

Abstract
We need better medicines to control acute and chronic pain. Fatty acid amide hydrolase (FAAH) and soluble epoxide hydrolase (sEH) catalyze the deactivating hydrolysis of two classes of bioactive lipid mediators - fatty acid ethanolamides (FAEs) and epoxidized fatty acids (EpFAs), respectively - which are biogenetically distinct but share the ability to attenuate pain responses and inflammation. In these experiments, we evaluated the antihyperalgesic activity of small-molecule inhibitors of FAAH and sEH, administered alone or in combination, in two pain models: carrageenan-induced hyperalgesia in mice and streptozocin-induced allodynia in rats. When administered separately, the sEH inhibitor 1-trifluoromethoxyphenyl-3-(1-propionylpiperidine-4-yl)urea (TPPU) and the peripherally restricted FAAH inhibitor URB937 were highly active in the two models. The combination TPPU plus URB937 was markedly synergistic, as assessed using isobolographic analyses. The results of these experiments reveal the existence of a possible functional crosstalk between FAEs and EpFAs in regulating pain responses. Additionally, the results suggest that combinations of sEH and FAAH inhibitors might be exploited therapeutically to achieve greater analgesic efficacy.
Keywords: fatty acid amide hydrolase, soluble epoxide hydrolase, fatty acid ethanolamide FAE, epoxidized fatty acid EpFA, anti-nociception, acute and chronic pain
Graphical Abstract
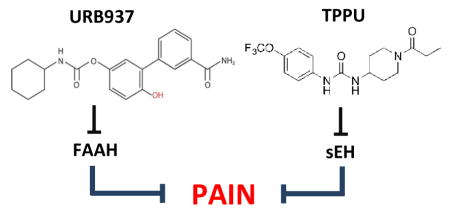
1. Introduction
Various bioactive lipid mediators regulate nociceptive pain and inflammation in peripheral tissues by interacting with receptor systems on primary sensory neurons and neighboring host-defense cells, such as macrophages, mast cells and keratinocytes [1]. In this context, two classes of long-chain fatty acid derivatives have been studied with particular attention: the fatty acid ethanolamides (FAE) [1] and the epoxidized fatty acids (EpFAs) [2,3]. These lipids demonstrate marked analgesic and anti-inflammatory properties but are subject to rapid deactivating metabolism, with the primary route for both being enzymatic hydrolysis [1–3]. The FAEs are degraded primarily by fatty acid amide hydrolase (FAAH) while the EpFAs are metabolized by soluble epoxide hydrolase (sEH). Both enzymes are hydrolases, but they are in different protein families. Because of the roles of the FAE and EpFAs in pain modulation [1] FAAH and sEH have been intensely studied to develop pharmacological inhibitors that are able to sustain endogenous levels of these analgesic and anti-inflammatory mediators.
FAAH is an intracellular serine hydrolase that belongs to the amide signature hydrolase superfamily of enzymes [4,5]. Preferred FAAH substrates include endogenous agonists of cannabinoid receptors, such as anandamide (arachidonoylethanolamide) [6,7], and type-α peroxisome proliferator-activated receptors (PPAR-α), such as oleoylethanolamide (OEA) [8] and palmitoylethanolamide (PEA) [9]. sEH is a bifunctional enzyme with an N-terminal phosphatase activity, whose physiological function is unclear, and a C-terminal hydrolase activity that catalyzes the conversion of EpFAs into dihydroxy fatty acids [10]. The epoxide hydrolase domain of sEH belongs to the α/β-hydrolase fold family of proteins [11]. Most of the activity of sEH is attributed to this domain, because EpFAs are known to activate large-conductance potassium (BK) channels [12], nuclear receptors (e.g. PPAR-α and PPAR-γ) [13,14], transient receptor potential (TRP) channels [15–17] as well as to inhibit the pro-inflammatory transcription factor NF-κB [18,19]. While there is evidence for the existence of EpFA receptors [20,21], the identities of these putative receptors remain undefined [22]. By contrast, the endocannabinoid anandamide is a known agonist of two G protein-coupled receptors, namely the CB1 and CB2 cannabinoid receptors [23]. Interestingly, the epoxyeicosatrienoic acids (EETs) weakly displace radioligand binding at CB2, but not CB1 receptors [24]. The significance of this weak CB2 agonism remains to be determined, but it is of interest due to the suggested role of CB2 receptors in peripheral pain modulation [25]. Little is known about the interaction of EpFAs derived from n-3 polyunsaturated fatty acids (e.g., docosahexaenoic acid and eicosapentaenoic acid) with the endocannabinoid system. However, EETs and n-3 EpFAs are robustly antihyperalgesic in acute and chronic models of pain [26,27]. The fact that EETs, as a subset, do not significantly activate the endocannabinoid receptor system suggests EpFAs and FAEs act through complementary rather than overlapping mechanisms.
