performed using a Turbo-DNA-free kit (Ambion, Austin TX) following the manufacturer's protocols.
2.5 Cell purification and culture
CD4+ or CD3+ T cells where indicated, were isolated from the spleens of transgenic mice by negative selection using magnetic beads (Miltenyi Biotec, Auburn CA) The cells were cultured in RPMI 1640 supplemented with 10% fetal calf serum, 2mM glutamine and penicillin / streptomycin, without or with doxy (2 μg/ml) for 18 h or 72 h. Where indicated, the cells were stimulated with 50 ng/ml PMA for 15 min at 37°C.
2.6 TCR stimulation
Purified CD4+ T cells were incubated at 107 cells / ml with anti-CD3ε (145-2C11, 5 mg/ml) plus anti-CD4 (GK1.5, 5 mg/ml) for 30 min at 4 °C. Both antibodies were kindly provided by Dr. G. Garcia (University of Michigan) [23]. Cells were washed and stimulated with goat anti- rat IgG to crosslink CD3 and CD4 determinants for 5 min at 37°C. Then cells were washed with PBS containing 5% BSA and the cell pellet was lysed to obtain the protein extract.
2.7 Protein isolation
Following culture and/or stimulation, the cells were centrifuged, resuspended in RIPA buffer (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1mM EDTA, 100 μg/ml PMSF, 100 μM sodium orthovanadate, 1mM DTT) and a protease and phosphatase inhibitor cocktail (Roche, Indianapolis IN), rotated at 4°C for 30 min The insoluble material was removed by centrifugation at 16000Xg for 30 min and the supernatant saved as whole cell lysate. The total protein was quantitated using the BCA Protein Assay (Pierce, Rockford, IL).
2.8 Antibodies
The following primary antibodies were used: rabbit polyclonal anti PKCαp-Thr638/641, anti-PKCθ p-Thr538, anti PKCδ p-Thr505, anti PKCδ, anti ERK p-Thr202/Tyr204 and anti ERK1/2 (Cell Signaling Tech., Beverly, MA). It was also used mouse anti-β actin (Sigma-Aldrich). Secondary antibodies included: anti-rabbit IgG horseradish peroxidase (1:2000, Cell Signaling Tech) and anti-mouse IgG horseradish peroxidase (1:4000, Amersham).
2.9 Immunoblotting
Studies for protein expression were performed as previously described [17]. Briefly, 20 μg of protein was subjected to electrophoresis in 10-12% SDS-polyacrylamide gels. The fractionated proteins were transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH) and blocked with 5% bovine serum albumin (Sigma-Aldrich). After a 16-hour incubation with the specific antibody followed by a horseradish peroxidase-linked secondary antibody, the proteins were visualized by chemiluminescence. For quantitative studies, the bands were analyzed with Image Quant 5.2 software (Amersham, Piscataway, NJ). Where indicated, blots were stripped and reprobed with the corresponding antibody. Values were normalized with respect to β-actin and/or total kinase content as indicated.
2.10 Real time RT-PCR
150 ng of RNA was converted to cDNA and amplified in one step using a Quanti-Tect SYBR Green RT-PCR kit (Qiagen, Venlo, Netherlands). CD70 transcripts were quantitated by real-time RT-PCR using a Rotor-Gene 3000 (Corbett Research, Sydney, Australia) and previously published protocols [17]. The amplification conditions were: 30 min at 50°C, 15 min at 95°C, 40 cycles of 15s at 94 °C, 20s at 56°C and 30s at 72°C followed by a final extension at 72° for 5 min. Transcript expression levels were normalized to GAPDH. The primers were: mouse GAPDH Fw: 5’-CAACGACCCCTTCATTGACCTC-3’, Rv: 5’-GCCTCACCCCATTTGATGTTAGTG-3’; mouse CD70 Fw: 5’-TGGCTGTGGGCATCTGCTC-3’, Rv:5’-ACATCTCCGTGGACCAGGTATG-3’; mouse DNMT1 Fw: 5’-GGAAGGCTACCTGGCTAAAGTCAAG-3’; Rv: 5’-ACTGAAAGGGTGTCACTGTCCGAC-3’; and mouse CD11a Fw: 5’-CAGATTGAAGATGGGGTTGTCG-3’, Rv: 5’-CGGGACGATTTTGTAACATAGGTC-3’ The PCR products were fractionated on a 2% agarose gel or 0.8% agarose gel where indicated, and stained with ethidium bromide.
Product quality was determined by melting curves. A series of five dilutions of one RNA sample were included to generate a standard curve, and this was used to obtain relative concentrations of the transcript of interest in each of the RNA samples. In each experiment, water was included as a negative control. GAPDH or β actin amplification, as described above, was used to confirmthat equal amounts of total RNA were added to each sample, and that the RNA was intact and equally amplifiable among all samples.
2.11 Flow cytometry
FACS analysis was used to study T cell activation. The following antibodies were used against lymphocyte surface markers: FITC anti-mouse CD62L (clone MEL-14, BioLegend, San Diego, CA); PE-anti mouse CD3ε chain (145-2C11), and PE-Cy5 anti-mouse CD44 (Pgp-1, Ly-24) were from BD Biosciences Pharmingen (San Diego, CA). The cells were stained with fluorochrome conjugated antibodies for 30 min on ice, washed, fixed in 2% paraformaldehyde, and analyzed using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ) as previously described [24].
