

Abstract
The inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) type 2 (IP3R2) is an intracellular Ca2+-release channel located on the endoplasmic reticulum (ER). It displays in many cell types a predominantly perinuclear or even nuclear localization. IP3R2 is characterized by a high sensitivity to both IP3 and ATP and is biphasically regulated by Ca2+. Interestingly, ATP stimulates IP3R2 independently of the cytosolic [Ca2+]. Furthermore, IP3R2 is modulated by phosphorylation events mediated by e.g. protein kinase A, Ca2+/calmodulin-dependent kinase II and protein kinase C. In addition to its regulation by protein kinase A, IP3R2 forms a complex with adenylate cyclase 6 and is directly regulated by cAMP, thereby linking in a new way Ca2+-dependent and cAMP-dependent signalling. Finally, in the ER, IP3R2 is less mobile than the other IP3R isoforms, while its functional properties appear dominant in heterotetramers. These properties make the IP3R2 a Ca2+ channel with exquisite properties for setting up intracellular Ca2+ signals with unique characteristics. IP3R2 plays a crucial role in the function of secretory cell types (e.g. pancreatic acinar cells, hepatocytes, salivary gland, eccrine sweat gland). In cardiac myocytes, the role of IP3R2 appears more complex, because, together with IP3R1, it is needed for normal cardiogenesis, while its aberrant activity is implicated in cardiac hypertrophy and arrhythmias. Moreover, IP3R2 expression is driven by IP3-induced Ca2+ release leading to a self-perpetuating system of cardiac hypertrophy. Most importantly, its high sensitivity to IP3 makes IP3R2 a target for anti-apoptotic proteins (e.g. Bcl-2) in B-cell cancers. Disrupting IP3R/Bcl-2 interaction therefore leads in those cells to increased Ca2+ release and apoptosis. Intriguingly, IP3R2 is not only implicated in apoptosis but also in the induction of senescence, another tumour-suppressive mechanism. These results were the first to unravel the physiological and pathophysiological role of IP3R2 and we anticipate that further progress will soon be made in understanding the function of IP3R2 in various tissues and organs.
Keywords: Apoptosis, ATP, Ca2+, Cancer, IP3, Heart, Kinases, Liver, Pancreas, Secretion, Senescence
1. Introduction
The inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) are ubiquitously expressed intracellular Ca2+-release channels. These channels are tetrameric in structure and predominantly localized in the endoplasmic reticulum (ER). IP3, produced by phospholipase C after cell activation by hormones, growth factors or neurotransmitters, diffuses into the cytosol, binds to and activates the IP3R, leading to Ca2+ release from the ER. This Ca2+ release is instrumental in the formation of the spatio-temporal Ca2+ signals, fundamental for the regulation of multiple cellular processes, including proliferation, differentiation, metabolism, secretion, cell fate and memory [1-3].
In all vertebrate organisms, three different genes encode IP3Rs, leading to three main types of IP3Rs, IP3R1 (first fully cloned in 1989 [4]), IP3R2 (first fully cloned in 1991 [5]) and IP3R3 (first fully cloned in 1993 [6]). Each monomer is about 2700 amino acids in length and consequently has a predicted molecular mass of approximately 300 kDa. The IP3R proteins are structurally and functionally divided into 5 distinct domains: the N-terminal coupling domain (a.k.a. suppressor domain), the IP3-binding core, the internal coupling domain (a.k.a the modulatory and transducing domain), the channel domain and the C-terminal coupling domain (a.k.a. the gatekeeper domain) [7]. The three IP3R isoforms share only 60-80 % overall similarity at the amino acid level, but the similarity is much higher in certain defined regions (e.g. the IP3-binding site, the 5th and 6th transmembrane regions, putative ER-retention signals) while much lower in others, allowing for the existence of very distinct properties between the isoforms (see section 2). Finally, a further diversity can result from alternative splicing and the formation of heterotetramers [1, 8, 9].
The vast majority of cell types express more than one IP3R isoform but their relative proportion can be highly variable [10-14]. Moreover, in various cases it was shown that the relative expression levels depended on the differentiation or developmental state of the cells or could be modulated by specific treatments (e.g. [14-21]).
The participation of the IP3 Rs in establishing distinct patterns of Ca2+ signals resulting in different cellular outcomes depends therefore on the complement of the various IP3R (splice) isoforms expressed, their intracellular location, the presence of regulatory factors, including associated proteins, and their phosphorylation status [1, 14, 22-25].
On the basis of their almost exclusive expression of a single IP3R isoform, some cell types have been used as model system for the analysis of the role of the different IP3R isoforms [8]. However, only a rather limited set of cell types expresses predominantly IP3R2 (Table 1).
Table 1.
Cell types or tissues predominantly expressing IP3R2.
| Cell type/tissues (in alphabetical order) |
Relevant references | Specific remarks |
|---|---|---|
| AR42J | [11, 12] | Pancreatoma cell line |
| Cardiac myocytes | [12, 13, 136, 189] | |
| Glia | [117, 190] | |
| Hepatocytes | [11-13, 29] | Polarized expression IP3R2 |
| Intercalated cells of renal collecting duct |
[191] | |
| Pancreatic acinar cells | [12] | IP3R3 ≈ IP3R2 |
| RBL-2H3 | [11, 115, 192] | Mucosal mast cell line |
Interestingly, in many cells IP3R2 is expressed at a different subcellular location than the other IP3R isoforms. For example, in bovine aortic endothelial cells, bovine adrenal glomerulosa cells and COS-7 cells [26], the HepG2 liver cell line [27] and the hippocampal cell line HT22 [28] IP3R2 displayed a predominantly nuclear localization, while in hepatocytes IP3R2 is confined to the apical pole of the cell, near the canalicular membrane [29].