Preclinical studies have documented the anti-inflammatory and anti-hyperalgesic effects of FAAH [1] and sEH inhibitors [3]. In the present study, we asked whether these two classes of agents act in an additive or super-additive (synergistic) manner to attenuate pain responses in rodent models. We used the peripherally restricted FAAH inhibitor URB937, which has no access to the brain and spinal cord yet causes pronounced antihyperalgesic effects that are due to its ability to elevate anandamide levels outside the brain and spinal cord [28]. sEH blockade was achieved employing the potent inhibitor 1-trifluoromethoxyphenyl-3-(1-propionylpiperidine-4-yl)urea (TPPU) [29]. As experimental models, we utilized a mouse model of acute inflammation (carrageenan) and a rat model of neuropathy (insulin-dependent diabetes evoked by streptozocin).
2. Materials and methods
2.1 Animals
We used male CD1 mice (25 to 30 g; Charles River, Calco, Italy) and Sprague–Dawley male rats (250 to 300 g; Charles River, Wilmington, MA, USA). All procedures performed in Italy were in accordance with the Ethical Guidelines of the International Association for the Study of Pain, Italian regulations on the protection of animals used for experimental and other scientific purposes (D.M. 116192), and European Economic Community regulations (O.J. of E.C. L 358/1 12/18/1986). Procedures and animal care performed at the University of California, Davis adhered to the guidelines of the National Institutes of Health guide for the care and use of Laboratory animals and were performed in accordance with the protocols approved by the Animal Use and Care Committee (IACUC) of the University of California, Davis. Great care was taken to minimize suffering of the animals and to reduce the number of animals used. Mice were housed in groups of 5 in ventilated cages containing autoclaved cellulose paper as nesting material and rats were housed 2 per cage on corncob bedding all with free access to food and water. They were maintained under a 12 h light/dark cycle (lights on at 08:00 a.m.), at controlled temperature (21 ± 1 °C) and relative humidity (55 ± 10%). The animals were randomly divided in groups of 6. Behavioral testing was performed between 9:00 a.m. and 5:00 p.m. Scientists running the experiments were not aware of the treatment protocol at the time of the test (blind procedure).
2.2 Chemicals
λ-Carrageenan, PEG 400 and TWEEN 80, were purchased from Sigma–Aldrich (Milan, Italy). The sEH inhibitors TPPU, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea, t-TUCB, trans-4-[4-(3-trifluoromethoxyphenyl-l-ureido)-cyclohexyloxy]-benzoic acid and the FAAH inhibitor URB937,3-(3-carbamoylphenyl)-4-hydroxy-phenyl] N-cyclohexylcarbamate, were synthesized and characterized in house as previously described [30–32]. URB937 was prepared immediately before use in 80% sterile saline solution/10% PEG 400/10% Tween 80 and orally administered to mice in a volume of 2.5 mL kg−1. TPPU, t-TUCB and URB937 administered to rats were prepared immediately before use in PEG 400 and intraperitoneally administered in a volume of 1 mL kg−1.
2.3 Carrageenan-induced inflammation
We induced paw edema by injecting λ-carrageenan (1% weight/vol in sterile water, 50 μL) into the left hind paw of lightly restrained adult male CD1 mice. Edema was measured with a plethysmometer (Ugo Basile, Comerio, Italy). Fresh inhibitor solutions were prepared immediately before use in 80% sterile saline solution/10% PEG-400/10% Tween 80 and orally administered in a volume of 2.5 mL kg−1. All experiments were performed in a quiet room, and experimenters were blinded to the treatment protocol at the time of the test. Mechanical hyperalgesia was evaluated by measuring the latency (in s) to withdraw the paw from a constant mechanical pressure exerted onto the dorsal surface [31]. A 15-g calibrated glass cylinder (diameter = 10 mm) drawn to conical point (diameter = 3 mm) was used to exert the mechanical force. The weight was suspended vertically between two rings attached to a stand and was free to move vertically. A cutoff time of 180 s was used. Heat hyperalgesia was assessed by the method of Hargreaves et al. [33], measuring the latency to withdraw the hind paw from a focused beam of radiant heat (thermal intensity: infrared 3.0) applied to the plantar surface in a plantar test apparatus (Ugo Basile). The cutoff time was set at 30 s.
2.4 Diabetic neuropathic pain model
Diabetic neuropathic pain was modeled using streptozocin, an antibiotic that selectively destroys pancreatic beta cells. The decrease in mechanical withdrawal thresholds (MWTs) measured with the von Frey assay (IITC, Woodland Hills, CA) develops within 5–7 days after streptozocin administration and persists for the lifetime of the animal. Baseline naive MWTs were established prior to induction of diabetes. For this, rats were acclimated to the laboratory and apparatus for 1 h and MWT were probed at least three times at 1-min intervals; the scores averaged per animal. After naïve MWTs were assessed, streptozocin (55 mg kg−1) in saline was injected via the tail vein as previously reported [34]. After one week, the MWTs of diabetic rats were measured and allodynia (a painful response to innocuous stimulation) was confirmed. Rats that scored 65% or lower of the original naïve baseline were considered allodynic and included in the experimental groups. For the bioassay, rats were assessed for their diabetic neuropathic MWTs and then injected intraperitoneally (i.p.) with TPPU or URB937 and MWT were measured 30, 60, 90, 120, 150, and 180 min after the injection. The procedure was followed for all doses (0.03, 0.1, 0.3, 1, 3 mg kg−1) of TPPU, 3 mg kg−1 t-TUCB and (0.03, 0.1, 0.3, 1, 3 mg kg−1) URB937. The diabetic neuropathic pain baselines and subsequent MWTs are reported as grams of force needed to elicit hind paw withdrawal.