2.12 Anti-dsDNA antibody quantitation
Serum anti-dsDNA IgG antibodies were measured by ELISA as previously described [13]. Briefly, microtiter plates (Costar, Corning, NY) were coated overnight at 4° C with 10 μg plasmid dsDNA, then 5 μL of mouse sera were added to each well in 100 μl of buffer and incubated overnight at 4° C. Bound anti-dsDNA antibody was detected by chemiluminescence using HRP-goat anti-mouse IgG (Bethyl Lab Inc. Montgomery, TX) at 450 nm in a spectrophotometer equipped with Softmax Pro software (Molecular Devices, Sunnyvale, CA). Murine monoclonal anti-dsDNA antibody (Chemicon, Billerica, MA) was used for the standard curve.
2.13 Histopathology
Double transgenic dnPKCδ/CD2rtTA mice were given 2mg/ml doxy/5% sucrose in their drinking water for 20 wks. Single transgenic CD2rtTA mice receiving 2mg/ml doxy/5% sucrose or double transgenic animals receiving 5% sucrose were used as controls. At the indicated times, the mice were sacrificed and their kidneys and lungs removed. The kidneys were bisected and one half embedded in O.C.T. (Thermo Fisher) and frozen in liquid nitrogen. Five micron sections were cut from the frozen tissue and fixed for 10 minutes in ice cold acetone. 10% horse serum/PBS was used to block non-specific sites and the sections were stained with a 1:50 dilution of biotin-goat anti-mouse IgG (Fc specific) antibody (USBiologicals)/FITC-Streptavidin (BD Pharmingen) to detect IgG depositions. The other kidney half and the lung were fixed in 10% formalin, paraffin embedded, sectioned, and stained with hematoxylin and eosin as previously described [14].
2.1 Statistics
Statistical analyses were performed by Student's t-test, linear regression or ANOVA as appropriate to determine significance between groups using Systat software. P-values ≤0.05 were considered statistically significant.
3. RESULTS
3.1 Generation of dnPKCδ double transgenic mice
We first generated a dnPKCδ/CD2-rtTA double transgenic mouse strain that expresses a dominant negative PKCδ (dnPKCδ) selectively in T cells. . The rtTA only binds the tetracycline response element recognition sequence in the presence of doxy. Administering doxy in the drinking water induces expression of the dnPKCδ transgene specifically in T cells (Fig 1).
Fig. 1.

Schematic representation of the Tet-On transgenic mouse model. This double transgenic mouse (dnPKCδ+/CD2-rtTA+) results from the breeding of two different strains of mice. One strain expresses the reverse tetracycline-controlled transactivator (rtTA) which comprises the TetR repressor and the VP16 transactivation domain. This vector is driven by the CD2 promoter (CD2+ cell-specific, PCD2), which causes rtTA expression only in CD2+ cells. The other strain expresses the dnPKCδ (PKCδ cDNA) under the control of a tetracycline-responding element (TRE). As consequence, the double transgenic mouse responds with transcription of the transgene only in the presence of the tetracycline derivative, doxycycline (doxy). In the absence of doxycycline, the transcription cannot be completed.
3.2 Leakiness and inducibility
DnPKCδ/CD2rtTA double transgenic mice were given doxy/sucrose or sucrose alone in the drinking water for two weeks. The mice were then sacrificed and dnPKCδ mRNA expression was compared in the heart, lung, liver, brain, spleen (sp), lymph nodes (LN) and thymus by RT-PCR. Figure 2A shows that the transgene is normally transcriptionally silent in these mice, and is induced by doxy only in lymph nodes, spleen and thymus, consistent with selective expression in CD2+ T cells.
Fig.2.

Inducibility of dnPKCδ expression in different tissues and cell populations. A. DnPKCδ expression was determined by RT-PCR in different tissues as indicated in the figure from four double transgenic mice that were given doxycycline (2mg/ml) with 5% sucrose (doxy) in the drinking water for two weeks. Expression was compared with same tissues from four control animals that only received 5% sucrose (no doxy) in the drinking water. Values represent mean ± SEM. * p≤0.05 vs no doxy. B. Representative experiment showing the expression of dnPKCδ in splenic T and B cells isolated from dnPKCδ/CD2rtTA double transgenic (+/+), dnPKCδ single transgenic (+/−) or, wild type (−/−) mice. Cells were cultured and treated (+) or not (−) with 1 mg/ml doxy for 18 h, where indicated. mRNA was purified, reversed transcribed and PCR amplified by using dnPKCδ specific primers. PCR products were analyzed by electrophoresis on 0.8% agarose gel. Similar results were obtained from 4 additional mice per strain. GAPDH was used as housekeeping gene.
3.3 Selective and specific dnPKCδ expression in T cells
The expression of the transgene was directed to the T cell compartment using the human CD2 promoter. Because CD2 is also a B cell marker, we analyzed dnPKCδ expression in both T and B cells. Both lymphocyte populations were isolated from the spleens of dnPKCδ/CD2 double transgenic (+/+); dnPKCδ single transgenic (+/−); and wild-type control (−/−) mice, treated or not with doxy for 18 h. cDNA was analyzed by PCR and the products were resolved on 0.8% agarose gel. Figure 2B shows expression of dnPKCδ only in dnPKCδ (+/+) doxy-treated T cells. T cells from single transgenic (+/−) or wild type (−/−) mice, treated or not with doxy; T cells from double transgenic mice but non-doxy treated; as well as B cells regardless of mouse strain or treatment, did not express the transgene. Therefore, dnPKCδ expression is restricted to T cells and inducible by doxy.
