

SUMMARY
Transforming growth factor-β (TGFβ) regulates the expression of genes supporting breast cancer cell in bone but little is known about prostate cancer bone metastases and TGFβ. Our study reveals that the TGFBR1 inhibitor SD208 effectively reduces prostate cancer bone metastases. TGFβ upregulates in prostate cancer cells a set of genes associated with cancer aggressiveness and bone metastases, and the most upregulated gene was PMEPA1. In patients, PMEPA1 expression decreased in metastatic prostate cancer and low Pmepa1 correlated with decreased metastasis-free survival. Only membrane-anchored isoforms of PMEPA1 interacted with R-SMADs and ubiquitin ligases, blocking TGFβ signaling independently of the proteasome. Interrupting this negative feedback loop by PMEPA1 knockdown increased prometastatic gene expression and bone metastases in a mouse prostate cancer model.
INTRODUCTION
Prostate cancer (PCa) is one of the most frequently diagnosed cancers in men and can be an indolent malignancy. However in advanced disease, most PCa patients will have bone metastases that are associated with hypercalcaemia, intractable pain, fracture or nerve compression syndrome, causing significant morbidity. Despite of current FDA-approved treatments, bone metastases from PCa remain incurable and further research is needed to understand the mechanisms of bone metastases. Recent evidence indicates that TGFβ supports the development of PCa bone metastases (Hu et al., 2012; Wan et al., 2012).
TGFβ plays a crucial role in regulating cell proliferation and differentiation. The TGFβ signaling pathway controls many normal physiological processes, including embryogenesis, immune responses and bone remodeling (Mohammad et al., 2009). TGFβ has a complex and sometimes paradoxical role in cancer: in early-stage it inhibits cell growth and is a tumor suppressor, while in later stages TGFβ promotes invasion and metastasis, in particular metastasis to bone (Pickup et al., 2013). Cancer cells in bone disrupt the bone remodeling process by altering bone resorption and bone formation. In turn the bone microenvironment supports cancer cell growth and survival via osteoblast-produced growth factors embedded in the mineral bone matrix and released during osteoclastic bone resorption (Weilbaecher et al., 2011). TGFβ is one of the most abundant growth factors in bone and is released during osteoclastic bone resorption. TGFβ signaling is activated in bone metastases samples from breast cancer (BCa) patients (Kang et al., 2005) and preclinical models have confirmed that bone resorption increases TGFβ signaling in BCa cells in bone (Korpal et al., 2009).
The canonical TGFβ signaling pathway uses receptor-activated SMAD (R-SMAD) proteins 2 and 3, which form complexes in the nucleus with DNA-binding co-factors such as SP1 and with transcriptional coactivators or corepressors to regulate gene expression. TGFβ signaling can be turned off by inhibitors such as HECT type E3 ubiquitin ligases including NEDD4, AIP4 and SMURF2, in a proteasome dependent or independent manner (Zhang et al., 2001; Lallemand et al., 2005; Tang et al., 2011). In BCa bone metastases, TGFβ controls the expression of multiple genes including CXCR4, MMP1, IL11, JAG1 or PTHRP that promote bone metastases (Yin et al., 1999; Kang et al., 2003; Kang et al., 2005; Sethi et al., 2011). In BCa and melanoma models, TGFβ signaling is essential for the formation of bone metastases (Yin et al., 1999; Javelaud et al., 2007), and it has been well established that anti-TGFβ therapies significantly reduce the development and progression of the associated bone metastases in mice (Juarez and Guise, 2010). Recent studies showed that inhibition of TGFβ signaling in PCa cells inhibit the development of bone metastases (Hu et al., 2012; Wan et al., 2012) but the underlying molecular mechanisms involved have not been determined. Characterizing the role of TGFβ-regulated genes associated with PCa bone metastases could identify therapeutic targets and diagnostic markers. Therefore in this study we sought to characterize the role of TGFβ signaling and TGFβ-regulated genes on the development of bone metastases of PCa.
RESULTS
The TGFBR1 inhibitor SD208 Inhibits Osteolytic Bone Metastases from PCa Cells in Mice
SD208 is a small molecule inhibitor of the kinase activity of TGFBR1 (EC50 = 48nM) (Uhl et al., 2004), we tested its efficacy on the human PCa cells, PC-3, in vitro and in vivo in a murine model of bone metastasis. In vitro treatment of PC-3 cells, with SD208 abrogated SMAD2 phosphorylation induced by TGFβ (Fig 1A). TGFβ also increased the expression of bone-metastatic genes PTHRP, IL11 and CTGF, in PC-3 cells while SD208 prevented the increase of all 3 mRNAs (Fig 1B). The effects of SD208 were not cytotoxic as assessed by MTT assay of proliferation of PC-3 cells in the presence or absence of TGFβ (Fig 1C).
Figure 1. SD208 inhibits TGFβ signaling in PC-3 cells in vitro and reduces the development of bone metastases from PC-3 in mice.
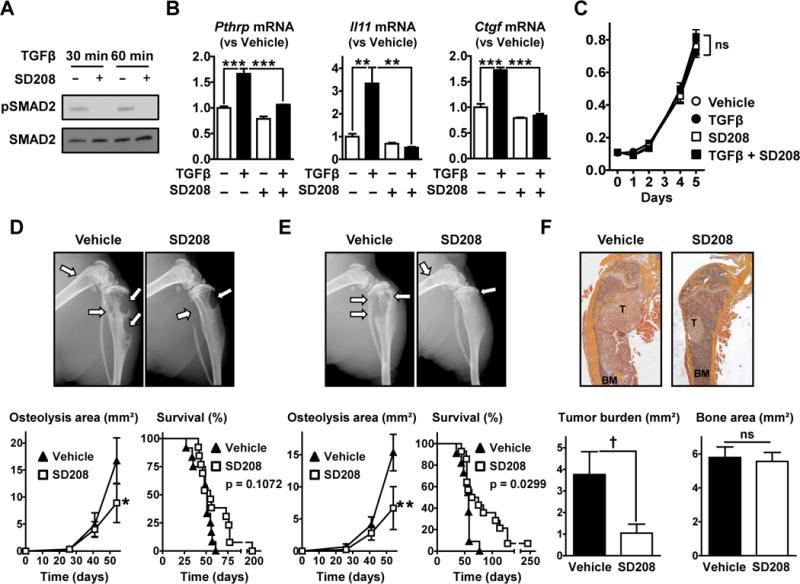
A. Western blot analysis of SMAD2 phosphorylation in PC-3 cells pre-treated ±SD208 (1 μM, 1 hr) and further cultured with TGFβ (5 ng/mL, 30–60 min) before lysis. B. Pthrp, Il11 and Ctgf mRNA measured by RT-qPCR in PC-3 cells cultured ±TGFβ (5 ng/mL) and ±SD208 (1 μM) for 24hr (n = 3). C. Growth of PC-3 cells cultured in the presence or absence of TGFβ (5 ng/mL) and SD208 (1 μM) assessed by MTT assay (n = 6). D,E&F. Nude mice inoculated with PC-3 cells received SD208 (50 mg/kg/day) or its vehicle (D) in a therapeutic manner (starting on day 26) or (E&F) in a preventive manner (starting 3 days before cell inoculation) (n = 11 to 14 mice per group). D&E. (Upper) Representative radiographs (arrows indicate osteolytic lesions). (Lower) Quantification of the osteolysis area on radiographs and Kaplan-Meyer analysis of mouse survival with a log-rank (Mantel-Cox) test. F. (Upper) Representative H&E stained sections of femurs (T, tumor; BM, bone marrow) and (Lower) histomorphometric analysis of tumor burden and bone areas. Results are expressed as the mean ± SEM. ns, not significant, * p < 0.05, ** p < 0.01 and *** p < 0.001 using a 2-way ANOVA with Bonferroni’s posttest. † p < 0.05 using a Student’s t test.
To test the efficacy of SD208 in vivo, we used a mouse model of bone metastases in which PC-3 cells are inoculated into the left cardiac ventricle of male nude mice. First, we tested the efficacy of a treatment regimen with SD208 (50 mg/kg/day) being administered starting on day 26 when osteolysis was first detected by x-ray. After 4 weeks of treatment, SD208 significantly decreased area of osteolysis measured on radiographs by 47% when compared to vehicle-treated mice (8.9±3.6 vs 16.7±4.3 mm2, respectively) (Fig 1D). However SD208 did not improve mouse survival when compared to control (55 vs 51 days of median survival, respectively) (Fig 1D).
In a prevention protocol where treatment began 3 days prior to tumor inoculation, SD208 (50 mg/kg/day) significantly decreased osteolysis area by 56% when compared to vehicle-treated mice (6.7±3.3 vs 15.3±2.8 mm2, respectively), after 54 days of treatment (Fig 1E). Furthermore, SD208 significantly increased mouse survival compared to vehicle treated mice (57 vs 69 days of median survival for vehicle- and SD208-treated mice) (Fig 1E). Histomorphometric analysis confirmed that SD208 significantly decreased the tumor burden in bones (Fig 1F). There were no differences in total bone area when mice received SD208 (Fig 1F), which may be due to the fact that the bones were harvested at different times since we were measuring mouse survival. Overall our data show that the TGFβ signaling inhibitor SD208 significantly decreased the progression of PC-3 osteolytic metastases.