2.5 Isobolographic analyses
The interaction between TPPU and URB937 was characterized by isobolographic analysis assuming that the combinations were constituted by equally effective doses of the individual drugs. Thus, from the dose–response curves of each individual agent, the median effective dose (ED50) was determined at time point of 4 hours in the carrageenan model and 1 hour in the diabetic neuropathic pain model. The ED50 were plotted on the x- and y-axes. Subsequently, dose–response curves were obtained by concurrent delivery of both drugs (URB937 and TPPU) in fixed ratios, based on the ED50 values of each individual agent in the four different measurements. To construct this curve, groups of animals received one dose of one of the following combinations: (1) (URB937 ED50 + TPPU ED50); (2) (URB937 ED50 + TPPU ED50)/2; (3) (URB937 ED50 + TPPU ED50)/4; or (4) (URB937 ED50 + TPPU ED50)/8. The experimental ED50 value for the combination was calculated from each curve at time point of 4 hours in the carrageenan model and 1 hour in the diabetic neuropathic pain model. The theoretical additive ED50 was estimated from the dose–response curve of each drug administered individually, which presupposes that the observed effect of the combination is the sum of the effects of each individual drug. This theoretical ED50 value was then compared with the experimentally derived ED50 value to determine if there was a statistically significant difference [35–37]. Drug combinations producing pharmacological synergism with potentiation generate experimental points that fall below the line joining the two axes corresponding to the theoretical additive line (isobole). Conversely, drug combinations producing antagonism generate experimental points that fall above the isobole.
2.6 In vitro inhibition assay
For the recombinant human (hsEH), mouse (msEH) and rat (rsEH) sEH, the in vitro IC50 values were determined using a previously reported fluorescence method using cyano(2-methoxynaphthalen-6-yl)methyl(3-phenyloxiran-2-yl)methyl carbonate (CMNPC) as substrate [38]. The recombinant sEHs were incubated with the inhibitors for 5 min in 100 mM sodium phosphate buffer (200 μL; pH 7.4) at 30 °C before fluorescent substrate (CMNPC) introduction ([S] = 5 μM). The rates of formation of the fluorescent product were assessed and were linear for the duration of the assay. It has been previously demonstrated that the sEHI IC50 values obtained with the fluorescence assay correlate extremely well (linear correlation coefficient R2=0.9) with the natural substrate (14,15 EET per a LC–MS method)[39]. For the recombinant human FAAH (hFAAH), N-(6-methoxypyridin-3-yl) octanamide ([S]= 50 μM) was used as substrate as previously described [40]. The enzyme was incubated in sodium phosphate buffer (0.1 M pH 8.0) containing 0.1 mg/mL of BSA for 5 min with the inhibitor before substrate introduction. The activity was followed kinetically for 10 min at 30C by following the appearance of the fluorescent product. The 2-AG-activity was measured in rat brain microsomes using a colorimetric assay as previously described [41].
2.7 Statistical analyses
Results are expressed as the mean ± SEM, or 95% confidence limits (95% CL). Effective doses were determined by linear regression analysis of dose–response curves. Individual slopes of the dose–response curves were compared by Student’s t-test, according to the test of parallelism, and isobolographic analyses were performed using the Prism software (GraphPad Software, San Diego, CA). The data from mechanical and heat hyperalgesia and mechanical allodynia were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons.
3. Results
3.1 Antihyperalgesic effects of TPPU, URB937 and synergy in a model of acute inflammation
To evaluate the antihyperalgesic activity of TPPU, which has not been previously reported, we tested the compound in the carrageenan model of acute inflammation in CD1 mice. Oral administration of the compound (0.1–10 mg kg−1) produced a dose-dependent and persistent suppression of carrageenan-induced edema (Fig. 1A). When TPPU was administered at its highest dosage (10 mg kg−1), the effect was still statistically detectable 24 hours after application (Fig. 1A, P< .001). The median effective dose (ED50) for TPPU was 0.3 mg kg−1 (CL 95% = 0.0087–0.13 mg kg−1). The CD1 mouse model was used previously to evaluate antihyperalgesic effects of FAAH inhibitors including URB937, whose ED50 on edema was 0.5 mg kg−1 (Fig. 1B) (CL 95% = 0.038–0.47 mg kg−1) [31]. The sEH inhibitor and FAAH inhibitor were also effective against mechanical hyperalgesia (Fig. 1C–D), and heat hyperalgesia (Fig. 1E–F). On mechanical hyperalgesia, the ED50 value for TPPU was 1 mg kg−1 (CL 95% = 0.032–0.55 mg kg−1) and for URB937 was 0.8 mg kg−1 (CL 95% = 0.021–0.43 mg kg−1); on heat hyperalgesia the ED50 for TPPU was 0.5 mg kg−1 (CL 95% = 0.049–0.51 mg kg−1) and for URB937 was 0.2 mg kg−1 (CL 95% = 0.058–0.46 mg kg−1). To assess possible anti-hyperalgesic synergy with coadministration of sEH and FAAH inhibitors, we investigated the effects of combinations of TPPU plus URB937 (Fig. 2). Co-administration of TPPU and URB937 in four oral fixed ratios resulted in dose- and time-dependent anti-inflammatory effects in the carrageenan model (Fig. 2C and E). The isobolographic analysis of the data supported that TPPU and URB937 acted synergistically against both types of hyperalgesia (Fig. 2D and F). The results suggest that TPPU prevents both edema and the development of acute pain responses evoked by carrageenan in mice. Additionally, TPPU and URB937 act synergistically to attenuate acute pain-related responses evoked by carrageenan.