These results strongly demonstrate that T-cell specific and tetracycline inducible expression of dnPKCδ is achieved by our model.
3.4 T cell ERK phosphorylation is decreased in dnPKCδ/rtTA mice receiving doxycycline
We tested if dnPKCδ/rtTA mice receiving doxy had decreased ERK activity. CD4+ T cells were isolated from the spleen of wild type (−/−),dnPKCδ single transgenic (+/−) and dnPKCδ /CD2rtTA double transgenic (+/+) mice receiving doxy in vivo for two weeks. Figure 3A shows a representative immunoblot comparing unstimulated and PMA-stimulated ERK phosphorylation. Reduced ERK phosphorylation is seen only in T cells from doxy-treated double transgenic mice relative to wild type (mean ± SEM = 0.63±0.13 vs 1.45±0.18) and to single transgenic controls (mean ± SEM = 1.73±0.20) following PMA stimulation (p≤0.02, n=4). A similar decrease in PMA-stimulated ERK signaling was observed in CD4+ splenic T cells from 4 dnPKCδ/CD2rtTA mice receiving doxy relative to 4 mice receiving only sucrose in vivo, and in T cells from 5 mice cultured in vitro with or without doxy for 18 h (Fig 3B). These experiments demonstrate that T cells lacking PKCδ activity have reduced ERK signaling resembling human lupus T cells [17]. We also investigated whether the status of the ERK signaling was also affected in response to T cell receptor activation. T cells from double and single transgenic mice, as well as wild type treated with doxy, were stimulated through CD3 and CD4 cell surface receptors by crosslinking with anti-rat IgG, and compared to unstimulated cells. Figure 3C shows a pattern of ERK activation similar to PMA stimulated T cells, in which ERK phosphorylation is impaired in T cells from double transgenic mice treated with doxy. However the magnitude of the activation was lower, which supports our previous observation of a lesser PKCδ activation through TCR complex compared to PMA [17].
Fig. 3.

Decreased p-ERK levels in mouse T cells expressing dnPKCδ. A. Representative experiment showing the levels of ERK phosphorylation inCD4+ T cells isolated from wild type (−/−), dnPKCδ single transgenic (+/−) or dnPKCδ / CD2rtTA double transgenic (+/+) mice that were given doxy in the drinking water. Cells were isolated from the spleen and unstimulated (−) or stimulated (+) with 50 ng/ml PMA for 15 min. Lysates were subjected to SDS-electrophoresis followed by immunoblotting with an antibody against phospho-ERK. The blot was then stripped and reprobed with anti ERK antibody as loading control. Results are representative of 4 independent experiments. B. Bar graph shown is a quantitative densitometric analysis of p-ERK and ERK in the lysates of CD4+ T cells isolated from four double transgenic mice doxy-treated (+) or not (−) for two weeks, and PMA-stimulated (in vivo). The ratio p-ERK/ERK in CD4+ T cells isolated from spleens of double transgenic mice and treated (+) or not (−) with doxyduring 18h and then PMA-stimulated for 15 min (in vitro) it is also shown. Values are the mean ± SEM of 5 independent experiments. * p≤0.05 vs non-doxy (−).C. Splenic T cells from dnPKCδ/CD2rtTA double transgenic (+/+), dnPKCδ single transgenic (+/−) or wild type (−/−) mice were treated with doxy for 18h and stimulated (+) or not (−) with anti-CD3/anti-CD4 where indicated. After stripping, the blot was reprobed with anti-total ERK antibody. p-ERK/ERK ratio corresponding to each sample is shown below. Results are representative of three independent experiments D. Protein extract from splenic T cells from doxy treated wild type animals that were stimulated (+) or not (−) with PMA; dnPKC δ (+/−) and double (+/+) transgenic mice that were doxy treated (+) or not (−) and PMA- stimulated were resolved by electrophoresis. Blot was probed with anti pPKCδ antibody, stripped and reprobed with anti-total PKCδ antibody. Data shown are representative of three experiments. pPKC δ/PKCδ ratio is shown below each sample. E. Splenic T cells from dnPKCδ/CD2rtTA double transgenic (+/+), dnPKCδ single transgenic (+/−) or wild type (−/−) mice were untreated or treated with doxy for 18h where indicated, and PMA stimulated for 15 min. Phosphorylation of PKCθ and PKCα was analyzed by immunoblotting with anti PKCθ p-Thr538 and anti-PKCα p-Thr638/641 antibodies. Beta actin was used as loading control. Results are representative of 4-5 mice per strain.
It is important to note that phosphorylation of PKCδ in PMA-stimulated T cells from double transgenic doxy-treated mice is decreased compared to those non-doxy treated (mean ± SEM=1.83 ± 0.09 vs 2.63 ± 0.17, p≤0.005, n=3). These results are similar to our previous observation in T cells transfected with dnPKCδ. On the contrary, PKCδ phosphorylation was not altered after PMA stimulation in doxy treated wild type mice nor in PMA-stimulated T cells from single transgenic mice regardless of the treatment (Fig 3D).