The nuclear localization of IP3R2 is particularly interesting with respect to the role of Ca2+ signalling in the nucleus, e.g. for gene transcription. Although the presence of IP3Rs in the inner leaflet of the nuclear envelope remains the subject of debate [30-32], there is at least strong evidence that a subset of the IP3R2 is facing the nucleoplasm in HepG2 cells [27], in SKHep1 cells [33] and in atrial myocytes [34] where they can control Ca2+ release directly into the nucleus.
Information concerning the regulation of IP3R2 expression is likewise still quite limited. The 5′-flanking region of murine IP3R2 has been sequenced and contained at least 7 transcription initiation sites with an upstream promoter containing no conventional TATA box but a GC box [35]. To the best of our knowledge, only two recent studies described pathways involved in the regulation of IP3R2 expression. First, in the heart, direct binding of nuclear factor of activated T cells (NFAT) c1 to the IP3R2 promoter drives IP3R2 expression [36]. Second, in dendritic cells, IP3R2 expression is controlled by the transcription factor ETS1, which itself depends on protein kinase B (AKT/PKB) 2 [37]. Finally, although not yet understood at the mechanistic level, in the HT22 cell line, oxidative stress leads to a specific upregulation of IP3R2 [38] (see section 5).
Splicing of IP3R2 is much less documented than that of IP3R1, but two different splice variants have been described. One appears to be muscle-specific and is limited to the N-terminal 175 amino acids of IP3R2 supplemented with 6 additional amino acids; although a regulatory role has been proposed, its function has not yet been elucidated. [39]. The second splice variant uses the same splice acceptor site and is lacking amino acids 176-208. The deletion is localized fully within the suppressor domain, which plays an important role in both the regulation of IP3 binding and in the coupling of the IP3R N-terminus to the channel region [40]. As a consequence, the resulting protein is defective in both IP3 binding and Ca2+ release. However, its expression in cells prevents the agonist-dependent clustering of the endogenous IP3Rs, probably via heterotetramerization, and can therefore impact intracellular Ca2+ signalling [41].
Finally, also at the protein level, IP3R2 levels appear to be regulated in a different manner when compared to the other isoforms. While all IP3R isoforms are downregulated under conditions of chronic stimulation [12, 42, 43], IP3R2 appeared the least susceptible [12].
In spite of the unique molecular properties displayed by IP3R2 (see sections 2.1-2.5), this protein is much less investigated than IP3R1 or indeed, even IP3R3. This apparent lack of progress was probably in retrospect multifaceted. The low general abundance of IP3R2 and the lack of good model systems for its investigation partially explain the fewer studies directed specifically towards the IP3R2. Additionally, but no less importantly, it appeared that it was historically difficult to make IP3R2 expression constructs. Moreover, the quality of the antibodies raised against IP3R2 was also generally poor. Finally, a recent analysis of classically used IP3R inhibitors demonstrated that IP3R2 expressed in DT40 triple knockout (DT40 TKO) chicken B lymphocytes was the least sensitive of all the IP3R isoforms to heparin, caffeine and 2-aminoethoxydiphenyl borate [44]. This latter observation may explain why pharmacological approaches to discern IP3R2 function have been largely unsuccessful.
Notwithstanding these issues, recent work has begun to unravel the significance of IP3R2 in a number of physiological settings. The aim of this review is therefore to highlight these important functions of IP3R2 and to so stimulate further research in the field.
2. Specific molecular and cellular properties of IP3R2
IP3R2 has a high sequence and structural homology with the other IP3R isoforms and consequently shares a large number of properties with IP3R1 and IP3R3. In this chapter, we will therefore focus on specific properties in which the IP3R2 clearly differs from the other isoforms (Figure 1).
Figure 1. Linear representation of the IP3R2 based on the human sequence demonstrating the interaction sites for its major regulators.
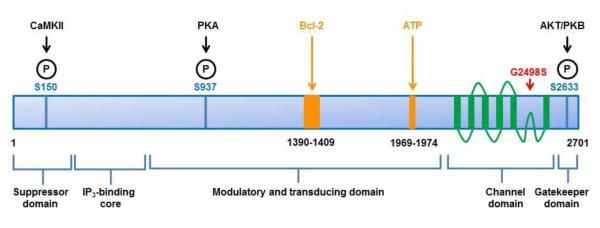
The IP3R2 is represented in blue; the 5 functional domains are indicated. In the channel domain, the 6 transmembrane helices as well as the connecting loops are depicted in green. The specific regulatory mechanisms discussed in the text are shown: identified phosphorylation sites are represented in dark blue, interaction sites for ATP and for Bcl-2 are in orange and the recently described Gly2498Ser mutation in the pore domain affecting IP3R2 function [126] is depicted in red.
2.1 IP3 affinity
Binding of IP3 to the IP3-binding core is the key step needed for the induction of IP3-induced Ca2+ release. As stated above (see section 1), the three IP3R isoforms all have a very similar structure [45]. For example, all IP3R isoforms contain their IP3-binding core towards their N-terminus, preceded by the so-called suppressor domain [46].