Identification of TGFβ Target Genes in PCa Cells
To identify genes regulated by TGFβ in PCa cells, we compared PC-3 cells cultured with or without TGFβ using Affymetrix gene chips HU-133A, which can detect 18,100 different transcripts of 14,500 genes. 271 different genes were significantly upregulated (>1.5 fold increase, p < 0.05) and 67 downregulated (<−1.5 fold decrease, p < 0.05), representing 2.33% of the genes analyzed (Tables 1, S1 & S2). The results of this microarray were validated by measuring mRNA levels of selected genes using RT-qPCR to confirm that TGFβ increases the expression of Pmepa1, Pthrp, Ctgf, Mmp13, Adam19, Thbs1, Nedd9, Dkk1, Col1a1, Vegfa mRNAs (Fig S1 & 2A). A survey of the current literature indicated that many of the upregulated genes have been linked to cancer and metastases (Table 1). For example, Nedd9, Mmp13, Upa increase cell invasion; Itgav increases the homing and adhesion of cancer cells to bone; Vegfa, Ctgf and Mmp13 support angiogenesis and Pthrp, Adam19 and Ctgf increase osteolysis (Table 1). Overall this microarray shows that TGFβ increases the expression of genes in PCa cells that regulate multiple steps of the metastatic cascade to bone.
Table 1.
Representative genes upregulated by TGFβ in PC-3 PCa cells and associated with cancer and bone biology (selected from supplementary Tables S1 & S2). See also Figure S1.
| Gene symbol | Gene name | Fold change | P value | Association with cancer | Reference |
|---|---|---|---|---|---|
| PMEPA1 | Prostate transmembrane protein, androgen induced 1 | 23.15 | 0.029 | Inhibits TGFβ and AR signaling | (Li et al., 2008; Watanabe et al., 2010) |
| IGFBP5 | Insulin-like growth factor binding protein-5 | 11.52 | 0.009 | Accelerate progression to androgen independence | (Miyake et al., 2000) |
| COL1A1 | Collagen type 1 α1 | 4.05 | 0.038 | Most abundant protein in the bone matrix & expressed by cancer cells homing to bone | (Koeneman et al., 1999) |
| NEDD9 | Neural precursor cell expressed, developmentally down-regulated 9, HEF1 | 4.04 | 0.045 | Promotes invasion and epithelial-mesenchymal transition | (Morimoto et al., 2014) |
| THBS1 | Thrombospondin 1 | 3.81 | 0.015 | Activator of TGFβ | (Crawford et al., 1998) |
| MMP13 | Matrix metallopeptidase 13 (collagenase 3) | 3.49 | 0.034 | Support cancer cell invasion and tumor angiogenesis & increases bone metastases | (Kudo et al., 2012; Shah et al., 2012) |
| PTHRP | Parathyroid hormone-related protein | 3.31 | 0.040 | Increases RANKL/OPG ratio & bone metastases | (Yin et al., 1999) |
| ID1 | Inhibitor of differentiation 1, inhibitor of DNA binding 1 | 3.09 | 0.039 | Increases prostate cell survival & proliferation | (Schmidt et al., 2010; Stankic et al., 2013) |
| CTGF | Connective tissue growth factor, CCN2 | 2.93 | 0.032 | Increases angiogenesis, osteoclastogenesis & bone metastases | (Kang et al., 2003; Shimo et al., 2006) |
| ADAM19 | ADAM metallopeptidase domain 19 | 2.73 | 0.043 | Solubilizes pro-osteoclastic RANKL | (Chesneau et al., 2003) |
| ITGAV | Integrin αV (vitronectin receptor) | 2.72 | 0.022 | Increases homing of cancer cells to bone | (Pécheur et al., 2002) |
| VEGFA | Vascular endothelial growth factor A | 2.29 | 0.032 | Increases angiogenesis | (Roberts et al., 2013) |
| UPA | Urokinase-type plasminogen activator | 1.81 | 0.030 | Pro-invasive protease & tumor marker | (Crippa, 2007) |
| DKK1 | Dickkopf 1 homolog | 1.52 | 0.0001 | Inhibits bone formation & increases osteolytic metastases | (Clines et al., 2007; Hall et al., 2010) |
The most highly upregulated gene, Pmepa1 (prostate transmembrane protein androgen induced-1) was increased 23.2-fold by TGFβ in the microarray (Table 1 & S1) and RT-qPCR confirmed that TGFβ quickly and durably increased Pmepa1 mRNA in PC-3 cells (Fig 2A). The PMEPA1 gene encodes 3 protein isoforms: a and b contain a type I transmembrane domain at the N-terminus and localize in the Golgi apparatus while isoform c lacks this transmembrane domain and is cytosolic (Xu et al., 2003) (Fig 2B). To determine which isoforms are expressed in cancer cells, we cloned the different PMEPA1 isoforms with a V5 epitope tag at the C-terminus. Each isoform was expressed in COS-7 cells and lysates analyzed in parallel with different cancer cell lines. In absence of TGFβ, only A549 lung cancer cells express detectable quantities of PMEPA1c (Fig 2C). In the presence of TGFβ, all prostate (PC-3 and DU145), breast (MDA-MB-231) and lung (A549) cancer cells tested expressed both membrane-bound PMEPA1a and cytosolic PMEPA1c (Fig 2C). The isoform b of PMEPA1 was not detected. Despite activation of TGFβ signaling as shown by phosphorylated Smad3, HepG2 cells did not express detectable amount of any PMEPA1 isoform (Fig 2C). DNA methylation can prevent induction of PMEPA1 gene by androgens (Sharad et al., 2014). Thus we cultured HepG2 with the methyltransferase inhibitors 5-azacytidine or 5-aza-2-deoxycytidine which restored expression of PMEPA1a and c in the presence of TGFβ (Fig 2D). The results indicate that TGFβ induces the expression of PMEPA1 a and c in multiple cancer cell lines and that PMEPA1 expression can be epigenetically regulated by methylation.
Figure 2. TGFβ increases the expression of membrane-bound and cytosolic PMEPA1 in cancer cells.
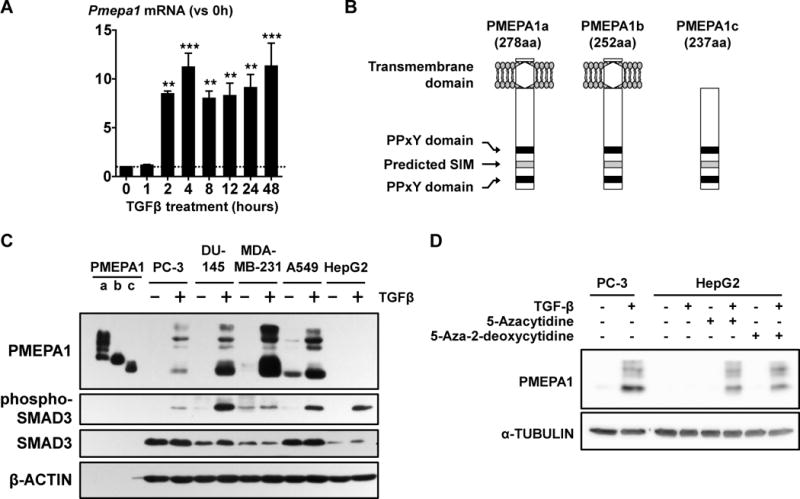
A. Levels of Pmepa1 mRNA assessed in PC-3 cells treated with TGFβ (5ng/mL). ** p < 0.01 and *** p < 0.001 using a 1-way ANOVA with Bonferroni’s posttest. B. Schematization of PMEPA1 isoforms. C. Western blot analysis of PMEPA1 expression and SMAD3 phosphorylation in prostate (PC-3, DU-145), breast (MDA-MB-231) and lung (A549) cancer cells and hepatocarcinoma cells (HepG2) treated ±TGFβ (5 ng/mL, 24 hr). Lysates of COS-7 cells transfected to express specific isoforms of PMEPA1 protein were used as standard. D. PMEPA1 expression in HepG2 cells treated ±TGFβ (5 ng/mL, 24 hr) after culture ±5-azacytidine or 5-aza-2-deoxycytidine (10 μM, 12 days).
Low Expression of Pmepa1 Is Associated with Metastases and Decreased Survival
The role of PMEPA1 in PCa and bone metastases has not been determined. To investigate the clinical significance of PMEPA1 in cancer, we compared PMEPA1 expression between normal tissue and primary tumors using Oncomine. PMEPA1 expression was significantly increased in the primary tumor of patients with PCa in multiple independent studies (Fig 3A & S2A). Similarly PMEPA1 expression was increased in the primary tumor of breast and lung cancer patients when compared to normal tissue (Fig 3B, 3C, S2B & S2C). We used the Yu dataset to compare PMEPA1 expression at different stages of PCa progression (Chandran et al., 2007). When compared to prostate tissue from healthy donors, Pmepa1 mRNA was significantly increased in the primary tumor as well as in normal tissue adjacent to the tumor that can contain higher levels of TGFβ (Fig 3D). However, in distant non-osseous metastases, PMEPA1 expression was unchanged when compared to normal prostate and thus was significantly decreased compared to primary tumors (Fig 3D). To compare PMEPA1 expression between different metastatic sites, we used the Zhang datasets of BCa metastases (Zhang et al., 2009). PMEPA1 expression tended to be higher in bone metastases compare to all other sites of metastases and was significantly higher than in brain and lung metastases in datasets GSE14017 and GSE14018, respectively (Fig 3E).