We tested phosphorylation of other PKC family members to exclude competition for second messengers or for others PKC-binding proteins that could affect their catalytic activity. We analyzed PKCα and PKCθ as representatives of the PMA-activated PKC isoforms. The former, belongs to the conventional calcium-dependent isoforms. PKCα is one of the most abundant isoforms in human and murine T lymphocytes [25] and a critical factor in Ag receptor signaling leading to T cell proliferation [26]. PKCθ , as well as PKCδ, is a member of the novel isoforms whose activation is calcium independent and PKCδ exhibits the highest homology to PKCθ [27]. PKCθ is selectively enriched in T cells and regulates T cell activation and survival [28]. The third group comprises the atypical PKC isoforms and was not studied because their activation is PMA-independent [29]. Splenic T cells from dnPKCδ/CD2rtTA double transgenic (+/+), dnPKCδ single transgenic (+/−) or wild type (−/−) mice were isolated and left untreated or treated with doxy for 18h where indicated, followed by PMA stimulation for 15 min. Lysates were analyzed by immunoblotting with specific anti-PKCθ phospho-Thr538 and anti-PKCα phospho-Thr638/641 antibodies, which recognize phosphorylation sites required for their catalytic activity [30] [31]. Immunoblotting showed that their phosphorylation remained unmodified in T cells from double and single transgenic mice with respect to wild type and regardless of treatment (Fig 3E). These results indicate that the transgene is not affecting the activity of other PKC isoforms.
3.5 DnPKCδ expression decreases T cell Dnmt1 levels and increases methylation sensitive gene expression
The decreased ERK pathway signaling in T cells from patients with active lupus may contribute to the development of autoimmunity by decreasing Dnmt1 levels, resulting in DNA demethylation and consequent overexpression of genes normally suppressed by methylation. To test this hypothesis, dnPKCδ/CD2rtTA double transgenic mice were given doxy/sucrose or sucrose alone in vivo for two weeks. Then, T cells from the spleen were isolated and Dnmt1, CD70, and CD11a mRNA expression were measured by RT-PCR. CD70 and CD11a were chosen because TNFSF7 (CD70) and LFA1 (CD11a) are methylation-sensitive genes in mouse and human T cells and are demethylated and overexpressed in T cells from patients with active lupus [13; 32]. Figure 4A shows an approximately 45% decrease in Dnmt1 transcripts that correlates with an increase in CD70 expression in CD3+ T cells from animals treated with doxy when compared to control animals. In a second set of experiments, T cells from dnPKCδ/CD2rtTA double transgenic mice were cultured in the presence of doxy and compared to T cells from animals expressing only the CD2rtTA transgene (−/+). After 72 h, Dnmt1 and CD70 mRNA levels were measured by real time RT-PCR. As expected, we detected decreased levels of Dnmt1 mRNA in correlation with increased CD70 expression in T cells from these double transgenic (+/+) mice relative to those expressing only the CD2-rtTA transgene (Fig. 4B). CD11a, was measured in CD3+ splenic T cells from double transgenic (+/+), single transgenic (+/−) or wild type mice, treated or not with doxy as indicated (Fig. 4C). A correlative increase is observed only in animals expressing dnPKCδ. Double transgenic mice given doxy significantly increased CD11a relative expression when compared to those non-doxy treated (mean ± SEM= 3.23 ± 0.18 vs 1.22 ± 0.15, p ≤ 0.016). These experiments clearly demonstrate that not only is the presence of both genes necessary but doxy is also required for those genes to be overexpressed and cause a phenotype similar to lupus T cells.
Fig. 4.

Decreased DNMT1 expression correlates with higher CD70 and CD11a mRNA levels in dnPKCδ transgenic T cells. A. Double transgenic animals were given doxy (doxy, n=8) or sucrose alone (no doxy, n=7) in the drinking water for two weeks. CD3+ T cells were then isolated from spleens and Dnmt1 and CD70 mRNA levels were measured relative to β-actin by RT-PCR. *p≤0.03 vs no doxy. Results represent mean ± SE of 4 independent experiments. B. CD3+ splenic T cells were isolated from four CD2rtTA single transgenic animals (−/+) or from five dnPKCδ/ CD2rtTA double transgenic animals (+/+) treated in vitro with doxy for 72 h. Dnmt1 and CD70 mRNA levels were then measured relative to β-actin by RT-PCR as in A. *p≤0.05 vs −/+. C. CD3+T cells isolated from wild type (−/−), single transgenic (+/−) or double transgenic (+/+) mice treated with doxy for two weeks were compared to non-treated animals. mRNA was purified and the expression of dnPKCδ and CD11a were analyzed by PCR amplification using specific primers. The PCR products were resolved by electrophoresis on 0.8% agarose gel. β actin was used as housekeeping gene. Data represent four independent experiments.
3.6 T cell activation in dnPKCδ expressing mice
To determine whether the overexpression of immune related genes observed in this mouse model were in correlation to T cell activation, we examined surface activation markers on murine T cells. Naïve T cells express low density of CD44 and high density of the L-selectin CD62L, which downregulates upon T cell activation while CD44 increases [33]. Splenic T cells from three double transgenic mice treated with doxy or given sucrose alone for three weeks were analyzed by flow cytometry and compared with non-treated animals. Fig 5 shows an approximate 50% increase in memory cells as measured by CD44hiCD62Llo cells in mice treated with doxy in comparison to those non-treated (mean ± SD: 31.3±1.7 vs 19.1±3.1 respectively, n=3, p≤0.001). Concomitantly, CD62Lhi cells decreased in mice that had been treated with doxy, in agreement with the increase in memory T cell subset. Although the extent of downregulation in CD62L is lower than the increase in CD44, it was significantly different between groups (p=0.035).
Fig 5.