Figure 1.

TPPU and URB937 show antiedematogenic and antihyperalgesic effects in a carrageenan model of acute inflammation in mice. (A) TPPU (0.1–10 mg kg−1, oral) and (B) URB937 (0.1–3 mg kg−1, oral) produced a robust reduction in paw volume. The two inhibitors reduced mechanical (C–D) and heat (E–F) hyperalgesia. The compounds were administered orally before intraplantar injection of carrageenan. Paw volume, mechanical and heat hyperalgesia were measured before (0 h) or 2, 4, 6, 24 and 48 h after TPPU (0.3–10 mg kg−1) and URB937 (0.1–3 mg kg−1) administration and were significantly different compared to vehicle treated groups. Results are expressed as mean ± SEM (n=6, each group). The data were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. * p<0.05, ** p< 0.01 and *** p<0.001 vs. vehicle.
Figure 2.

TPPU synergizes with URB937 in reducing paw volume, mechanical and heat hyperalgesia. (A, C and E) Combinations in fixed ratios of TPPU with URB937 produce a decrease of paw volume and a reduction of mechanical and heat hyperalgesia in dose- and time-dependent manner. Paw volume, mechanical and heat hyperalgesia were measured immediately before (0 h) or 2, 4, 6, 24 and 48 h after the combination. (B, D and F) The synergy was supported by an isobolographic analysis that revealed a substantial difference in the experimental value below the isobole which indicates pharmacological synergism with potentiation. Results are expressed as mean ± SEM (n=6, each group). The data were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. * P<0.05, ** P< 0.01 and *** P<0.001 vs. vehicle.
3.2 Inhibitor efficacy and synergy in a model of chronic neuropathic pain
We next tested whether TPPU and URB937 are able to alleviate the mechanical allodynia associated with a diabetic neuropathic pain model. We produced peripheral neuropathy in rats using streptozocin, which results in the development of allodynia (Fig. 3). We administered TPPU (0.03–3 mg kg−1) intraperitoneally to diabetic rats and measured mechanical allodynia. As shown in Fig. 3A, a single administration of TPPU was sufficient to cause a rapid reversal of established allodynia (P < 0.001 for 3 mg kg−1). The TPPU ED50 was 0.1 mg kg−1 (CL 95% = 0.053–0.11 mg kg−1). Figure 3B shows that the peripheral FAAH inhibitor URB937 also inhibited mechanical allodynia in diabetic rats, with an ED50 of 0.3 mg kg−1 (CL 95% = 0.064–0.43 mg kg−1). The average naïve baseline for groups that were induced for the diabetic model was 78.6±0.9 SEM grams to elicit a withdrawal. This value declined with the diabetic neuropathy to a diabetic baseline of 45.2±0.7 SEM. However with the 3 mg kg−1 i.p. dose of TPPU the pre diabetic average was met at the 60 and 90 minute time points (77.7±1.1 and 79.4±2.7 grams respectively). Lower doses of TPPU were significant in elevating thresholds over the diabetic baselines, though did not meet naïve baselines. The effects of TPPU and URB937 (3 mg kg−1) were similar to those of another sEH inhibitor, t-TUCB (3 mg kg−1i.p., Fig. 3C), which previously demonstrated efficacy in the diabetic neuropathy model [42]. Thus, two structurally distinct sEH inhibitors produced potent antihyperalgesia in this chronic pain model. Interestingly, the duration of action of t-TUCB was greater than those of TPPU or URB937 (Fig. 3C). However, because of the in vitro selectivity of the individual inhibitors on sEH and FAAH (Table I), TPPU was selected for co-administration with the FAAH inhibitor to probe for synergistic effects in vivo. When TPPU and URB937 were administered together, the combination displayed improved efficacy over the time course of the diabetic neuropathy model (Fig. 4A). An isobolographic analysis of the data shows that TPPU and URB937 were robustly synergistic (Fig. 4B).
Figure 3.