A striking property of the IP3R2 is its much higher affinity for IP3 when compared with the two other IP3R isoforms. This was first observed in IP3-binding experiments, which under various conditions demonstrated a rank-order of IP3 affinities IP3R2 > IP3R1 > IP3R3 [5, 10, 47]. Subsequent studies conclusively demonstrated that the IP3-binding cores of each of the IP3R isoforms display a similar affinity for IP3 (about 2 nM), but as demonstrated by the analysis of normal and chimeric N-terminal domains of the different IP3R isoforms the presence of the suppressor domain determines the specific IP3 affinity of the isoform (50, 14 and 163 nM for IP3R1, IP3R2 and IP3R3, respectively). These results underpin the importance of the suppressor domain for IP3R function [46].
The difference in IP3 affinity is also reflected in functional experiments. Analysis of IP3-induced Ca2+ release in DT40 TKO cells expressing a single IP3R isoform demonstrated that DT40 TKO cells heterologously expressing a single IP3R isoform could sustain Ca2+ oscillations for an extended period after stimulation by an anti B-cell receptor antibody only if the expressed isoform was IP3R2 [48]. Very similar results were obtained in vascular myocytes, whereby only the cells expressing IP3R2 in addition to IP3R1 displayed a Ca2+ oscillation pattern [49, 50]. Comparison of native IP3R1 (from cerebellum) and IP3R2 (from heart) [51] or the comparative analysis of each of the three IP3R isoforms heterologously expressed in Sf9 insect cells [52] also confirmed the rank-order of the sensitivity of channel opening towards IP3 as IP3R2 > IP3R1 > IP3R3, when measured after incorporation in planar lipid bilayers.
It must however be pointed out that a number of studies found a rank-order of affinities that is different from that mentioned above [53, 54]. Although this discrepancy has never been fully clarified, it can be assumed that variability in the experimental conditions (e.g. pH, [Ca2+]) as well as in the state of the IP3Rs (e.g. redox state, phosphorylation state, associated proteins, existence of heterotetramers) could explain this variability [1].
Taken together, most of the available evidence points to IP3R2 as being the most sensitive IP3R isoform. This observation raises the intriguing potential that IP3R2 can be active in the presence of basal, resting IP3 levels.
2.2. Regulation by cytosolic Ca2+
It has long been recognized that IP3R activity can be biphasically regulated by cytosolic Ca2+, meaning that a relatively low [Ca2+] (usually less than 0.3 μM) potentiate IP3-induced Ca2+ release, while higher [Ca2+] lead to an inhibition of the IP3R. Plotting IP3R activity against [Ca2+] therefore leads to a typical bell-shaped curve. The original observations were obtained in smooth muscle [55], neurons [56, 57] and oocytes [58], all tissues later shown to be particularly rich in IP3R1. It was therefore a long standing question whether this property was uniquely related to this isoform or whether IP3R2 and IP3R3 shared this property.
Although the stimulatory effect of Ca2+ on IP3R2 (and IP3R3) activity has never been in doubt, the inhibitory action of a high [Ca2+] was not always clearly observed. The group of Mignery published single-channel data demonstrating that both IP3R2 endogenously expressed in heart [51] or recombinantly expressed IP3R2 [59] displayed a much broader bell-shaped dependence towards Ca2+, meaning that on the one hand the stimulatory phase starts at a much lower [Ca2+] than for IP3R1, and on the other hand that IP3R2 remains active at a [Ca2+] already fully inhibiting IP3R1. In contrast to these reports, the group of Bezprozvanny found quite similar (and narrow) [Ca2+] response curves for each of the three IP3R isoforms heterologously expressed in Sf9 insect cells and investigated in planar lipid bilayers [60].
As will be further explained below (see section 2.3), the sensitivity of the IP3R2 towards Ca2+ is not dependent on the presence of ATP, but the latter will increase the likelihood of IP3R2 being in an open state at all [Ca2+] (Figure 2).
Figure 2. Modulation of IP3R2 single-channel activity by IP3, Ca2+ and ATP.

Representative single channel recordings of IP3R2 expressed in DT40 TKO cells using the “on-nucleus” configuration of the patch-clamp technique. In A, channel activity was stimulated with a maximal [IP3] (10 μM) at the indicated [Ca2+] and [ATP] (10 μM, in blue; 5 mM, in black). The pooled data in B reveal that channel activity stimulated by maximal [IP3] is modulated by [Ca2+] in a biphasic manner and that this relationship is unaffected by increasing the [ATP]. In C, channel activity was stimulated with a sub-maximal [IP3] (1 μM) at the indicated [Ca2+] and [ATP] (10 μM, in blue; 5 mM, in black). The pooled data in D demonstrate that while channel activity is also biphasically regulated by [Ca2+] at sub-maximal [IP3], the maximally achievable open probability, at each [Ca2+], is, in contrast to what happens at a maximal [IP3], markedly potentiated in the presence of a high [ATP]. Modified from [79], with permission.
Taken together, these results indicate that IP3R2, similarly to the other IP3R isoforms, is regulated in a biphasic way by the cytosolic [Ca2+], though that depending on the exact state of the receptor, different sensitivities to Ca2+ can be observed.