Figure 3. Low expression of PMEPA1 is associated to with poor prognosis of cancer patients.
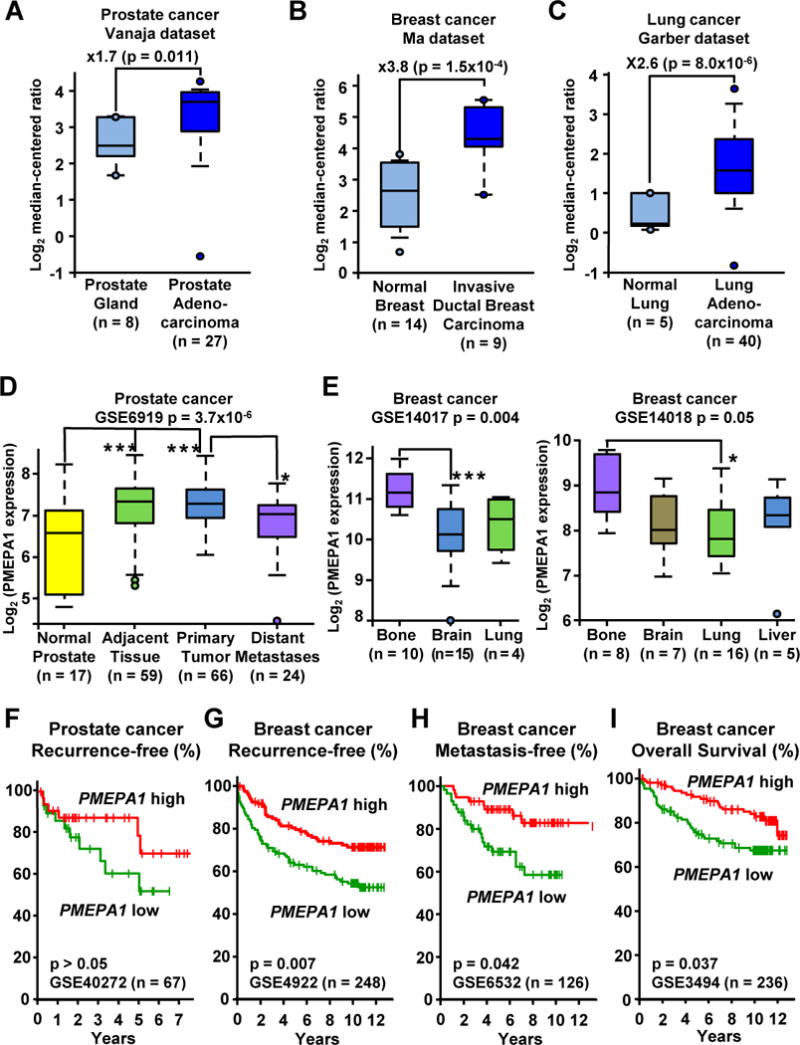
A,B&C. PMEPA1 expression levels in normal tissue and primary tumor of prostate (A), breast (B) and lung (C) cancer patients (Garber et al., 2001; Vanaja et al., 2003; Ma et al., 2009). Expression levels are presented as box-plots and were compared using unpaired Student’s t test. D. PMEPA1 expression in normal prostate and in samples from PCa patients (GSE6919 dataset). E. PMEPA1 expression in bone, brain, lung and liver metastases of BCa patients (GSE14017 and GSE14018 datasets). F,G,H&I. Kaplan-Meyer analysis of (F) recurrence-free survival in the Gulzar dataset (GSE40272), (G) Recurrence-free survival in the Loi dataset (GSE4922), (H) Metastasis-free survival in the Ivshina dataset (GSE6532) and (I) Overall survival in the Miller dataset (GSE3494) based on PMEPA1 expression in the primary tumor of prostate or BCa patients. Survival analysis was performed using log rank test. * p < 0.05 and *** p < 0.005 using a 1-way ANOVA with Tukey’s posttest. See also Figure S2.
We used the PROGgene database to determine the prognostic value of PMEPA1 expression in primary tumors (Goswami and Nakshatri, 2013). The Gulzar dataset (Gulzar et al., 2013) suggests that PCa recurrence is higher in patients with low PMEPA1 expression, compared to those with high PMEPA1 (Fig 3F). Despite the separation between the populations after 2 years, the difference was not significant, which could be due to the small number of patients (n = 67). In the absence of other available PCa cohorts, we analyzed PMEPA1 in BCa cohorts. The Loi dataset revealed that BCa patients with lower Pmepa1 mRNA had a significantly earlier recurrence (Fig 3G) (Loi et al., 2007). Lower expression of PMEPA1 was also significantly associated with decreased time to metastases and decreased survival for BCa patients in the Ivishna and Miller datasets, respectively (Fig 3H & 3I) (Miller et al., 2005; Ivshina et al., 2006). These findings show clinical significance for PMEPA1 in cancer and validate the need to further understand the regulation of PMEPA1 expression and its function.
TGFβ Activates PMEPA1 Promoter and Increases PMEPA1 Transcription
To characterize how TGFβ regulates PMEPA1 expression, we measured Pmepa1 mRNA using RT-qPCR. The transcription inhibitor, actinomycin D, abrogated TGFβ-induction of Pmepa1, while the translation inhibitor, cycloheximide, did not prevent the Pmepa1 increase suggesting that TGFβ increases PMEPA1 transcription without requiring de novo protein synthesis (Fig 4A). The TGFBR1 inhibitor SD208 completely prevented Pmepa1 induction by TGFβ, while inhibitors of p38, JNK or MEK kinase had no effect (Fig 4A & 4B). This suggests that the non-canonical TGFβ signaling pathway does not regulate PMEPA1 transcription which is likely to be regulated by the SMAD canonical pathway.
Figure 4. TGFβ increases the transcription of PMEPA1 and activates a 1.5 kb fragment of PMEPA1 promoter independently of the Smad Binding Elements.
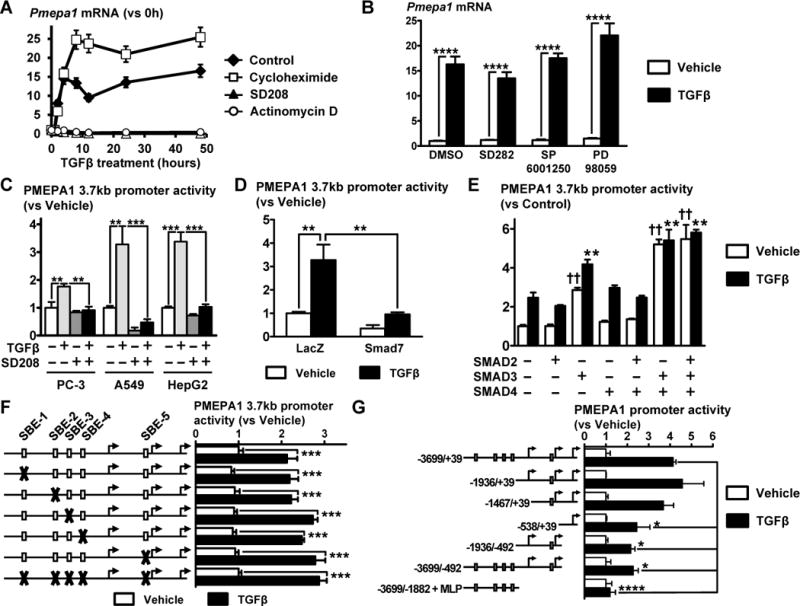
A. Levels of Pmepa1 mRNA assessed in PC-3 cells treated with TGFβ (5 ng/mL) in the presence or absence of SD208 (1 μM), cycloheximide (1 μM) or actinomycin D (1 μg/mL) using RT-qPCR (n = 3). B. Measure of Pmepa1 mRNA in PC-3 cells treated ±TGFβ (5 ng/mL, 24 hr) in the presence or absence of kinase inhibitors specific for p38 (SD282, 1 μM), JNK (SP6001250, 5 μM) or MEK (PD98059, 25 μM) using RT-qPCR (n = 3). C. Transfection of PC-3, A549 and HepG2 cells with a pGL3 plasmid containing a 3.7 kb fragment (−3699/+39) of PMEPA1 promoter. Cells were then treated ±TGFβ (5 ng/mL) and ±SD208 (1 μM) for 24 hr before measuring dual-luciferase activity normalized using renilla luciferase (n = 4). D. Ectopic expression of SMAD7 in A549 cells transfected with pGL3-hPMEPA1(−3699/+39) and treated ±TGFβ (5 ng/mL, 24 hr) before measuring dual-luciferase activity (n = 4). E. Ectopic expression of SMAD2, 3 or 4 in A549 cells transfected with pGL3-hPMEPA1(−3699/+39) and treated ±TGFβ (5 ng/mL, 24 hr) before measuring dual-luciferase activity (n = 4). F. Effect of SBE mutations (represented by X) on the activity of PMEPA1(−3699/+39) promoter transfected in A549 cells cultured ±TGFβ (5 ng/mL, 24 hr) before measuring dual-luciferase activity (n = 4). G. Activity of fragments of PMEPA1 promoter measured by dual-luciferase assay in A549 cells cultured ±TGFβ (5 ng/mL, 24 hr) (n = 4). Results are expressed as the mean ± SEM. ns, not significant, * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001 and using a 2-way ANOVA with Bonferroni’s posttest. See also Figure S3.