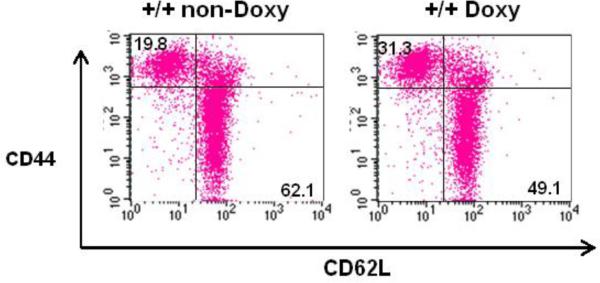
Increase in memory T cell population in mice expressing dnPKCδ. Flow cytometric analysis for CD44 and CD62L expression on CD3+ splenic cells from double transgenic mice treated or not with doxy. Percentages are shown in each quadrant. Data are representative of three independent experiments.
3.7 Inducing dnPKCδ expression in T cells causes a lupus-like autoimmunity
Since T cells from mice lacking PKCδ activity functionally resemble T cells from patients with active lupus, in which PKCδ is inactivated by oxidative damage [20], we determined if the double transgenic animals develop anti-dsDNA antibodies when given doxy. The double transgenic mice were given doxy/sucrose or sucrose alone in their drinking water, and IgG anti-dsDNA antibodies were measured serially over time by ELISA. Fig. 5 shows that mice receiving doxy, developed significantly higher anti-dsDNA levels than the controls (p=0.044 by ANOVA)
3.8 Glomerulonephritis and histopathology in double transgenic mice
Next, we determined if impaired T cell PKCδ signaling causes glomerulonephritis in this lupus model. DnPKCδ/CD2rtTA mice were given sucrose or doxy / sucrose in their drinking water and CD2rtTA single transgenic (−/+) mice were given doxy for 6 months. The mice were then sacrificed and the kidneys and lungs examined histologically. The animals receiving doxy had a diffuse glomerulonephritis with hypercellularity and perivascular leukocytic infiltration as well as increased mesangial matrix and nuclear karyorrhexis (Fig. 7 A ) when compared to those non-doxy treated (Fig 7B). Kidneys were not affected by the presence of the CD2rtTA gene even after doxy treatment (Fig. 7C). Double transgenic mice treated with doxy showed evidence of pneumonitis with perivascular leukocytic infiltration (Fig. 7D) that was not observed in those animals without doxy treatment (Fig. 7E) or in single transgenic mice (Fig. 7F).
Fig.7.

Glomerulonephritis, pneumonitis and IgG deposition in mice lacking T cell PKCδ activity. Hematoxylin and eosin staining of sections of kidneys from A. dnPKCδ/CD2rtTA double transgenic mice (+/+) treated or B. no treated with doxy or; C. CD2+rtTA single transgenic mice (−/+) treated with doxy for 6 months. A glomerulonephritis with leukocyte infiltration is seen in A. Second row: D. ungs from the same doxy treated double transgenic animals show pneumonitis with leukocyte infiltration as well E. Absence of cell infiltration in animals non-doxy treated or, F. in single transgenic mice treated with doxy. Data are representative of 5 to 7 different animals. Magnification: 400X. G. Representative immunohistochemical staining of kidneys from six dnPKCδ/rtTA double transgenic mice receiving doxy. Intense IgG deposition in all the glomeruli along capillary walls and in the mesangial region is seen. H. Similar staining of representative kidney sections from double transgenic animals receiving only sucrose (n=5) and, I. from single CD2 rtTA transgenic animals receiving doxy (n=5) were used as controls. In both controls, the blank areas are glomeruli with no immune complex deposition. Magnification: 400X.
Immunohistochemical analysis revealed the presence of immunoglobulin in the kidneys of the dnPKCδ/CD2rtTA animals receiving doxy. Figure 7G shows intense IgG deposition in a mesangial and capillary wall pattern. In contrast, double transgenic mice receiving only sucrose (Fig. 7H), or single transgenic mice given doxy (Fig. 7I), had no evidence of antibody deposition. Kidney disease was accompanied by proteinuria (30 to 100 mg/dl) in 8 out of 10 double transgenic mice receiving doxy. No protein was observed in the urine of single transgenic mice receiving doxy or double transgenic mice receiving sucrose (not shown).
4. DISCUSSION
These studies characterize a new transgenic mouse model in which controlled inhibition of T cell PKCδ induces ERK pathway signaling defects and altered gene expression resembling those reported in lupus patients. The signaling anomalies caused a serological and histological pattern consistent with human lupus, suggesting that inactivating PKCδ in T cells causes lupus-like autoimmunity.
The double transgenic mice express a dnPKCδ under the control of a CD2 promoter. Although mice express CD2 on all B cells in similar quantities to that on T cells, it was observed that CD2 has no function and does not transduce a signal in murine B cells [34]. This agrees with the absence of dnPKCδ expression in B cells we observed in our model. Also, in other mouse models, transgenes under the control of a CD2 promoter were not expressed in B cells [35], nor in CD3− cell population [13]. Thus, our double transgenic mice respond to doxycycline with transcription and expression of the dnPKCδ transgene only in T cells, providing tightly regulated inducible gene expression.
It is interesting that the decrease in ERK signaling was observed after both direct stimulation with a phorbol ester and activation of the TCR complex by crosslinking. Although in both cases the expression of the dnPKCδ was required.