2.3. Regulation by ATP
2.3.1. Regulation of IP3R2 is distinct from other IP3Rs
Adenine nucleotides were recognized by early studies [61-68] as important regulators of IP3-induced Ca2+ release, raising the attractive possibility that channel activity could be fine-tuned to match the metabolic status of the cell. The diversity of cell types in which ATP modulates IP3-induced Ca2+ release is consistent with a regulation affecting all IP3R family members. However, two initial studies reported that in contrast to IP3R1 and IP3R3, IP3R2 was not subject to modulation by adenine nucleotides [48, 52]. Specifically, studying individual mammalian isoforms reconstituted in planar lipid bilayers, Bezprozvanny and colleagues reported that under conditions optimal for channel activity IP3R2 had no requirement for ATP [52]. This observation was independently confirmed studying Ca2+ release from DT40 cells expressing a single IP3R isoform following genetic ablation of the other family members [48]. These reports are, in hindsight, important because they provide the first indication that ATP regulation of IP3R2 was distinct from the other family members. A subsequent detailed analysis of individual mammalian isoforms expressed in the DT40 TKO IP3R null background confirmed these earlier reports [69]. IP3R2 was indeed, in contrast to other IP3R family members, insensitive to ATP at maximal [IP3]. Nevertheless, it was demonstrated that IP3R2 activity, measured as Ca2+ release, or at the single channel level in “on-nucleus” patch clamp recordings, was markedly enhanced at sub-saturating [IP3] [69]. Moreover, the sensitivity of ATP regulation of IP3-induced Ca2+ release also differed between individual isoforms under identical conditions with IP3R2 being strikingly more sensitive than IP3R1 or IP3R3 (EC50 40 μM, 100 μM and 500 μM for IP3R2, IP3R1 and IP3R3 respectively) [69, 70]. An issue posed by these data is the concentration range of ATP that might be expected to dynamically regulate IP3R activity. In turn, this raises the question whether modulation occurs at physiological levels of nucleotides or is only relevant under pathological conditions when ATP is depleted. Given the cellular levels of MgATP (~1 mM) and “free” ATP3− and ATP4− (10-100 μM) the answer is fundamentally dependent on the “species” of ATP that regulates IP3R channel activity. Several studies have addressed this issue and have reached disparate conclusions [67, 71-73] and thus this important issue as well as the consequences of the high functional affinity of IP3R2 remains to be resolved.
2.3.2. Putative peptide motifs in IP3R2 responsible for ATP regulation
Modulation of IP3R activity is widely believed to occur by ATP binding to glycine-rich domains (Gly-Xaa-Gly-Xaa-Xaa-Gly), reminiscent of Walker type A repeats, present in a number of proteins that utilize ATP in a catalytic manner [74-77]. Consistent with this idea, a number of studies using either photo-affinity or fluorescent ATP probes have demonstrated binding to regions of IP3R or glutathione S-transferase-recombinant fragments harbouring these putative recognition sites [69, 75-77]. The primary sequence of IP3R2 contains one such motif, Gly-Leu-Gly-Leu-Leu-Gly, spanning amino acids 1969-1974, which has been termed the “ATPB” site (Figure 1). Mutagenesis of three Gly residues to Ala in the motif eliminated binding of ATP and nucleotide regulation of Ca2+ release, confirming the functional importance of the ATPB site in IP3R2 [69]. Moreover, in cells expressing IP3R2 with an ATP binding-deficient ATPB motif, the frequency and amplitude of B cell receptor-activated Ca2+ oscillations were markedly reduced compared with wild-type IP3R2, suggesting strongly that nucleotide regulation of Ca2+ release is at least, constitutively required to shape cytosolic Ca2+ signals at physiologically relevant ATP levels [69]. Unexpectedly, mutations of all known Walker A motifs in IP3R1 and IP3R3 failed to abrogate nucleotide modulation [70]. The somewhat surprising conclusion is therefore, that ATP regulation of IP3R1 and IP3R3 is independent of known ATP-binding motifs, and thus the identity of molecular sites of nucleotide regulation in these IP3R remains to be elucidated. Consequently, the ATPB site in IP3R2 is unique as the only molecular locus for regulation of IP3R family members by adenine nucleotides that is defined unequivocally.
2.3.3. Mechanism of ATP regulation of IP3R2
Several studies have investigated the biophysical basis for ATP regulation of IP3R channel activity. In accordance with the singular features of IP3R2 when compared with other family members, it also appears that ATP regulates IP3R2 in a similarly distinctive manner. Using “on-nucleus” patch clamp single channel recordings of both endogenous Xenopus IP3R1 or rat IP3R1 expressed in DT40 TKO cells, elevating ATP levels increased the channel open probability (Po) by modulating the sensitivity of the channel to both activating and inhibitory [Ca2+], essentially left-shifting the bell-shaped [Ca2+] versus Po relationship at a given [IP3] [78, 79].