TPPU and URB937 in single administration reduce diabetic neuropathic pain in rat. (A) The TPPU dose dependent reduction of allodynia was highly significant in diabetic neuropathic rats (P ≤0.001 0.1–3 mg kg−1). (B) URB937 also significantly improved mechanical withdrawal thresholds in neuropathic rats (P ≤0.05 0.1 mg kg−1, P ≤0.001 0.3–3 mg kg−1). The average naïve baseline for groups that were induced for the diabetic model was 78.6±0.9 SEM grams of force to elicit a withdrawal. This value declined with the diabetic neuropathy to a diabetic baseline of 45.2±0.7 SEM. The withdrawal data were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. (C) The 3 mg-kg−1 doses of TPPU and URB937 are compared with the sEH inhibitor t-TUCB. The sEH inhibition showed similar efficacy at the same dose with two structurally diverse small molecule inhibitors in the chronic pain model. Results are expressed as mean ± SEM (n=6, each group). The data were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. * P<0.05, ** P< 0.01 and *** P<0.001 vs. vehicle.
Table I.
Comparative in vitro efficacy of FAAH and sEH inhibitors
| Structure | hFAAH | hsEH | msEH | rsEH | 2-AG hydrolysis | |
|---|---|---|---|---|---|---|
|
| ||||||
| IC50(nM) | ||||||
| FAAHI |
URB937 |
17 ± 4 | 7,180 ± 550 | 17,800 ± 1,200 | 16,600 ± 1,200 | >100,000 |
| sEHI |
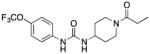 TPPU |
6,610 ± 880 | 0.9 ± 0.2 | 7.4 ± 1.0 | 4.6 ± 0.4 | >100,000 |
|
t-TUCB |
260 ± 80 | 0.4 ± 0.1 | 3.1 ± 0.8 | 11.9 ± 1.8 | >100,000 | |
The IC50 of URB937 on the FAAH enzyme was previously determined to be 26.8 ± 4.9 nM in rat. The median inhibitory activity of URB937 on non-target monoacylglycerol lipase (MAGL) was >100 nM in mice [28]. The results presented in the table are (n=3) averages ±standard deviation with differences of 2-fold or greater considered significant [40].
Figure 4.
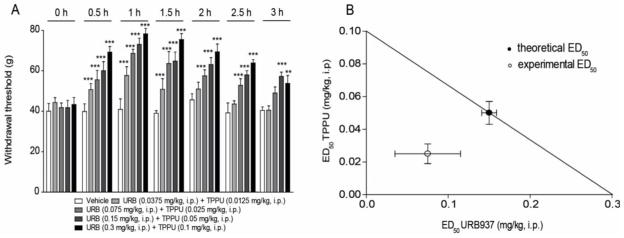
TPPU synergizes with URB937 against diabetic neuropathic pain in rat. (A) The combinations of URB937 and TPPU effectively blocked allodynia in the diabetic neuropathy model. The combination of the inhibitors in one i.p. administration was more efficacious (P ≤0.001) than vehicle over several hours. (B) The isobolographic analysis of the URB937 and TPPU combination supports the efficacy is synergistic against chronic neuropathic pain. Filled circles represent the theoretical ED50 with 95 % confidence limits (CL). Open circles the experimental ED50 with 95 % CL. Results are expressed as mean ± SEM (n=6, each group). These groups are included in the average scores for pre-diabetic (78.6±0.9 SEM grams) and pretreatment diabetic (45.2±0.7 SEM grams) baselines. Results are expressed as mean ± SEM (n=6, each group). The data were compared using two-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. ** P< 0.01 and *** P<0.001 vs. vehicle.
4. Discussion
In this study we determined the effect of combining a FAAH inhibitor with a sEH inhibitor in two rodent models of pain: carrageenan-induced hyperalgesia in mice and streptozocin-induced allodynia in rats. Our rationale was to enhance the concentration and thus the effective activity of the lipid mediators that are degraded by these enzymes (namely, FAE by FAAH and EpFAs by sEH), which are known to produce analgesic effects through independent mechanisms. Thus, we hypothesize that these mediators are synergistic at the physiological level. The results show that FAAH and sEH inhibitors are individually active, as previously demonstrated [31,42], and that their activity is synergistically enhanced by co-administration. This synergy may be exploited therapeutically to achieve better pain control at lower drug dosages or to develop dual agents that simultaneously inhibit both FAAH and sEH activities.
sEH inhibitors (sEHI) such as t-TUCB have demonstrated anti-hyperalgesic activity in the streptozocin model, both in rats and mice, and in clinical equine laminitis, a chronic neuropathy with inflammatory origins [42–44]. In the present study we tested the compound TPPU, which had not been tested in pain models before, for several reasons: (1) it is highly potent (Ki = 0.91 nM) [45] and selective for sEH over FAAH (Table 1); (2) it has improved water solubility (60 μg/ml) and lower melting point (198.2 C) compared to other sEHI; and (3) has a longer koff rate on recombinant human sEH, as assessed by FRET displacement [45]. TPPU is a competitive, tight-binding reversible inhibitor more potent than sEHI previously shown to elevate both plasma and tissue (including nervous tissue) levels of epoxy fatty acids with concurrent improvement of nociceptive thresholds [42,46]. Our results show that TPPU is highly effective in reducing mechanical and heat hyperalgesia evoked by carrageenan (Fig. 1), and attenuating allodynia in the rat streptozocin model (Fig. 3).