Other PKC isoforms do not contribute to the defective ERK pathway signaling observed in T cells from dnPKCδ/rtTA double transgenic, the same as that observed in human lupus. This strongly suggests that a defective PKCδ is causing the decreased ERK signaling which correlates with a lower Dnmt1 gene expression. The decrease in Dnmt1 also correlated with a higher expression of methylation-sensitive genes, TNFSF7 and LFA1. TNSF1 gene encodes CD70, a B cell costimulatory molecule that contributes to antibody production. CD70 is overexpressed in experimentally demethylated T cells and in T cells from patients with lupus and it contributes to cause B cell IgG overproduction [36]. On the other hand, LFA1 encodes CD11a, which stabilizes the T cell receptor- MHC complex interaction. LFA1 overexpression decreases the threshold for TCR-self-antigen-class II MHC molecule interactions, causing autoreactivity. CD4+ T cells, by overexpressing LFA1 cause autoreactivity in vitro and in vivo by adoptive transfer into syngeneic mice [37]. On the other hand, it is interesting that the same sequences in the LFA1 [38] and TNSF7 [32; 39; 40] promoters demethylate in T cells experimentally demethylated with hydralazine or other inhibitors of DNMT expression; in T cells from lupus patients; and in T cells from MRLlpr lupus-prone mice as they develop autoimmunity. In this mouse model, the overexpression of methylation sensitive genes caused T cell activation as was observed by the increase in memory T cells. The lower extent of CD62L downregulation could be due to the retained expression of CD62L by activated cells as was observed in other models [41] There is also a possibility that some memory cells persist as naïve cells, without losing their naïve phenotype [42].
SJL mice contain lupus susceptibility genes and they developed a glomerulonephritis and inflammatory lung disease that is similar to human lupus. However, the SJL strain is a low IgG Ab responder [43], what may explain the antibody titers we observed in this mouse model. These results indicate a relevant role for the PKCδ - ERK signaling pathway in causing autoimmunity through the regulation of DNA methylation and gene expression.
PKCδ plays a critical negative role in cellular function by inhibiting proliferation and promoting cell death [44]. A greater proliferation was observed in PKCδ-deficient B cells from PKCδ knockout mice [45]. Further, PKCδ-deficient T cells have a reduced threshold for activation by cell bound allogenic MHC molecules in vitro, suggesting that the PKCδ signaling pathway is necessary for T cell attenuation [46] and is considered a T cell negative regulator [47]. A similar response was observed in demethylated T cells that became autoreactive [3]. All these observations are consistent with this mouse model, which developed a lupus-like disease due to the lack of PKCδ activity in T cells, and correlate with our findings in T cells from patients with active lupus [17], a disease characterized by exaggerated cellular and humoral immune responses. Recently, a rare isolated case of familial lupus was described in three siblings with juvenile SLE due to a missense mutation in PRKCD (PKCδ) [48], which also points to PKCδ as a key molecule in the development of lupus.
The term exposome refers to both environmental factors exogenous and endogenous to which organisms are exposed in a lifetime [49; 50]. Thus, the exposome interacts with the genome in the development of autoimmune diseases by influencing epigenetic profiles. Although the molecular mechanisms underlying the etiology of lupus are not yet fully understood, it is nevertheless clear that the production of reactive intermediates and their oxidative products play an important role in initiating and exacerbating the lupus phenotype [51; 52]. We observed an impaired PKCδ activity in T cells from patients with active lupus due to oxidation causing post-translational modifications that prevents phosphorylation of PKCδ at the activation loop [20], which is required for PKCδ activation. Therefore, the decreased ERK pathway signaling observed in lupus T cells causing epigenetic changes may be explained by the deficient PKCδ activity, which is an upstream regulator of ERK in CD4+ T cells [17].
4.1 Conclusions
This new mouse model demonstrates that the environmentally-induced PKCδ inhibition in T cells that was observed in patients with active lupus, may cause a lupus-like disease in genetically predisposed organisms.
Highlights.
Impairment in T cell PKCδ in this mouse model is causing deficient ERK signaling, as observed in SLE.
Decreased T cell PKCδ signaling results in DNA demethylation, overexpression of TNSF7 (CD70) and LFA1 (CD11a) genes, and autoimmunity.
Abnormalities in T cell PKCδ deficient transgenic mice resemble human active lupus
Decreased ERK signaling due to PKCδ inactivation may have a causative role in human lupus
Fig.6.

Serum ds-DNA autoantibody production when T cell PKCδ activity is decreased. Blood was obtained from C57BL/6 × SJL mice carrying the mutant dnPKCδ and CD2rtTA genes and given 2 mg/ml doxy/5% sucrose (doxy) or 5% sucrose alone (no doxy) in their drinking water during the indicated times. Serum concentrations of IgG anti ds-DNA were determined by ELISA. Values are the mean ± SEM of 6 independent experiments. Comparison between both groups by ANOVA regression. Results are the mean ± SEM of 4-6 mice per point; p=0.044.
ACKNOWLEDGEMENTS
We are very thankful to Dr. S.H.Yuspa for his generous gift of the PKCδK376R mutant and to Dr. Rose Zamoyska for providing the CD2-rtTA mice. We are also thankful to Maggie Van Keuren at the University of Michigan Transgenic Core for generating the TRE2-dnPKCδ transgenic mouse and to Dr. Thomas Saunders for his helpful guidance. We are thankful to Dr. Faith Strickland for her help with the histology sample preparations.