In contrast, while IP3R2 displays an identical biphasic Ca2+ sensitivity when exposed to saturating [IP3] (conditions in which IP3R2 is insensitive to ATP), at low [IP3], the Ca2+ sensitivity of mouse IP3R2 was not altered by increasing ATP [79]. Elevating ATP simply dramatically enhanced Po, resulting in a marked increase in activity. A detailed kinetic analysis of the channel gating also indicated that IP3R2 displayed “bursting” activity with properties distinct from IP3R1 (Figure 2). Specifically, with elevated ATP, the number of bursting episodes of relatively constant duration was increased, while IP3R1 bursts simply lengthened in time. By analogy to a gear change in a car, we have termed this the transition from ‘park’ into a ‘drive’ mode. A minimal scheme to describe the channel kinetics at sub-saturating [IP3] suggests that both channels transition between single open and closed states during drive mode with relatively constant kinetics and then are “parked” in a longer-lived closed state in the interburst intervals [79]. In the case of the IP3R1, increasing Ca2+ and ATP facilitates bursting by facilitating both the transition out of the parked state and also by decreasing the likelihood it will return to this state. In contrast, [Ca2+] does not influence the time the IP3R2 spends in drive mode but simply destabilizes the parked state to initiate activity (Figure 3). Increasing ATP then appears to markedly increase overall channel Po by prominently decreasing the amount of time in the parked state [79]. This unique property results in dissociation of the modulation of IP3R2 activity by ATP from the [Ca2+] in its immediate environment and likely allows added flexibility for tuning Ca2+ signals to the needs of the cell.
Figure 3. “Park and Drive” model for IP3R1 and IP3R2 gating.

An increase in IP3R1 (A) and IP3R2 (B) channel activity in the presence of activating ligands is characterized by an increase in channel “bursting” without altering the intraburst kinetics. The bursts have subtype specific characteristics. A gating scheme for both channels can minimally be described by three states; one open state (in green) and two closed states (in red). Bursting activity is represented by rapid transitions between the open state (O) and a short-lived closed state (C1) representing the “Drive Mode” of the channel. In the interburst intervals, the channel is effectively “Parked” in a long-lived closed state (C2). For both IP3R1 (A) and IP3R2 (B) increasing the concentrations of activating ligands solely alters the transition from C2 to C1. However, ligands both increase the likelihood that IP3R1 will leave the parked state to drive mode, as well as reciprocally decreasing the chances it will return to this state, thus extending the period of bursting (A). In the case of IP3R2, ligands only destabilize the parked state resulting in an increase in bursting episodes of relatively constant duration (B).
2.3.4. Dominance of IP3R2 ATP regulatory characteristics
An important question exists as to how the distinct features of individual IP3R subtypes are reflected in the overall characteristics of Ca2+ release from heterotetrameric channels. Specifically, are the properties simply a blended integration of the individual subtypes or can a particular subtype dominate the overall characteristics? Studies investigating ATP regulation of Ca2+ release in cells expressing multiple IP3R isoforms indicate that the latter possibility occurs, specifically when IP3R2 is expressed. For example, the characteristics of ATP regulation of IP3R2 (albeit the lack of regulation at saturating [IP3]) were observed in DT40 cells engineered to express only IP3R2, or in cells expressing both IP3R2 and IP3R1 or IP3R3 [48]. Similarly, in salivary and pancreatic acinar cells [80, 81], which natively express IP3R2 and IP3R3 to approximately equal extents, the features of ATP regulation precisely match those documented for IP3R2 stably expressed in isolation in either DT40 TKO [69] or in AR42J pancreatoma [80] cells (i.e. absence of regulation at saturating [IP3] and EC50 for ATP ~40 μM). Notably, similar experiments in pancreatic and parotid acinar cells prepared from IP3R2 null animals revealed identical properties to IP3R3 (i.e. regulation at all [IP3] and EC50 for ATP ~500 μM) [80, 81]. Conversely, “rescue” experiments ectopically expressing IP3R2 in RINm5F insulinoma cells which predominately express IP3R3, converted IP3R3 characteristics to IP3R2 [80]. While these data clearly indicate the dominant influence of IP3R2 expression and are consistent with this occurring as a function of heterotetramer formation, these data could also formally be explained by an intermolecular interaction between clusters of homotetrameric IP3R. This issue has recently been tackled by generating tetrameric IP3R from concatenated IP3R dimers connected by short flexible linkers [82]. Expression of dimers results in the assembly of tetramers where the subunit composition can be unequivocally defined. Expression of dimers of IP3R1 or IP3R2 exhibited the distinctive properties of ATP regulation typical of channels assembled from their respective monomeric parent subtype. Remarkably when heterodimers of IP3R1 and IP3R2 were expressed, resulting in assembly of channels consisting of equal numbers of IP3R1 and IP3R2 subunits, ATP regulation was indistinguishable from IP3R2, thus recapitulating the dominant effects seen in cells expressing native receptors [82]. These data indicate that IP3R2 in the context of a heterotetrameric channel exerts a dominant influence. Further work is needed to establish the number of monomers of IP3R2 necessary to exert this influence and whether IP3R2 similarly is the principle monomer that dictates the overall channel properties when subjected to other forms of regulation.
2.4. Regulation by phosphorylation
Like for many other ion channels, phosphorylation/dephosphorylation reactions provide a versatile, reversible form of acute regulation of IP3R activity. IP3Rs have been shown to be biochemical substrates for numerous families of serine/threonine and tyrosine directed kinases. In a more limited number of cases, a comprehensive documentation of the phosphorylation event, including location of the substrate motif and the subsequent functional consequences have been detailed. These studies have largely focused on IP3R1 as a template. With some notable exceptions, for example the AKT/PKB site conserved in the C-termini of each IP3R [83, 84], the amino acid motifs subject to phosphorylation events are not generally preserved between IP3R subtypes (Figure 1). Therefore, this form of regulation has the capacity to provide modulation of activity in an IP3R sub-type specific manner. Below we highlight reports that have specifically focused on regulation of IP3R2 activity. The interested reader is directed to Vanderheyden et al. [23] and Betzenhauser and Yule [85] for detailed discussion of IP3R phosphorylation and its functional consequences.