To identify the region of PMEPA1 promoter that is activated and the TGFβ response elements within it, we cloned 3.7 kb of the human PMEPA1 promoter, from nt −3699 to +39, relative the transcription start site (NM_020182) into a pGL3 luciferase reporter plasmid. TGFβ significantly increased the activity of the PMEPA1 promoter in the TGFβ responsive PC-3 (PCa), A549 (lung cancer) and HepG2 cells (hepatocarcinoma) (Fig 4C). The TGFBR1 inhibitor, SD208, prevented promoter activition by TGFβ (Fig 4C). To confirm the role of SMAD proteins, we tested the effect of ectopic expression of SMAD2, 3, 4 or 7 on PMEPA1 promoter in A549 cells. Overexpression of SMAD7 significantly decreased PMEPA1 promoter activity induced by TGFβ (Fig 4D). SMAD2 or 4 alone or combined had not effect on TGFβ-induced activity of PMEPA1 promoter (Fig 4E). However overexpression of SMAD3 significantly increased PMEPA1 promoter activity in the presence or absence of TGFβ and the activity of the promoter was maximal when SMAD3 and 4 were both overexpressed (Fig 4E). This result shows that TGFβ-induced activity of PMEPA1 3.7 kb promoter fragment is mediated by SMAD3 and not SMAD2.
We found 5 different SMAD3/4 binding elements (SBE), 5′-CAGACA-3′ (Dennler et al., 1998), on the minus and plus strands, at positions −717, −2008, −2306, −2629 and −3284. However inactivating mutations of any single or all 5 SBEs did not affect PMEPA1 promoter activity (Fig 4F). Deletion analysis of the PMEPA1 promoter to identify TGFβ-response regions indicated that deletions upstream of nucleotide −538 or downstream of nucleotide −492 significantly decreased TGFβ-induced promoter activity (Fig 4G). The smallest fragment that had a TGFβ-activation similar to the full fragment is a 1.5 kb fragment (−1467/+39) (Fig 4G). Analysis of this sequence indicated the presence of multiple putative sites for the transcription factor SP1 within GC-rich regions (69 and 79% GC; Fig S3) where SMADs interacting with SP1 can bind to DNA (Shi and Massague, 2003).
Membrane-bound PMEPA1 Inhibits TGFβ/SMAD Signaling via HECT E3 Ubiquitin Ligases
PMEPA1 controls negative feed-back loops in androgen receptor (AR) and TGFβ signaling (Xu et al., 2003; Watanabe et al., 2010). Therefore we studied how the different PMEPA1 isoforms affect TGFβ signaling in cancer cells. Specific isoforms of PMEPA1 were expressed in HepG2 cells that lack endogenous PMEPA1 and TGFβ signaling was monitored with a synthetic promoter (CAGA)9 reporter in a dual luciferase assay (Fig 5A). Membrane-bound PMEPA1a and b significantly decreased promoter activity induced by TGFβ (Fig 5A) in a dose-dependent manner (Fig S4A). Cytosolic PMEPA1c had no effect on (CAGA)9 promoter activity induced by TGFβ (Fig 5A).
Figure 5. Only membrane-bound PMEPA1 inhibits TGFβ signaling pathway by interacting with SMAD2/3 and HECT E3 ubiquitin ligases.
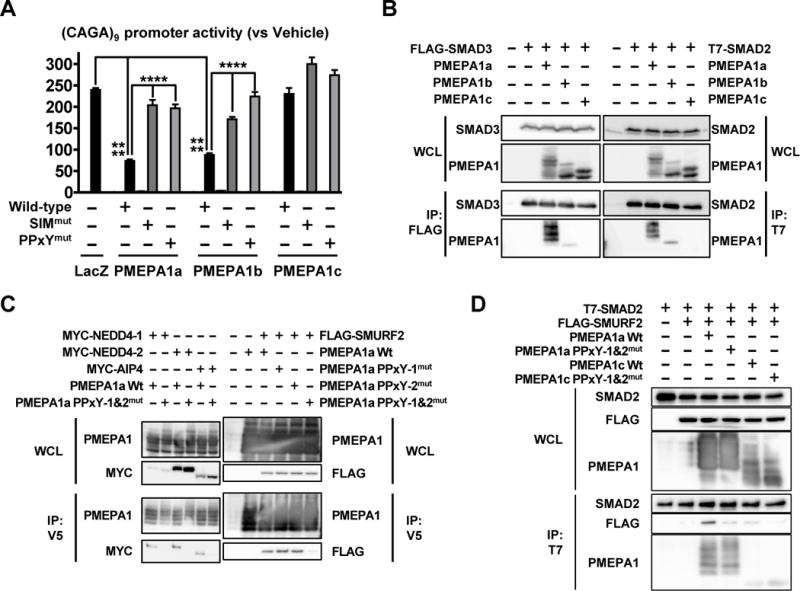
A. (CAGA)9 promoter activity in HepG2 transfected to ectopically express PMEPA1 isoforms with or without mutations in the SIM and PPxY domains and cultured ±TGFβ (5 ng/mL, 24 hr). Results are expressed as the mean ± SEM (n = 4). **** p < 0.0001 using 2-way ANOVA with Bonferroni’s posttest. B. COS-7 cells were transfected with the indicated constructs and subjected to anti-FLAG or T7 immunoprecipitation before Western blot analysis of whole cell lysate (WCL) and immunoprecipitated fractions (IP). C. Transfection of COS-7 cells to express the indicated constructs before immunoprecipitation with an anti-V5 antibody and immunoblot analysis. D. COS-7 cells were transfected to express the indicated construct before immunoprecipation with an anti-T7 antibody and Western blot. See also Figure S4.
To understand how PMEPA1 inhibits TGFβ signaling, we introduced inactivating mutations in the PPxY domains and the SMAD interaction motif (SIM) in the C-terminus of all PMEPA1 isoforms (Xu et al., 2003; Watanabe et al., 2010) (Fig 2B). PMEPA1a and b with a mutated SIM did not inhibit (CAGA)9 promoter activity compared to the wild type proteins (Fig 5A). Similarly, inactivating mutation of both PPxY domains reversed TGFβ signaling inhibition. PMEPA1a or b proteins with only one of the PPxY domains mutated still inhibited TGFβ signaling when compared to cells transfected to express LacZ (Fig S4B). PMEPA1c with mutated SIM and PPxY domain had no effect on TGFβ signaling (Fig 5A). These results indicate that only membrane-bound PMEPA1 isoforms inhibit TGFβ signaling via their SIM and PPxY domains.
To explore further how membrane-bound PMEPA1 regulates TGFβ signaling, we assessed the PMEPA1-SMAD interaction. COS-7 cells were co-transfected to express a PMEPA1 isoform and SMAD2 or 3, followed by SMAD immunoprecipitation. Membrane-bound PMEPA1a and b were co-immunoprecipitated with SMAD2 and 3 (Fig 5B). PMEPA1c that lacks the transmembrane domain was not co-immunoprecipitated with either SMAD2 or 3 (Fig 5B). The lack of interaction between PMEPA1c and R-SMADs may explain why PMEPA1c does not inhibit TGFβ signaling (Fig 5A). Next, we tested the interaction of PMEPA1a with different HECT E3 ubiquitin ligases in COS-7 cells. PMEPA1 was initially identified as a protein interacting with NEDD4-1. We confirmed that NEDD4-1 is co-immunoprecipitated with PMEPA1a, which was prevented by mutated PPxY domains (Fig 5C). PMEPA1a also interacted via its PPxY domains with the HECT E3 ubiquitin ligases NEDD4-2, AIP-4 and SMURF2, negative regulators of TGFβ signaling (Fig 5C). E3 ubiquitin ligases bind to SMAD2/3 so we tested whether PMEPA1 affected this interaction. When COS-7 cells were co-transfected to express SMAD2 and SMURF2, SMURF2 co-immunoprecipitating with SMAD2 was hardly detectable (Fig 5D). However expression of PMEPA1a increased the amount of SMURF2 precipitated with SMAD2 (Fig 5D). The increased SMAD2-SMURF2 interaction was prevented by mutated PPxY PMEPA1a, which cannot interact with SMURF2. Co-expression of PMEPA1c that did not TGFβ signaling had little effect on the SMAD2-SMURF2 interaction (Fig 5D).
Different E3 ubiquitin ligases decrease TGFβ signaling by proteasome-dependent (Zhang et al., 2001) or -independent mechanisms (Lallemand et al., 2005; Tang et al., 2011). Since PMEPA1-E3 ubiquitin ligase interaction is critical for PMEPA1 function, we tested whether it is mediated by the proteasome using the proteasome inhibitor MG132. PMEPA1a expression significantly decreased TGFβ signaling (Fig S4C) but proteasome inhibition by MG132 (confirmed by accumulation of ubiquitinated proteins, data not shown) did not prevent the PMEPA1a effect on (CAGA)9 promoter activity (Fig S4C). MG132 significantly decreased TGFβ signaling (Fig S4C), so we tested the effect of catalytically inactive E3 ubiquitin ligases that can not transfer ubiquitin to their substrates. AIP4(C830A) and SMURF2(C716A) did not reverse PMEPA1a-induced inhibition of TGFβ signaling (Fig S4D). Overall our results demonstrate that only membrane-bound PMEPA1 inhibits TGFβ signaling by recruiting SMAD2/3 and HECT E3 ubiquitin ligases, independently of ubiquitination and proteasome degradation.