The transgenic Core support was provided by the University of Michigan Multipurpose Arthritis Center, NIH grant AR20557, and the Nathan Shock Center, NIH grant P30AG013283. This work was supported by NIH grants AR4525, AG25877, ES015214 and a Merit grant from the Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Sawalha AH, Wang L, Nadig A, Somers EC, McCune WJ, Hughes T, Merrill JT, Scofield RH, Strickland FM, Richardson B. Sex-specific differences in the relationship between genetic susceptibility, T cell DNA demethylation and lupus flare severity. J Autoimmun. 2012;38:J216–22. doi: 10.1016/j.jaut.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun. 2009;33:3–11. doi: 10.1016/j.jaut.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson B. Effect of an inhibitor of DNA methylation on T cells. II. 5-Azacytidine induces self-reactivity in antigen-specific T4+ cells. Hum Immunol. 1986;17:456–70. doi: 10.1016/0198-8859(86)90304-6. [DOI] [PubMed] [Google Scholar]
- 4.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33:1665–73. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 5.Yung RL, Richardson BC. Drug-induced lupus. Rheum Dis Clin North Am. 1994;20:61–86. [PubMed] [Google Scholar]
- 6.Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988;140:2197–200. [PubMed] [Google Scholar]
- 7.Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, Richardson BC. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest. 1993;92:38–53. doi: 10.1172/JCI116576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, Richardson BC. Procainamide inhibits DNA methyltransferase in a human T cell line. J Rheumatol. 1991;18:530–4. [PubMed] [Google Scholar]
- 9.Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem. 2005;280:40749–56. doi: 10.1074/jbc.M505593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, Richardson B. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003;48:746–56. doi: 10.1002/art.10833. [DOI] [PubMed] [Google Scholar]
- 11.Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, Hanash SM, Richardson BC. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 12.Gorelik G, Richardson B. Key role of ERK pathway signaling in lupus. Autoimmunity. 2010;43:17–22. doi: 10.3109/08916930903374832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, Osban J, Knowlton N, Johnson K, Richardson B. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–78. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strickland FM, Hewagama A, Lu Q, Wu A, Hinderer R, Webb R, Johnson K, Sawalha AH, Delaney C, Yung R, Richardson BC. Environmental exposure, estrogen and two X chromosomes are required for disease development in an epigenetic model of lupus. J Autoimmun. 2012;38:J135–43. doi: 10.1016/j.jaut.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sestak AL, Nath SK, Sawalha AH, Harley JB. Current status of lupus genetics. Arthritis Res Ther. 2007;9:210. doi: 10.1186/ar2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boackle SA. Advances in lupus genetics. Curr Opin Rheumatol. 2013;25:561–8. doi: 10.1097/BOR.0b013e328363eb4e. [DOI] [PubMed] [Google Scholar]
- 17.Gorelik G, Fang JY, Wu A, Sawalha AH, Richardson B. Impaired T cell protein kinase C delta activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J Immunol. 2007;179:5553–63. doi: 10.4049/jimmunol.179.8.5553. [DOI] [PubMed] [Google Scholar]
- 18.Oates JC. The biology of reactive intermediates in systemic lupus erythematosus. Autoimmunity. 2010;43:56–63. doi: 10.3109/08916930903374683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF. Markers of oxidative and nitrosative stress in systemic lupus erythematosus: Correlation with disease activity. Arthritis Rheum. 2010;62:2064–2072. doi: 10.1002/art.27442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorelik GJ, Yarlagadda S, Richardson BC. Protein kinase Cδ oxidation contributes to ERK inactivation in lupus T cells. Arthritis and rheumatism. 2012;64:2964–74. doi: 10.1002/art.34503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol Cell Biol. 1999;19:8547–58. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Legname G, Seddon B, Lovatt M, Tomlinson P, Sarner N, Tolaini M, Williams K, Norton T, Kioussis D, Zamoyska R. Inducible Expression of a p56Lck Transgene Reveals a Central Role for Lck in the Differentiation of CD4 SP Thymocytes. Immunity. 2000;12:537–546. doi: 10.1016/s1074-7613(00)80205-8. [DOI] [PubMed] [Google Scholar]
- 23.Garcia GG, Miller RA. Increased Zap-70 Association with CD3ζ in CD4 T Cells from Old Mice. Cellular Immunology. 1998;190:91–100. doi: 10.1006/cimm.1998.1394. [DOI] [PubMed] [Google Scholar]
- 24.Lu Q, Wu A, Ray D, Deng C, Attwood J, Hanash S, Pipkin M, Lichtenheld M, Richardson B. DNA Methylation and Chromatin Structure Regulate T Cell Perforin Gene Expression. The Journal of Immunology. 2003;170:5124–5132. doi: 10.4049/jimmunol.170.10.5124. [DOI] [PubMed] [Google Scholar]
- 25.Gorelik G, Barreiro Arcos ML, Klecha AJ, Cremaschi GA. Differential expression of protein kinase C isoenzymes related to high nitric oxide synthase activity in a T lymphoma cell line. Biochimica et biophysica acta. 2002;1588:179–88. doi: 10.1016/s0925-4439(02)00163-1. [DOI] [PubMed] [Google Scholar]
- 26.Pfeifhofer C, Gruber T, Letschka T, Thuille N, Lutz-Nicoladoni C, Hermann-Kleiter N, Braun U, Leitges M, Baier G. Defective IgG2a/2b class switching in PKC alpha−/− mice. J Immunol. 2006;176:6004–11. doi: 10.4049/jimmunol.176.10.6004. [DOI] [PubMed] [Google Scholar]