2.4.1. Regulation of IP3R2 by protein kinase A (PKA)
Historically, perhaps the most exhaustive investigation of IP3R modulation relates to PKA phosphorylation of IP3R1. Indeed, IP3R1 was identified as a major brain phosphorylated substrate even prior to the protein being appreciated as the receptor for IP3 [86]. Subsequent studies demonstrated that IP3R1 is phosphorylated at serine residues within two canonical consensus motifs (Bas-Bas-Xaa-Ser/Thr, where Bas = a basic residue) [87, 88] and phosphorylation is associated with markedly enhanced Ca2+ release [89, 90]. To complete the strong case for IP3R1 being a functionally important PKA substrate, mutation of Ser1589 and Ser1755 to non-phosphorylatable alanine residues completely abrogates phosphate incorporation and the functional effects of PKA activation [90-92]. However, while PKA activation in cells that predominately express IP3R2 such as hepatocytes, parotid acinar cells and AR42J similarly results in enhanced Ca2+ release [93-95], the PKA substrate motifs present in IP3R1 are not conserved in IP3R2 [5]. In addition, while PKA activation results in IP3R2 phosphorylation, phosphate incorporation is non-stoichiometric and indeed much reduced in comparison to IP3R1 [96]. Nevertheless, IP3R2 contains approximately 30 serine or threonine residues, which constitute minimal PKA consensus motifs consisting of basic residues preceding the phosphorylated amino acid at the −2 and −3 positions (Bas-Bas-Xaa-Ser/Thr). Using an approach based on expressing consecutive domains of IP3R2 with N-terminal epitope tags, it was shown that PKA could only specifically phosphorylate in vitro a peptide fragment consisting of amino acids 920-1583 [97]. Mutation of Ser937 to alanine (Ser937Ala) abrogated all phosphorylation of the fragment, pinpointing this residue as the PKA target site (Figure 1). Subsequently, it was shown that an antibody raised against phospho-Ser937 recognized IP3R2 after forskolin treatment in cells expressing IP3R2 but not cells expressing a mutant full-length receptor harbouring a Ser937Ala mutation [97]. Notably, Ser937 was independently identified as a phosphorylated residue in a proteomic screen of hepatocytes [98]. Importantly, PKA activation markedly potentiated IP3-induced Ca2+ release in DT40 TKO cells expressing IP3R2 but not Ser937 Ala IP3R2 thus establishing this motif as likely solely responsible for the PKA-mediated phospho-regulation of IP3R2 [97]. The Ca2+ signalling machinery is a rich source of substrates responsible for cross-talk between cAMP and Ca2+ signalling which ensure fine-tuning of the Ca2+ signal and appropriate activation of effectors [99]. PKA phosphorylation of IP3R2 likely is an important site of this interaction in cells such as astrocytes, cardiac myocytes, hepatocytes and acinar cells that prominently express this family member.
2.4.2. Regulation of IP3R2 by Ca2+/calmodulin-dependent protein kinase II (CaMKII)
CaMKII are a family of serine/threonine kinases assembled as either homo- or heteromultimers derived from the products of four closely related genes [100]. As a Ca2+/calmodulin-regulated enzyme, this kinase is an important primary effector of IP3R-induced Ca2+ release and accordingly plays prominent roles in regulating various signal transduction pathways including the translocation of transcription factors and activity of ion channels [101, 102]. Notably, IP3Rs are substrates for the kinase, which provides a regulatory loop following Ca2+ release. Early work suggested that IP3R1 was a substrate for CaMKII in vitro and that the sites were distinct from those phosphorylated by PKA [103], however the functional consequences were poorly defined. Subsequently, studies based largely on pharmacology, concluded that Ca2+ release from Xenopus oocytes and HeLa cells was attenuated following CaMKII activation [104, 105]. More recently, a thorough characterization of the molecular sites and functional consequences of the CaMKII-mediated phopho-regulation of IP3R2 has been reported. Using a similar approach to that used to identify PKA sites, the ability of CaMKII to phosphorylate IP3R2 fragments in vitro was assessed. It was initially demonstrated that a candidate residue was present within a fragment encompassing the initial 1078 amino acids [106] and further refinement narrowed the potential phosphor-acceptor residue to within residues 134-338 [107]. Subsequent mutagenesis of potential serines/threonine residues in CaMKII consensus motifs (Ser/Thr-Xaa-Asp) within this region [107] identified Ser150 as phosphorylated by CaMKII (Figure 1). This site is conserved in mammalian IP3R family members and ryanodine receptor (RyR) 2, suggesting a common mode of regulation in these channels. When incorporated in bilayers, CaMKII phosphorylation reduced the Po of IP3R2 and this effect was reversed by the CaMKII inhibitor KN62. Importantly, the reduced channel activity was absent in Ser150Ala IP3R2, indicating that the site was functionally relevant [107]. Notably, CaMKII colocalizes and interacts with IP3R2 in the nuclear envelope of cardiac myocytes [106, 108]. This interaction has been proposed to be functional important for cardiac remodelling during hypertrophy [109] (see section 4).