Knockdown of PMEPA1 Increases TGFβ Signaling in Cancer Cells
To assess the role of endogenous PMEPA1 in cancer cells, we knocked down all Pmepa1 isoforms using 2 siRNAs. Transfection of these siRNAs, individually or pooled, prevented the induction of detectable PMEPA1 protein by TGFβ (Fig 6A). We tested the effect of PMEPA1 knockdown on TGFβ signaling. In PC-3 cells, Pmepa1 siRNAa decreased SMAD2 phosphorylation compared to Control (Fig 6B). The Pmepa1 siRNA had no effect on total SMAD2 protein measured by Western blot (Fig 6B), confirming that PMEPA1 inhibition of TGFβ signaling is independent of protein degradation. Similarly, siPmepa1 increased SMAD2 phosphorylation without changing total SMAD2 protein in other prostate (DU145), breast (MDA-MB-231) and lung (A549) cancer cells (Fig S5A). In HepG2 cells that do not express PMEPA1, siPmepa1 had no effect on SMAD2 phosphorylation (Fig S5A). We also analyzed the nuclear translocation of SMAD2. After TGFβ treatment, phosphorylated SMAD2 was not detected in the non-nuclear fraction regardless of the siRNA transfected (Fig 6C). In the nuclear fraction, knockdown of PMEPA1 increased the amount of phosphorylated SMAD2 as well as prolonged the retention of SMAD2 in the nucleus for up to 2 hours (Fig 6C & S5B). We measured the mRNAs of TGFβ-regulated genes. In TGFβ-treated PC-3 cells, PMEPA1 knockdown significantly increased Il11, Mmp13 and Adam19 mRNA (Fig 6D).
Figure 6. PMEPA1 knockdown increases TGFβ signaling in PC-3 cells in vitro and the development of PC-3 bone metastases in mice.
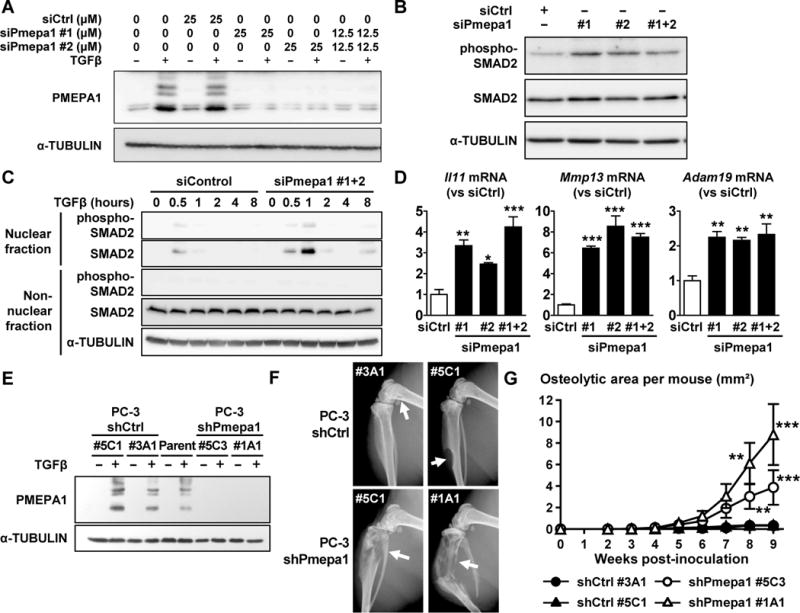
A. PC-3 cells transfected with a non-targeting siRNA (siControl) or different siRNA against PMEPA1 (siPmepa1 #1 and #2) were treated ±TGFβ (5 ng/mL) before measuring PMEPA1 expression by Western blot. B. Immunoblot analysis of SMAD2 phosphorylation in PC-3 cells transfected with siControl or siPmepa1#1+2 (25 μM) and treated with TGFβ (5 ng/mL, 4 hr). C. Analysis of SMAD2 phosphorylation in the nuclear and non-nuclear fractions of PC-3 cells transfected with siControl or siPmepa1 (25 μM) and treated with TGFβ (5 ng/mL). D. Il11, Mmp13 and Adam19 mRNA levels measured by RT-qPCR in PC-3 cells transfected with siControl or siPmepa1 (25 μM) and treated with TGFβ (5 ng/mL). Results are expressed as the mean ± SEM relative mRNA level (n = 3). * p < 0.05, ** p < 0.01 and *** p < 0.001 vs siCtrl using a 1-way ANOVA with a Dunnett’s posttest. E. Expression of PMEPA1 in parental PC-3 cells or after stable transfection to a non-targeting shRNA (shCtrl) or an shRNA against PMEPA1 (shPmepa1) and cultured ±TGFβ (5 ng/mL, 24 hr). F&G. Nude mice were inoculated in the left cardiac ventricle with PC-3 shCtrl (#3A1 or #5C1) PC-3 shPmepa1 (#5C3 or #1A1) cells and surveyed by x-ray over time. (F) Representative radiographs (arrows indicate osteolytic lesions). (G) Area of osteolysis measured on radiographs. Results are expressed as the mean area per mouse ± SEM. ** p < 0.01 and *** p < 0.001 compared to shCtrl #3A1 or #5C1 using a 2-way ANOVA with Bonferroni’s posttest. See also Figure S5.
PMEPA1 was previously shown to regulate cancer cell growth, either inhibiting PCa cell proliferation or preventing TGFβ-induced growth arrest via p27 and p21 depending on the study (Xu et al., 2003; Li et al., 2008). We tested the effect of siPmepa1 on the cell cycle inhibitors p21 and p27 and of the cell cycle regulator CYCLIN D1 in PC-3 and DU145 cells as well as in E7 and RWPE-1 prostate epithelial cells. Although siPmepa1 increased p21 in PC-3 and decreased p21 and p27 in DU145, PMEPA1 knockdown had little effect on CYCLIN D1 (Fig S5E). In prostate epithelial cells PMEPA1 knockdown had no effect on p21, p27 or CYCLIN D1 expression (Fig S5E). PMEPA1 knockdown in the presence or absence of TGFβ did not markedly affect the cell cycle distribution or the growth of PC3, DU145, E7 or RWPE-1 cells (Fig S5F&G). These results demonstrate that PMEPA1 knockdown increases TGFβ signaling and the expression of pro-metastatic gene but does not affect the growth of PCa cells.
PMEPA1 Knockdown Increases PC-3 Bone Metastases in Mice
To test the role of PMEPA1 in bone metastases, PC-3 cells were stably transfected with plasmids expressing either an shRNA control or one knocking down all of the variants of Pmepa1. Single cell clones with knockdown of PMEPA1 were selected and transfection stability tested by culturing the cells for 70 days in absence of antibiotic. Two clones (shPmepa1 #5C3 and #1A1) with a >75% decrease of Pmepa1 mRNA compared to untransfected parental PC-3 cells (data not shown) and undetectable levels of PMEPA1 protein in the presence of TGFβ were selected (Fig 6E). We also selected 2 PC-3 clones expressing shRNA control (shCtrl #5C1 and #3A1) that expressed levels of PMEPA1 protein similar to parental PC-3 cells when cultured with TGFβ (Fig 6E). We also compared the growth of the different PC-3 clones using an MTT assay over 5 days. There were no differences in the proliferation between the parental PC-3 cells and the shCtrl or shPmepa1 clones in absence or presence of exogenous TGFβ (Fig S5C & S5D), similar to the results observed using siRNA.
Male nude mice were inoculated in the left cardiac ventricle with either a control clone (shCtrl #3A1 or 5C1) or a PMEPA1 knockdown clone (shPmepa1 #5C3 or 1A1). The progression of osteolytic lesions on radiographs was assessed over 9 weeks. The area of osteolysis on radiographs was significantly increased in mice inoculated with the PC-3 shPmepa1 clones compared to mice inoculated with the PC-3 shCtrl clones (Fig 6F & 6G). Our results indicate that loss of expression of PMEPA1 increases PCa bone metastases.
DISCUSSION
Most men with advanced PCa have incurable bone metastases, a major source of morbidity and mortality. It is therefore critical to understand the mechanisms of PCa bone metastases to identify therapeutic targets and develop efficient treatments. Another critical challenge with PCa is to discriminate aggressive from indolent PCa since many patients with low risk disease are overtreated.
In this study, we demonstrated that SD208, an inhibitor of TGFBR1 reduced bone metastases from PC-3 PCa cells in mice. SD208 was more effective when given as a preventive therapy, which is consistent with previous reports in melanoma and glioma models (Uhl et al., 2004; Mohammad et al., 2011). As a preventive agent, SD208 also improved survival. The results further demonstrate the potential of SD208 to treat or prevent bone metastases.