The kinetically irreversible peripheral FAAH inhibitor URB937 was previously tested in the mouse carrageenan model [31]. In those experiments, URB937 showed efficacy at 0.3 mg kg−1 in reducing paw volume and increasing thermal and mechanical paw withdrawal latencies. The same study also tested URB937 in a sciatic nerve constriction model of neuropathy, where the compound also successfully elevated withdrawal thresholds. Multiple FAAH inhibitors and specifically URB937 has been shown to elevate anandamide in vivo [28]. Here, the 0.3 mg kg−1 dose of URB937 attenuated allodynia in neuropathic rats (Fig. 3B). The efficacy of URB937 in the constriction injury model of neuropathy in mice was similar to the results in the diabetic neuropathy model in rat [31]. However, the duration of the effect of URB937 and TPPU may have been affected by the induced drug metabolizing enzyme activity in the streptozocin model [46].
The combination of URB937 and TPPU showed significant synergistic efficacy in the two pain models investigated here (Figures 2 and 4). sEH blockade is known to synergize with other inhibitors targeting the arachidonic acid cascade. The improved nociceptive outcome with co-administration of cyclooxygenase (COX) inhibitors was an early demonstration of this effect [47,48]. Administration of sEHI correlates with a decrease in COX-2 protein and mRNA expression levels [49,50]. In fact, combining indomethacin, a COX inhibitor, with either FAAH or sEH inhibitors revealed synergy in inflammatory pain models [31,47]. Given the demonstrated effect of COX in metabolizing endocannabinoids [51,52] and the potential for the prostamide metabolites of COX to increase neuronal excitability [53], it is possible that this mechanism contributed to the synergy of combined FAAH and sEH inhibitors. There has been recent exploration of selective COX-2 inhibitors that block prostamide production but leave prostaglandins intact [54]. However, the sEHI mediated down regulation of induced COX-2 mRNA data are from spinal cord whereas URB937 is peripherally restricted, so there is likely a combined beneficial effect on the nociceptive endpoints. Despite this, the sEH inhibition can alter COX-2 protein in liver [49], a major site of metabolism, and therefore there may be a COX-2 related mechanism when TPPU is combined with the peripherally restricted FAAH inhibitor. Future studies will investigate the benefits of a strategy to combine inhibitors of all three of these enzymes the FAAH, sEH and COX in either a polypharmacological approach or with a designed multiple ligand molecule.
Alternatively, recent studies have addressed possible crosstalk between the cannabinoid and cytochrome P450 pathways exemplified by the epoxygenation of endocannabinoids. In experiments using human liver microsomes arachidonoylethanolamide (anandamide) was transformed in vitro into all four possible regioisomers of epoxidized eicosatrienoic ethanolamide (EET-EA) metabolites [55]. These metabolites were further transformed by epoxide hydrolase into their corresponding diols. The EET-EA metabolites were also formed in human brain microsomal and mitochondrial preparations [56]. Biologically, it was found that the EET-EA metabolites had low nanomolar IC50 on the CB2 receptors and specifically 5,6-EET-EA was more stable in mouse brain and had a higher affinity at the CB2 receptor than anandamide. Use of a sEH inhibitor in this study demonstrated that sEH was primarily responsible for the degradation of the EET-EA product and not FAAH [57]. Additionally, Chen et al. identified two epoxygenated arachidonyl glycerol regioisomers, 2-11,12-EG and 2-14,15-EG, that were produced in vivo. Both regioisomers of 2-EG bind to CB1 and CB2 receptors with slightly improved potency over 2-AG [58]. Thus, there is a possibility that blocking the degradation of the FAE with FAAH inhibitors in combination with sEH inhibitors elevates several classes of metabolites that cooperate to elicit antihyperalgesic and antiinflammatory effects. In future experiments it will be important to define the site of action of the putative metabolites and their relative concentrations as well as the in vivo efficacy.
5. Conclusion
The antihyperalgesic effects of FAE and EpFA are evident with in vivo inhibition of their principal degrading enzymes: FAAH and sEH respectively. This activity is observable in two distinct models of pain including acute inflammatory and diabetic neuropathic pain employed here. Moreover, coadministration of these enzyme inhibitors results in a synergistic reduction in pain behavior. These results are suggestive of a crosstalk between these lipid-derived signaling pathways.