2.4.3. Regulation of IP3R2 by protein kinase C (PKC)
Cerebellar IP3R was also initially identified as a substrate for PKC with phosphorylation sites independent of those sites modified by PKA [103]. Interestingly, however, it was demonstrated that phosphorylation by PKC was enhanced by prior PKA phosphorylation indicating a potential additional layer of cross-talk between these prominent cellular signalling systems [110]. Similarly to CaMKII, because at least conventional PKC family members are regulated by an elevation in Ca2+, PKC phosphorylation provides a potential feedback loop to regulate IP3R activity. To date however, the functional effects of PKC phosphorylation and the sites of phosphorylation are relatively poorly defined. Unfortunately despite the general appreciation of the amino acid motifs that constitutes a PKC consensus sequence and the presence of multiple such templates in IP3R, none have been experimentally defined. Furthermore, while PKC activation results in enhanced Ca2+ release from liver nuclei, presumably reflecting IP3R1 and IP3R2 activity [111], Ca2+ release is inhibited in AR42J cells, which predominately express IP3R2 [112]. These disparate findings may reflect subtype-specific regulation of IP3R given that the PKC consensus motif numbers and location are different in each family member [9]. However an additional consideration is that numerous proteins in the signalling pathway from plasma membrane receptor occupation to the generation of Ca2+ signals are substrates for PKC and thus caution must be taken in interpreting data generated from indirect measurements of IP3R activity. Our own experience is that IP3R2 single channel activity recorded in either DT40 cell nuclei or in DT40 plasma membranes was unaffected by phorbol ester treatment or recombinant PKC exposure (Wagner, Chandrasekhar and Yule; unpublished observations). These data might indicate that IP3R is not a direct substrate for PKC. Yet, we cannot formally exclude the possibility that a scaffolding or anchoring protein necessary for activity is absent from the DT40 system. Hence, further work is required to characterize the impact of PKC on IP3R activity in general and on IP3R2 in particular.
2.5. Other characteristics of IP3R2
All the characteristics discussed above (see sections 2.1-2.4) have been the subject of extensive investigations. There are however, a few less studied properties, which nevertheless might be very interesting for understanding the cellular function of IP3R2.
2.5.1. Regulation of IP3R2 by cAMP
Similarly to the other IP3R isoforms, many accessory proteins interact with, and modulate IP3R2 function. These proteins include regulatory and structural proteins, many of which were also reported to interact with IP3R1 and/or IP3R3 [1, 2, 14]. An interesting exception is the interaction described between IP3R2 and type 6 adenylate cyclase (AC6) [113]. In HEK 293 cells stably transfected with the type I parathyroid hormone receptor, a complex is formed between IP3R2, AC6 and Gαs [113, 114]. This close association facilitates an exquisite regulation of IP3R2 by cAMP, and in addition, Ca2+ released through IP3R2 may control AC6 in a negative feedback loop. Importantly, the regulation by cAMP does not require the canonical ATP-binding site or the activity of PKA. Moreover, although all IP3R isoforms are potentially sensitive to cAMP, only IP3R2 has been unequivocally linked to a cAMP-producing enzyme (AC6) [114]. This mechanism can be of great general importance, since it provides a novel example of cross-talk between the cAMP- and the Ca2+-dependent pathways.
2.5.2. Clustering and mobility of IP3R2
All IP3R isoforms, including IP3R2 [115] are known to cluster in an agonist-dependent way [116] but a punctate distribution of IP3R2 has also been observed for native IP3R2 [117] and heterologously expressed IP3R2 [41] in resting cells. This property can be correlated to the higher affinity of IP3R2 for IP3 (see section 2.1), which may allow the clustering to occur at basal, resting [IP3]. Interestingly, also other differences in behaviour were found between the different IP3R isoforms. A recent study performed in COS-7 cells confirmed that heterologously expressed IP3R2 showed a punctate distribution, in contrast to IP3R1 and IP3R3 that were uniformly distributed [118]. Moreover, IP3R2 appeared much less mobile than either the other IP3R isoforms or than other proteins involved in intracellular Ca2+ handling, such as RyR1 or sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) 1. In addition, its mobility depended on its intracellular localization with the IP3R2 located in the perinuclear region having the lowest mobility. As the IP3R2 has the highest sensitivity to IP3 (see section 2.1), its lesser mobility may determine the initiation sites for intracellular Ca2+ signals.
3. The function of IP3R2 in secretory cells
IP3R2 exhibits prominent expression in classical secretory cells (Table 1), including exocrine cells of the pancreas [119-121], salivary glands [81, 120-123], lacrimal gland [124], olfactory glands [125], liver [29], eccrine sweat glands [126] and the secretory epithelia of the biliary tree [127] and the intestine [18] and the goblet cells of the small intestine [128]. A common feature of these epithelial cells is that they are morphologically and functionally polarized to secrete fluid and protein across their apical pole into a lumen forming a duct. Notably, the Ca2+ signal is centrally important to the primary secretory function of these cells by virtue of directly activating ion channels and the exocytotic machinery necessary for vectoral fluid and protein secretion [129, 130]. In exocrine glands, IP3R2 and IP3R3 are expressed in approximately equal numbers [12] and both family members are co-localized to a region immediately below the apical plasma membrane [119, 120]. This region has been termed “the trigger zone” because Ca2+ signals are invariably initiated in this region prior to the signal spreading as a Ca2+ wave towards the basal aspects of the cell [131-133]. There appears to be substantial functional redundancy between IP3R2 and IP3R3 in exocrine cells as mice null for either IP3R in isolation have no obvious phenotype. However, compound knockouts of IP3R2 and IP3R3 have severe exocrine deficiency manifested as dry mouth [121], dry eye [124], pancreatic insufficiency [121] and attenuated mucus secretion [125]. As such, double knockouts are born normally but demise soon after weaning [121]. Indeed a detailed analysis of these mice have shown that Ca2+ signals in pancreatic, salivary, lacrimal and mucus glands are essentially unaltered in IP3R3 null mice [121, 124, 125] and only reduced to a modest degree at low [IP3] in IP3R2 null mice [80, 81, 121, 124, 125]. These data suggest that IP3R2 is not generally essential for overall exocrine function.