Our results suggest that inhibition of TGFβ signaling impairs the interactions of PC-3 cells with the bone microenvironment since SD208 had no effect on cell growth in vitro. These interactions drive a feed-forward “vicious cycle” that fuels tumor growth in bone as tumor cells stimulate bone destruction and the release of growth factors, such as TGFβ, into the bone microenvironment. SD208 decreased TGFβ-regulated genes such as PTHRP and IL11, which are critical for tumor-associated osteoclastic bone resorption and are likely responsible for the anti-bone metastasis activity of the drug (Yin et al., 1999; Kang et al., 2005). This is consistent with a previous study where SD208 was only efficient at preventing the growth of 1205Lu melanoma cells in bone, not in soft tissues (Mohammad et al., 2011). The microarray and PCR analyses confirmed that TGFβ increases the expression of multiple pro-osteolytic genes PTHRP, IL11, ADAM19. In addition TGFβ increased the expression of genes associated with different steps of the bone metastasis cascade such as angiogenesis (VEGFA, CTGF, MMP13) or homing to bone (ITGAV) and invasion (UPA, MMP13, NEDD9). Interestingly, many of these genes were also identified in BCa and melanoma studies (Kang et al., 2003; Dunn et al., 2009; Mohammad et al., 2011). Therefore TGFβ coordinates the expression of multiple genes that cooperate to promote PCa bone metastases.
The gene most increased by TGFβ identified in the microarray was PMEPA1 (also known as TMEPAI, STAG1 and N4wwBP4). In vitro TGFβ quickly and strongly induced the expression of PMEPA1 isoforms a and c in different prostate, breast and lung cancer cell lines. In samples from PCa patients, PMEPA1 is increased in the primary tumor as well as in the tissue adjacent to it, which contains high levels of TGFβ (Carstens et al., 2014). Increased resorption at sites of bone metastases also increases local TGFβ concentration (Korpal et al., 2009), and we found that PMEPA1 was also higher in bone metastases compared to other metastatic sites in samples from BCa patients. The design of the probes in the microarray did not permit discrimination between the different variants of Pmepa1 isoform mRNAs.
PMEPA1 regulates negative feedback loops in AR and TGFβ signaling (Li et al., 2008; Watanabe et al., 2010). We confirmed that overexpression of PMEPA1 in HepG2 cells decreased TGFβ signaling. However, unlike the findings of Watanabe et al. (2010), only the transmembrane PMEPA1 inhibited activation of the (CAGA)9 TGFβ-reporter. The cytosolic PMEPA1c isoform had no effect on TGFβ signaling when overexpressed in HepG2 cells, which could be due to the lack of interaction with R-SMADs. Despite an intact SIM at its C-terminus, PMEPA1c was not co-immunoprecipitated with SMAD2 or 3, suggesting that intracellular localization of PMEPA1 is important for its function. Interaction of PMEPA1 with R-SMADs is critical for the inhibition of TGFβ signaling as exemplified by PMEPA1c and by PMEPA1a with a mutated SIM. Similarly, interaction of PMEPA1 via its PPxY domains with HECT E3 ubiquitin ligase is critical for its function. The results suggest a model where PMEPA1a and b act as docking proteins to recruit E3 ubiquitin ligases and R-SMADs and target them for proteasomal degradation. However, PMEPA1 function was not linked to ubiquitination, and its knockdown did not increase SMAD2 levels: indicating that the inhibition of TGFβ signaling by PMEPA1 is proteasome-independent. It has previously been reported that SMAD7 inhibits TGFβ signaling by recruiting the E3 ubiquitin ligase AIP4 and TGFBR1 but without SMAD7 degradation (Lallemand et al., 2005). Our results suggest that, similarly, membrane-bound PMEPA1 recruit E3 ubiquitin ligases and SMAD2 or 3, preventing their phosphorylation and down-regulating TGFβ signaling. In prostate, breast and lung cancer cells, TGFβ induced expression of the cytosolic PMEPA1c and the membrane-bound PMEPA1a. Knockdown of PMEPA1 increased SMAD2 phosphorylation and the expression of TGFβ-regulated genes demonstrating that endogenous PMEPA1 is an inhibitor of TGFβ signaling in multiple cancer cells. Accordingly, stable knockdown of PMEPA1 in PC-3 prostate cancer cells increased the development of osteolytic bone metastases in mice, consistent with increased expression of pro-metastatic genes like IL11, ADAM19 and MMP13.
To determine if PMEPA1 expression is of clinical relevance, we interrogated databases of gene expression in patient samples. Multiple independent datasets of patient samples indicated that PMEPA1 expression was increased in the primary tumors of patients with prostate, breast or lung cancer. This is consistent with previous results showing that PMEPA1 is highly expressed by epithelial cells of the prostate gland and in tumor tissues (Xu et al., 2000). However levels of Pmepa1 mRNA have also been found to be decreased in PCa of higher grade (Xu et al., 2003). Considering that gene expression in microarray-based datasets are semi-quantitative and that we did not seek to compare the expression of PMEPA1 between different stages in the primary tumor, these results should be interpreted with this caveat in mind. These differences could be explained by the fact that PMEPA1 is expressed selectively in epithelial cells and epithelial cell content in tumors is variable.
Our results and other previously published data support that PMEPA1 is a negative regulator of TGFβ signaling (Watanabe et al., 2010). Since TGFβ acts as tumor suppressor in early stages of cancer, it is possible that, early on, induction of PMEPA1 expression by environmental factor (i.e., TGFβ, androgens) decreases TGFβ signaling, shielding cancer cells from its anticancer properties (Pickup et al., 2013) (Fig 7). However our study found no effect of PMEPA1 on the proliferation of PCa cells or prostatic epithelial cells. Additional experiments are needed to characterize the role of PMEPA1 during the early stages of cancer development. Even though Pmepa1 levels are increased in the primary tumor, we also found that expression of PMEPA1 was decreased in distant metastases of PCa patients when compared to the primary tumor. This result is consistent with observation from Xu et al. (2003) and report of loss of expression of Pmepa1 due to accumulated methylation in PMEPA1 promoter (Sharad et al., 2014). Methyltransferase inhibitors restored TGFβ-inducible expression of PMEPA1 in HepG2 cells suggesting that TGFβ response elements in PMEPA1 promoter can also be inactivated by methylation. Thus decreased expression of PMEPA1 in metastases could be due to epigenetic silencing. We identified a 1.5kb fragment of the human PMEPA1 promoter that is responsive to TGFβ. Further experiments could analyze the methylation profile of this promoter in cancer cells with high or low expression of Pmepa1 and ask whether methylation of the PMEPA1 promoter predicts patient outcomes.
Figure 7. Effect of PMEPA1 expression on metastases from PCa cancer.
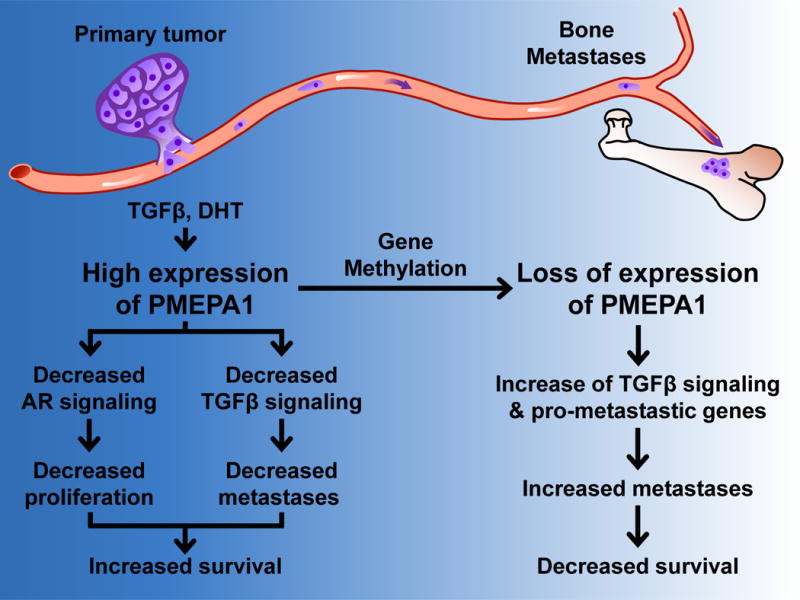
High expression of PMEPA1 in primary tumor decreases AR and TGFβ signaling preventing, progression of disease and increasing patient survival. Methylation of PMEPA1 promoter leads to decreased PMEPA1 expression and increased TGFβ signaling and TGFβ-regulated pro-metastatic genes ultimately causing an increase of metastases and a decrease of patient survival.
To test the prognostic value of PMEPA1 expression, we used the PROGgene database. The only PCa dataset available indicated that low Pmepa1 mRNA in the tumors of PCa patients were associated with shorter time to relapse. The difference was not significant perhaps due to small sample size. BCa datasets offer greater statistical power. Three different datasets indicated that low expression of Pmepa1 was significantly associated with decreased survival and shorter time to relapse or metastases. The data are consistent with increased bone metastases caused by Pmepa1 knockdown.