- 27.Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C delta (PKC delta): activation mechanisms and functions. J Biochem. 2002;132:831–9. doi: 10.1093/oxfordjournals.jbchem.a003294. [DOI] [PubMed] [Google Scholar]
- 28.Isakov N, Altman A. Protein kinase C(theta) in T cell activation. Annu Rev Immunol. 2002;20:761–94. doi: 10.1146/annurev.immunol.20.100301.064807. [DOI] [PubMed] [Google Scholar]
- 29.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–96. [PubMed] [Google Scholar]
- 30.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Graham C, Li A, Fisher RJ, Shaw S. Phosphorylation of the protein kinase C-theta activation loop and hydrophobic motif regulates its kinase activity, but only activation loop phosphorylation is critical to in vivo nuclear-factor-kappaB induction. Biochem J. 2002;361:255–65. doi: 10.1042/bj3610255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Q, Wu A, Richardson BC. Demethylation of the Same Promoter Sequence Increases CD70 Expression in Lupus T Cells and T Cells Treated with Lupus-Inducing Drugs. J Immunol. 2005;174:6212–6219. doi: 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- 33.Sprent J. Lifespans of naive, memory and effector lymphocytes. Curr Opin Immunol. 1993;5:433–8. doi: 10.1016/0952-7915(93)90065-z. [DOI] [PubMed] [Google Scholar]
- 34.Keogh MC, Elliot J, Norton T, Lake RA. A function for CD2 on murine B cells? Immunol Cell Biol. 1997;75:333–9. doi: 10.1038/icb.1997.51. [DOI] [PubMed] [Google Scholar]
- 35.Parsons MJ, Jones RG, Tsao MS, Odermatt B, Ohashi PS, Woodgett JR. Expression of active protein kinase B in T cells perturbs both T and B cell homeostasis and promotes inflammation. J Immunol. 2001;167:42–8. doi: 10.4049/jimmunol.167.1.42. [DOI] [PubMed] [Google Scholar]
- 36.Oelke K, Lu Q, Richardson D, Wu A, Deng C, Hanash S, Richardson B. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum. 2004;50:1850–60. doi: 10.1002/art.20255. [DOI] [PubMed] [Google Scholar]
- 37.Yung R, Powers D, Johnson K, Amento E, Carr D, Laing T, Yang J, Chang S, Hemati N, Richardson B. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866–71. doi: 10.1172/JCI118743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Q, Kaplan M, Ray D, Zacharek S, Gutsch D, Richardson B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002;46:1282–91. doi: 10.1002/art.10234. [DOI] [PubMed] [Google Scholar]
- 39.Kaplan MJ, Lu Q, Wu A, Attwood J, Richardson B. Demethylation of promoter regulatory elements contributes to perforin overexpression in CD4+ lupus T cells. J Immunol. 2004;172:3652–61. doi: 10.4049/jimmunol.172.6.3652. [DOI] [PubMed] [Google Scholar]
- 40.Sawalha AH, Jeffries M. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupus-prone mice. European Journal of Immunology. 2007;37:1407–1413. doi: 10.1002/eji.200636872. [DOI] [PubMed] [Google Scholar]
- 41.Sprent J. Immunological memory. Curr Opin Immunol. 1997;9:371–9. doi: 10.1016/s0952-7915(97)80084-2. [DOI] [PubMed] [Google Scholar]
- 42.Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–35. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, Longmate JA, Adamus G, Hargrave PA, Wakeland EK. Interval mapping of quantitative trait loci controlling humoral immunity to exogenous antigens: evidence that non-MHC immune response genes may also influence susceptibility to autoimmunity. J Immunol. 1996;157:2498–505. [PubMed] [Google Scholar]
- 44.Gschwendt M. Protein kinase C delta. Eur J Biochem. 1999;259:555–64. doi: 10.1046/j.1432-1327.1999.00120.x. [DOI] [PubMed] [Google Scholar]
- 45.Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, Tsukiyama T, Nagahama H, Ohno S, Hatakeyama S, Nakayama KI. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–9. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 46.Gruber T, Barsig J, Pfeifhofer C, Ghaffari-Tabrizi N, Tinhofer I, Leitges M, Baier G. PKCdelta is involved in signal attenuation in CD3+ T cells. Immunol Lett. 2005;96:291–3. doi: 10.1016/j.imlet.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Black AR, Black JD. Protein kinase C signaling and cell cycle regulation. Front Immunol. 2012;3:423. doi: 10.3389/fimmu.2012.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, Szynkiewicz M, Zabot MT, Rouvet I, Bhaskar SS, Daly SB, Dickerson JE, Mayer J, O'Sullivan J, Juillard L, Urquhart JE, Fawdar S, Marusiak AA, Stephenson N, Waszkowycz B, W.B. M, Biesecker LG, C.M.B. G, Rene C, Eliaou JF, Fabien N, Ranchin B, Cochat P, Gaffney PM, Rozenberg F, Lebon P, Malcus C, Crow YJ, Brognard J, Bonnefoy N. Protein kinase cdelta deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis Rheum. 2013;65:2161–71. doi: 10.1002/art.38008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wild CP. The exposome: from concept to utility. Int J Epidemiol. 2012;41:24–32. doi: 10.1093/ije/dyr236. [DOI] [PubMed] [Google Scholar]
- 50.Bogdanos DP, Smyk DS, Invernizzi P, Rigopoulou EI, Blank M, Pouria S, Shoenfeld Y. Infectome: a platform to trace infectious triggers of autoimmunity. Autoimmunity reviews. 2013;12:726–40. doi: 10.1016/j.autrev.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oates JC, Gilkeson GS. The biology of nitric oxide and other reactive intermediates in systemic lupus erythematosus. Clinical Immunology. 2006;121:243–250. doi: 10.1016/j.clim.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013;9:674–86. doi: 10.1038/nrrheum.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]