Acknowledgments
Research Funding: This study was partially supported by grants from the National Institutes on Drug Abuse (RO1-DA-012413, DP1DA031387 to D.P.). The work completed at the University of California Davis was supported by the National Institute of Environmental Health Sciences (NIEHS) Grant R01 ES002710, NIEHS Superfund Research Program P42 ES004699, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R21AR062866, National Institute of Neurological Disorders and Stroke (NINDS) U54 NS079202-01 and Grants NIEHS T32ES007059 and NIH 5T32DC008072-05 (to K.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. B.D.H. is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
Abbreviations
- BK
large-conductance potassium channels
- CB
cannabinoid receptor
- CL
confidence limits
- ED50
median effective dose
- EpFA
epoxidized fatty acids
- FAAH
fatty-acid amide hydrolase
- FAE
fatty acid ethanolamides
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- PPARα and PPARγ
peroxisome proliferator-activated receptors
- sEH
soluble epoxide hydrolase
Footnotes
Conflict of interest: D.P. is inventor in patent applications that describe peripheral FAAH inhibitors. The University of California holds patents on the sEH inhibitors used in this study as well as their use to treat inflammation, inflammatory pain, and neuropathic pain. BD Hammock and B Inceoglu are co-founders and K Wagner is an employee of EicOsis L.L.C., a startup company advancing sEH inhibitors into the clinic.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Piomelli D, Sasso O. Peripheral gating of pain signals by endogenous lipid mediators. Nature neuroscience. 2014;17:164–174. doi: 10.1038/nn.3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner K, Inceoglu B, Gill SS, Hammock BD. Epoxygenated fatty acids and soluble epoxide hydrolase inhibition: Novel mediators of pain reduction. J Agric Food Chem. 2011;59:2816–2824. doi: 10.1021/jf102559q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang G, Kodani S, Hammock BD. Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Progress in lipid research. 2014;53:108–123. doi: 10.1016/j.plipres.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annual Review of Biochemistry. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 5.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 6.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 7.Piomelli D. The molecular logic of endocannabinoid signalling. Nature reviews Neuroscience. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 8.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor ppar-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 9.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Molecular pharmacology. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 10.Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annual Review of Pharmacology and Toxicology. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decker M, Arand M, Cronin A. Mammalian epoxide hydrolases in xenobiotic metabolism and signalling. Arch Toxicol. 2009;83:297–318. doi: 10.1007/s00204-009-0416-0. [DOI] [PubMed] [Google Scholar]
- 12.Larsen BT, Miura H, Hatoum OA, Campbell WB, Hammock BD, Zeldin DC, Falck JR, Gutterman DD. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via bk(ca) channels: Implications for soluble epoxide hydrolase inhibition. American journal of physiology Heart and circulatory physiology. 2006;290:H491–499. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang X, Hu S, Watanabe T, Weintraub NL, Snyder GD, Yao J, Liu Y, Shyy JY, Hammock BD, Spector AA. Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. J Pharmacol Exp Ther. 2005;314:260–270. doi: 10.1124/jpet.105.085605. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: The role of ppargamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102:16747–16752. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate trpv4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 16.Earley S, Heppner TJ, Nelson MT, Brayden JE. Trpv4 forms a novel ca2+ signaling complex with ryanodine receptors and bkca channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- 17.Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R. Epoxyeicosatrienoic acids regulate trp channel dependent ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:2612–2618. doi: 10.1161/ATVBAHA.107.152074. [DOI] [PubMed] [Google Scholar]
- 18.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome p450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, Luria A, Hammock BD, Imig JD. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive goto-kakizaki rats. Clin Sci (Lond) 2009;116:61–70. doi: 10.1042/CS20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye D, Zhou W, Lu T, Jagadeesh SG, Falck JR, Lee HC. Mechanism of rat mesenteric arterial katp channel activation by 14,15-epoxyeicosatrienoic acid. Am J Physiol Heart Circ Physiol. 2006;290:H1326–1336. doi: 10.1152/ajpheart.00318.2005. [DOI] [PubMed] [Google Scholar]
- 21.Li PL, Campbell WB. Epoxyeicosatrienoic acids activate k+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ Res. 1997;80:877–884. doi: 10.1161/01.res.80.6.877. [DOI] [PubMed] [Google Scholar]
- 22.Spector AA, Kim H-Y. Cytochrome p450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2014 doi: 10.1016/j.bbalip.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG International union of pharmacology. Xxvii. Classification of cannabinoid receptors. Pharmacological Reviews. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 24.Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (eets) Prostaglandins Other Lipid Mediat. 2007;82:42–49. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guindon J, Hohmann AG. Cannabinoid cb2 receptors: A therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res. 2010;51:3481–3490. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner K, Vito S, Inceoglu B, Hammock BD. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins & Other Lipid Mediators. 2014 doi: 10.1016/j.prostaglandins.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature neuroscience. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sirish P, Li N, Liu JY, Lee KS, Hwang SH, Qiu H, Zhao C, Ma SM, Lopez JE, Hammock BD, Chiamvimonvat N. Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5618–5623. doi: 10.1073/pnas.1221972110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: Structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53:7067–7075. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanz G, Reggiani A, Piomelli D. Peripheral faah inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 34.Aley KO, Levine JD. Rapid onset pain induced by intravenous streptozotocin in the rat. J Pain. 2001;2:146–150. doi: 10.1054/jpai.2001.21592. [DOI] [PubMed] [Google Scholar]