Our results show that decreased PMEPA1 increases TGFβ signaling and expression of prometastatic genes, suggesting a model where decreased PMEPA1 expression suppresses a negative feedback loop and restores TGFβ signaling (Fig 7). This allows TGFβ to exert its prometastatic effects as cancer progresses – consistent with our observation that Pmepa1 knockdown increases bone metastases and that low expression of PMEPA1 is associated with bad patient prognosis. PMEPA1 not only inhibits TGFβ signaling, it also targets the AR for degradation (Li et al., 2008). It is possible that the increased time to relapse for PCa patients with higher expression of PMEPA1 in the primary tumor is due to decreased AR signaling (Fig 7). Pmepa1 could distinguish indolent from aggressive PCa and prevent overtreatment of PCa patients. Loss of expression of PMEPA1 as cancer progresses would restore AR expression, whose signaling remains active in a large subset of metastatic PCa patient, including castration resistant PCa (Taylor et al., 2010). Thus the overall role of PMEPA1 in cancer likely derives from its regulation of both TGFβ and AR signaling. However our experiments were conducted with TGFβ-sensitive PCa cells PC-3 and DU145 that are both AR-negative as the androgensensitive PCa cells like LNCap, VCaP or MDA-PCa-2b were not found to be TGFβ-responsive (data not shown). Therefore, it was not possible to test the effect of PMEPA1 loss of expression on both AR and TGFβ signaling. Despite that limitation, our results in PC-3 and DU145 have important implications about the role of increased TGFβ activity caused by PMEPA1 loss and metastatic progression in patients with AR-negative PCa.
It was recently reported that Pmepa1 knockdown decreased the growth of Calu3 lung cancer cells in mice (Vo Nguyen et al., 2014). Analysis of Pmepa1 expression in lung cancer patient datasets revealed that low levels of Pmepa1 are associated with a significantly increased survival or longer time before recurrence in 2 different cohorts of patients (Fig S2D & S2E). The pro- and anti-cancer functions of PMEPA1 are probably microenvironment and cancer-type dependent.
In conclusion, we have shown that TGFβ signaling in PCa cells increases the expression of numerous genes associated with the development of bone metastases and that TGFβ supports the development of bone metastases from PCa in mice. Consequently anti-TGFβ therapies with agents such as SD208 could be used as treatment or prevention. Our study also revealed how membrane-bound PMEPA1a recruits R-SMADs and E3 ubiquitin ligases to inhibit TGFβ signaling by a mechanism independent of the proteasome. Loss of PMEPA1 increased TGFβ signaling and development of bone metastases in mice. Importantly, our data are validated by evidence from human databases showing that low levels or loss of expression of Pmepa1 are associated with poor clinical outcome and could be used as a predictor of cancer progression in patients suffering from breast or prostate cancer.
EXPERIMENTAL PROCEDURES
Animal studies
All mouse experiments were approved by and performed following the guidelines of the Institutional Animal Care and Use Committee at the University of Virginia (Charlottesville, VA). For bone metastasis study, 4-week old athymic male mice (Harlan Sprague Dawley Inc) were anesthetized and inoculated into the left cardiac ventricle with 105 cancer cells in 100 μL of PBS. The development of bone lesions was surveyed by radiography using a Faxitron MX-20 with digital camera (Faxitron Bioptics, LLC). Mice were monitored daily for signs of discomfort, and were either euthanized all at one time or individually when presenting signs of distress for survival studies. More information about animal experiments and their analysis are presented in the Supplemental Experimental Procedures.
Cell Culture, Treatment, and Assays
PC-3 and DU145 human PCa cells, HepG2 human hepatocarcinoma cells, A549 human lung cancer cells, MDA-MB-231 human BCa cells, RWPE-1 normal epithelial prostatic cells and COS-7 kidney cells from green monkey were obtained from the American Type Culture Collection (ATCC). E7 prostate epithelial cells were generated by Drs Jerde, Ewald and Jarrard (Indiana University and University of Wisconsin) and were a kind gift from Dr Travis Jerde (Indiana University) (Schwarze et al., 2002). E7 cells were cultured in DMEM/F12 media supplemented with sodium pyruvate and FBS (5%) and used for MTT cell-proliferation, RNA extraction and RT-qPCR, dual-luciferase assay, immunoprecipitation and immunoblotting. More information on these methods including transfection, antibodies, reagents, plasmids are provided in the Supplemental Experimental Procedures.
Microarray Analysis and Data Mining
Microarray analysis with Affymetrix chips was performed by the University of Virginia Biomolecular Research Facility. See Supplemental Experimental Procedures for details.
Expression of PMEPA1 in normal and malignant samples of prostate, breast and lung tissues from patients was queried using the Oncomine database. The p-values presented were extracted directly from the Oncomine analysis and the tests have not been repeated manually. To compare PMEPA1 expression in patient samples, the datasets GSE6919, GSE14017 and GSE14018 were downloaded from Pubmed GEO and analyzed by the Center for Computational Biology and Bioinformatics (Indiana University). Prognostic value of Pmepa1 mRNA in the primary tumor of patients with prostate, breast or lung cancer was assessed using the PROGgene database (Goswami and Nakshatri, 2013). Relapse-free, metastasis-free or overall survival was compared between high and low PMEPA1 expression groups using median gene expression value as bifurcating point.
Statistical Analysis
Statistical analysis of the microarray data was performed using dChip software and Student’s t test. The significance criterion consisted of: 1) p ≤ 0.05; 2) fold change ≥1.5 or ≤-1.5; and 3) signal differential ≥Min (100, 10× 10% mean signal intensity of absent probe sets). Statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad software, Inc.). Comparisons of two groups were performed with a non-parametric Mann-Whitney’s U test and for comparisons of three or more groups we used a 1-way ANOVA test, with a Dunnett’s post-test when comparing to a control group or with a Bonferroni’s to compare selected pairs of group. For responses that are affected by two variables, a 2-way ANOVA with a Bonferroni post-test was used. To compare mouse survival, we used Kaplan-Meier analysis with a log-rank (Mantel-Cox) test. Analysis of patient survival data was done as described (Goswami and Nakshatri, 2013). Results are expressed as mean ± SEM and a p ≤ 0.05 was considered significant.
Supplementary Material
SIGNIFICANCE.
Our preclinical data reveal that in prostate cancer, TGFβ controls the expression of a prometastatic gene program similar to that seen in melanoma and breast cancer, supporting the use of TGFβ inhibitors to treat prostate cancer metastases to bone, which is an abundant source of TGFβ. PMEPA1 was the gene most increased by TGFβ in prostate cancer cells, where knocking it down increased bone metastases. The identification of PMEPA1 as a major target of TGFβ and negative feedback regulator of TGFβ signaling suggests that PMEPA1 might be a useful prognostic marker of metastases to TGFβ-rich sites and predictor of metastasis-free survival. Enhancing membrane-bound PMEPA1 activity could be used as treatment strategy against prostate cancer bone metastases.
HIGHLIGHTS.
TGFβ inhibition decreases prometastatic genes and prostate cancer bone metastases
PMEPA1 inhibits TGFβ signaling by a non-proteasomal mechanism
Clinically, low PMEPA1 correlates with poor metastasis-free survival
PMEPA1 knockdown increases prostate cancer bone metastases in a mouse model
Acknowledgments
The authors acknowledge financial support from the US Department of Defense PCRP (grants PC061185 to PGJF and PC040341 to TAG), the National Institutes of Health (R01CA069158 and U01CA143057 to TAG), the US Department of Veterans Affairs (JMC), the Prostate Cancer Foundation (TAG), the Jerry W. and Peggy S. Throgmartin Endowment of Indiana University (TAG), and Indiana Economic Development Fund (TAG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
Microarray data are available in the Gene Expression Omnibus (GEO) database under accession number GSE58698.
AUTHORS CONTRIBUTIONS
PGJF, JMC and TAG conceived the study, generated hypotheses, designed experiments and analyzed the data. PGJF designed and performed all in vitro experiments. PGJF, PJ, MN, HSK, HWW, XP, KSM, TAG performed and analyzed results from animal experiments. GJ and YL performed bioinformatics analyses of PMEPA1 expression in patient data. GAC and CDW contributed to experimental design, and reagent and sample generation. PGJF, PJ, GAC, KSM, CDW, JMC and TAG wrote, reviewed and/or revised the manuscript.
The authors do not have any potential financial conflict of interest.