- 35.Tallarida RJ. Drug synergism: Its detection and applications. J Pharmacol Exp Ther. 2001;298:865–872. [PubMed] [Google Scholar]
- 36.Tallarida RJ. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther. 2006;319:1–7. doi: 10.1124/jpet.106.104117. [DOI] [PubMed] [Google Scholar]
- 37.Tallarida RJ. Interactions between drugs and occupied receptors. Pharmacol Ther. 2007;113:197–209. doi: 10.1016/j.pharmthera.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem. 2005;343:66–75. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schebb NH, Huby M, Morisseau C, Hwang SH, Hammock BD. Development of an online spe-lc-ms-based assay using endogenous substrate for investigation of soluble epoxide hydrolase (seh) inhibitors. Analytical and bioanalytical chemistry. 2011;400:1359–1366. doi: 10.1007/s00216-011-4861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang H, Nishi K, Tsai H-J, Hammock BD. Development of highly sensitive fluorescent assays for fatty acid amide hydrolase. Analytical Biochemistry. 2007;363:12–21. doi: 10.1016/j.ab.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casida JE, Gulevich AG, Sarpong R, Bunnelle EM. S-arachidonoyl-2-thioglycerol synthesis and use for fluorimetric and colorimetric assays of monoacylglycerol lipase. Bioorganic & medicinal chemistry. 2010;18:1942–1947. doi: 10.1016/j.bmc.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 42.Wagner K, Inceoglu B, Dong H, Yang J, Hwang SH, Jones P, Morisseau C, Hammock BD. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. European Journal of Pharmacology. 2013;700:93–101. doi: 10.1016/j.ejphar.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inceoglu B, Wagner KM, Yang J, Bettaieb A, Schebb NH, Hwang SH, Morisseau C, Haj FG, Hammock BD. Acute augmentation of epoxygenated fatty acid levels rapidly reduces pain-related behavior in a rat model of type i diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11390–11395. doi: 10.1073/pnas.1208708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guedes AG, Morisseau C, Sole A, Soares JH, Ulu A, Dong H, Hammock BD. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet Anaesth Analg. 2013 doi: 10.1111/vaa.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee KS, Liu JY, Wagner KM, Pakhomova S, Dong H, Morisseau C, Fu SH, Yang J, Wang P, Ulu A, Mate CA, Nguyen LV, Hwang SH, Edin ML, Mara AA, Wulff H, Newcomer ME, Zeldin DC, Hammock BD. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. Journal of Medicinal Chemistry. 2014;57:7016–7030. doi: 10.1021/jm500694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inceoglu B, Wagner KM, Yang J, Bettaiebb A, Schebb NH, Hwang SH, Morisseau CHF, Hammock BD. Acute augmentation of epoxygenated fatty acid levels rapidly reduces pain-related behavior in a rat model of type i diabetes. Proceedings of the National Academy of Sciences of the United States of America; 2012. Available online June 25. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces lps-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79:2311–2319. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105:18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Telleria-Diaz A, Schmidt M, Kreusch S, Neubert AK, Schache F, Vazquez E, Vanegas H, Schaible HG, Ebersberger A. Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: Involvement of prostaglandins and endocannabinoids. Pain. 2010;148:26–35. doi: 10.1016/j.pain.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 52.Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: Novel targets for neuropathic and inflammatory pain. British journal of pharmacology. 2007;152:624–632. doi: 10.1038/sj.bjp.0707433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gatta L, Piscitelli F, Giordano C, Boccella S, Lichtman A, Maione S, Di Marzo V. Discovery of prostamide f2alpha and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. PLoS ONE. 2012;7:e31111. doi: 10.1371/journal.pone.0031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, Colbran RJ, Reese J, Marnett LJ, Patel S. Substrate-selective cox-2 inhibition decreases anxiety via endocannabinoid activation. Nature neuroscience. 2013;16:1291–1298. doi: 10.1038/nn.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snider NT, Kornilov AM, Kent UM, Hollenberg PF. Anandamide metabolism by human liver and kidney microsomal cytochrome p450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J Pharmacol Exp Ther. 2007;321:590–597. doi: 10.1124/jpet.107.119321. [DOI] [PubMed] [Google Scholar]
- 56.Snider NT, Sikora MJ, Sridar C, Feuerstein TJ, Rae JM, Hollenberg PF. The endocannabinoid anandamide is a substrate for the human polymorphic cytochrome p450 2d6. J Pharmacol Exp Ther. 2008;327:538–545. doi: 10.1124/jpet.108.141796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Snider NT, Nast JA, Tesmer LA, Hollenberg PF. A cytochrome p450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol Pharmacol. 2009;75:965–972. doi: 10.1124/mol.108.053439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen JK, Chen J, Imig JD, Wei S, Hachey DL, Guthi JS, Falck JR, Capdevila JH, Harris RC. Identification of novel endogenous cytochrome p450 arachidonate metabolites with high affinity for cannabinoid receptors. J Biol Chem. 2008 doi: 10.1074/jbc.M709873200. [DOI] [PMC free article] [PubMed] [Google Scholar]