References
- Carstens JL, Shahi P, Van Tsang S, Smith B, Creighton CJ, Zhang Y, Seamans A, Seethammagari M, Vedula I, Levitt JM, et al. FGFR1-WNT-TGF-beta signaling in prostate cancer mouse models recapitulates human reactive stroma. Cancer Res. 2014;74:609–620. doi: 10.1158/0008-5472.CAN-13-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran U, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon F. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64–64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesneau V, Becherer J, Zheng Y, Erdjument-Bromage H, Tempst P, Blobel C. Catalytic properties of ADAM19. J Biol Chem. 2003;278:22331–22340. doi: 10.1074/jbc.M302781200. [DOI] [PubMed] [Google Scholar]
- Clines G, Mohammad K, Bao Y, Stephens O, Suva L, Shaughnessy J, Fox J, Chirgwin J, Guise T. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol. 2007;21:486–498. doi: 10.1210/me.2006-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford S, Stellmach V, Murphy-Ullrich J, Ribeiro S, Lawler J, Hynes R, Boivin G, Bouck N. Thrombospondin-1 Is a Major Activator of TGF-β1 In Vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- Crippa M. Urokinase-type plasminogen activator. Int J Biochem Cell Biol. 2007;39:690–694. doi: 10.1016/j.biocel.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier J. Direct binding of Smad3 and Smad4 to critical TGF-β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn L, Mohammad K, Fournier P, McKenna C, Davis H, Niewolna M, Peng X, Chirgwin J, Guise T. Hypoxia and TGF-β drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One. 2009;4:e6896. doi: 10.1371/journal.pone.0006896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Troyanskaya O, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen G, Perou C, Whyte R, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C, Nakshatri H. PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinforma. 2013;3:22–22. doi: 10.1186/2043-9113-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulzar Z, McKenney J, Brooks J. Increased expression of NuSAP in recurrent prostate cancer is mediated by E2F1. Oncogene. 2013;32:70–77. doi: 10.1038/onc.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Zhang H, Baile S, Ljungman M, Kuhstoss S, Keller E. p21CIP-1/WAF-1 Induction Is Required to Inhibit Prostate Cancer Growth Elicited by Deficient Expression of the Wnt Inhibitor Dickkopf-1. Cancer Res. 2010;70:9916–9926. doi: 10.1158/0008-5472.CAN-10-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Gupta J, Zhang Z, Gerseny H, Berg A, Chen YJ, Zhang Z, Du H, Brendler C, Xiao X, et al. Systemic delivery of oncolytic adenoviruses targeting transforming growth factor-beta inhibits established bone metastasis in a prostate cancer mouse model. Hum Gene Ther. 2012;23:871–882. doi: 10.1089/hum.2012.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivshina A, George J, Senko O, Mow B, Putti T, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Mohammad K, McKenna C, Fournier P, Luciani F, Niewolna M, Andre J, Delmas V, Larue L, Guise T, Mauviel A. Stable Overexpression of Smad7 in Human Melanoma Cells Impairs Bone Metastasis. Cancer Res. 2007;67:2317–2324. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- Juarez P, Guise TA. TGF-β in cancer and bone: Implications for treatment of bone metastases. Bone. 2010;48:23–29. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Kang Y, He W, Tulley S, Gupta G, Serganova I, Chen C, Manova-Todorova K, Blasberg R, Gerald WL, Massague J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA. 2005;102:13909–13914. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel P, Shu W, Drobnjak M, Kakonen S, Cordon-Cardo C, Guise T, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Koeneman K, Yeung F, Chung L. Osteomimetic properties of prostate cancer cells: A hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246–261. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-β signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2009;15:960–966. doi: 10.1038/nm.1943. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Iizuka S, Yoshida M, Tsunematsu T, Kondo T, Subarnbhesaj A, Deraz E, Siriwardena S, Tahara H, Ishimaru N, et al. Matrix Metalloproteinase-13 (MMP-13) Directly and Indirectly Promotes Tumor Angiogenesis. J Biol Chem. 2012;287:38716–38728. doi: 10.1074/jbc.M112.373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand F, Seo S, Ferrand N, Pessah M, L’Hoste S, Rawadi G, Roman-Roman S, Camonis J, Atfi A. AIP4 Restricts Transforming Growth Factor-β Signaling through a Ubiquitination-independent Mechanism. J Biol Chem. 2005;280:27645–27653. doi: 10.1074/jbc.M500188200. [DOI] [PubMed] [Google Scholar]
- Li H, Xu L, Masuda K, Raymundo E, McLeod D, Dobi A, Srivastava S. A feedback loop between the androgen receptor and a NEDD4-binding protein, PMEPA1, in prostate cancer cells. J Biol Chem. 2008;283:28988–28995. doi: 10.1074/jbc.M710528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt A, Gillet C, Ellis P, Harris A, Bergh J, Foekens J, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009;11:R7. doi: 10.1186/bcr2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L, Smeds J, George J, Vega V, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu E, Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake H, Pollak M, Gleave M. Castration-induced up-regulation of insulinlike growth factor binding protein-5 potentiates insulin-like growth factor-I activity and accelerates progression to androgen independence in prostate cancer models. Cancer Res. 2000;60:3058–3064. [PubMed] [Google Scholar]
- Mohammad K, Chen C, Balooch G, Stebbins E, McKenna C, Davis H, Niewolna M, Peng X, Nguyen D, Ionova-Martin S, et al. Pharmacologic inhibition of the TGF-β type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS One. 2009;4:e5275. doi: 10.1371/journal.pone.0005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad K, Javelaud D, Fournier P, Niewolna M, McKenna C, Peng X, Duong V, Dunn L, Mauviel A, Guise T. TGF-β-RI Kinase Inhibitor SD-208 Reduces the Development and Progression of Melanoma Bone Metastases. Cancer Res. 2011;71:175–184. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Tanaka T, Nitta Y, Ohnishi K, Kawashima H, Nakatani T. NEDD9 crucially regulates TGFβ-triggered epithelial–mesenchymal transition and cell invasion in prostate cancer cells. The Prostate. 2014;74:901–910. doi: 10.1002/pros.22809. [DOI] [PubMed] [Google Scholar]
- Pécheur I, Peyruchaud O, Serre C, Guglielmi J, Voland C, Bourre F, Margue C, Cohen-Solal M, Buffet A, Kieffer N, Clézardin P. Integrin avb3 expression confers on tumor cells a greater propensity to metastasize to bone. Faseb J. 2002;16:1266–1268. doi: 10.1096/fj.01-0911fje. [DOI] [PubMed] [Google Scholar]
- Pickup M, Novitskiy S, Moses H. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Cossigny D, Quan G. The Role of Vascular Endothelial Growth Factor in Metastatic Prostate Cancer to the Skeleton. Prostate Cancer. 2013;2013:418340–418340. doi: 10.1155/2013/418340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Asirvatham A, Chaudhary J. Inhibitor of differentiation 1 (ID1) promotes cell survival and proliferation of prostate epithelial cells. Cell Mol Biol Lett. 2010;15:272–295. doi: 10.2478/s11658-010-0007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19:192–205. doi: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Huang D, Blick T, Connor A, Reiter L, Hardink J, Lynch C, Waltham M, Thompson E. An MMP13-selective inhibitor delays primary tumor growth and the onset of tumor-associated osteolytic lesions in experimental models of breast cancer. PLoS One. 2012;7:e29615. doi: 10.1371/journal.pone.0029615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharad S, Ravindranath L, Haffner M, Li H, Yan W, Sesterhenn I, Chen Y, Ali A, Srinivasan A, McLeod D, et al. Methylation of the PMEPA1 gene, a negative regulator of the androgen receptor in prostate cancer. Epigenetics. 2014;9:918–927. doi: 10.4161/epi.28710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shimo T, Kubota S, Yoshioka N, Ibaragi S, Isowa S, Eguchi T, Sasaki A, Takigawa M. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J Bone Miner Res. 2006;21:1045–1059. doi: 10.1359/jbmr.060416. [DOI] [PubMed] [Google Scholar]
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massague J, Benezra R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013;5:1228–1242. doi: 10.1016/j.celrep.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Yamashita M, Coussens N, Tang Y, Wang X, Li C, Deng C, Cheng S, Zhang Y. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. Embo J. 2011;30:4777–4789. doi: 10.1038/emboj.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma J, Almirez R, Mangadu R, Liu Y, Platten M, Herrlinger U, et al. SD-208, a novel transforming growth factor β receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64:7954–7961. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- Vanaja D, Cheville J, Iturria S, Young C. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–3882. [PubMed] [Google Scholar]
- Vo Nguyen T, Watanabe Y, Shiba A, Noguchi M, Itoh S, Kato M. TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci. 2014;105:334–341. doi: 10.1111/cas.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Li Z, Yingling J, Yang J, Starbuck M, Ravoori M, Kundra V, Vazquez E, Navone N. Effect of transforming growth factor-β (TGF-β) receptor I kinase inhibitor on prostate cancer bone growth. Bone. 2012;50:695–703. doi: 10.1016/j.bone.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Itoh S, Goto T, Ohnishi E, Inamitsu M, Itoh F, Satoh K, Wiercinska E, Yang W, Shi L, et al. TMEPAI, a transmembrane TGF-β-inducible protein, sequesters Smad proteins from active participation in TGF-β signaling. Mol Cell. 2010;37:123–134. doi: 10.1016/j.molcel.2009.10.028. [DOI] [PubMed] [Google Scholar]
- Weilbaecher K, Guise T, McCauley L. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Shi Y, Petrovics G, Sun C, Makarem M, Zhang W, Sesterhenn I, McLeod D, Sun L, Moul JW, Srivastava S. PMEPA1, an androgen-regulated NEDD4-binding protein, exhibits cell growth inhibitory function and decreased expression during prostate cancer progression. Cancer Res. 2003;63:4299–4304. [PubMed] [Google Scholar]
- Xu LL, Shanmugam N, Segawa T, Sesterhenn IA, McLeod DG, Moul JW, Srivastava S. A novel androgen-regulated gene, PMEPA1, located on chromosome 20q13 exhibits high level expression in prostate. Genomics. 2000;66:257–263. doi: 10.1006/geno.2000.6214. [DOI] [PubMed] [Google Scholar]
- Yin J, Selander K, Chirgwin J, Dallas M, Grubbs B, Wieser R, Massague J, Mundy G, Guise T. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chang C, Gehling D, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci USA. 2001;98:974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.