A possible exception to this idea has been highlighted by a recent study, which investigated the cause of a severe congenital sweating defect in a Pakistani family [126]. Ca2+ signalling is known to be important for sweat secretion and both IP3R2 and IP3R3 are expressed in the secretory cells of the sweat gland. A screen based on identifying regions of autozygosity in the genome of afflicted individuals revealed a mutation (Gly2498Ser) targeting an amino acid predicted to be critical to the function of the selectivity filter in the pore region of IP3R2. This mutation rendered the channel completely inactive when expressed in DT40 TKO cells, thus potentially explaining the defect observed in the patients. Consistent with this idea, subsequent studies showed that mice lacking IP3R2 exhibited a decreased ability to sweat although the effect was more modest than observed in humans. The differences in severity between the phenotype observed between mouse and human may reflect the relative levels of these subtypes in mouse versus human. Alternatively, the relatively mild phenotype in the mouse might be related solely to the knockout of IP3R2, reflecting some degree of compensation by the residual IP3R3. In this scenario the more severe effect in human could be due to the combined effect of ablation of IP3R2 pore function and a possible dominant negative effect of the mutant IP3R2 when incorporated into heterotetramers containing IP3R3.
The primary secretory function of hepatocytes is the secretion of bile and changes in intracellular Ca2+ play important regulatory roles in this process. Hepatocytes express predominantly IP3R2 (Table 1) with smaller amounts of IP3R1 and virtually no IP3R3. However, in contrast to exocrine acinar cells, each isoform exhibits a distinct sub-cellular localization and therefore the isoforms appear not to have redundant functions. IP3R2 is enriched at the canalicular membrane, whereas IP3R1 has a more uniform distribution throughout structures in the cytosol [29]. Consistent with the sensitivity of IP3R2, agonist-induced Ca2+ signalling is initiated through IP3R2 localized to the canalicular membrane [29, 134, 135] and Ca2+ release through this isoform is necessary for trafficking of the bile salt export pump to the canalicular membrane [134].
4. The function of IP3R2 in the heart
As indicated (Table 1), cardiomyocytes are one of the cell types in which IP3R2 are highly expressed. Both atria and ventricles express IP3R2 [136] and consequently IP3R2 channels have been implicated in both physiological and pathophysiological signalling in the heart.
At the physiological level, Mikoshiba and co-workers showed that IP3R2 channels, together with IP3R1 channels, are critical for normal cardiogenesis [137]. Consistent with this, IP3R2 and IP3R1 are co-expressed in different parts of the embryonic heart, including atria, ventricle and atrioventricular canal, and in different cell types, including endothelial cells and cardiomyocytes, although timing differences in the appearance of IP3R2 versus IP3R1 exist. IP3R1/IP3R2 double knockout mice die in utero at embryonic stage E11.5 with major heart defects at the level of the ventricles (thin myocardial wall and poor trabeculation) and the atrioventricular canal (reduced number of cells). These defects were associated with a decrease in endocardial and myocardial cell proliferation. Furthermore, mesenchymal cells were lacking at the level of the developing atrioventricular canal. The authors hypothesized that this phenotype was due to the absence of IP3R-mediated Ca2+ signalling, downstream of the activation of the Ca2+/calmodulin-dependent phosphatase calcineurin and of the translocation of NFATc to the nucleus. Indeed, in an ex vivo epithelial-mesenchymal transition (EMT) assay, the defect in EMT in atrioventricular explants derived from IP3R1/IP3R2 double knockout mice could be restored by transducing constitutively active calcineurin. Moreover, the phenotype of these mice resembled well the phenotypes of mice knockouts for calcineurin B [138] or for NFATc3/NFATc4 [139]. Interestingly, the defect in endocardial cells could also be observed in developing zebrafish exposed to calcineurin inhibitors, such as FK506 or cyclosporine A. Hence, from this study, it is clear that IP3R1 or IP3R2 channels are needed for activating calcineurin/NFATc signalling and endocardial cell proliferation in vertebrates. It is important to note that the presence of either IP3R1 or IP3R2 is sufficient to drive normal cardiac development, indicating redundant functions for these channels in this process. In addition to the cardiac crescent, or first heart field, giving rise to a linear beating tube, there is a second source of myocardial cells, which is termed the second heart field. However, with respect to the latter, it appears that there is a redundant role for IP3R1 and IP3R3 [140]. IP3R1/IP3R3 double knockout mice are characterized by hypoplasia of the outflow tract and the primitive right ventricle at E8.5-9.5, probably due to a defective Mef2c-Smyd1 transcriptional pathway.
At the functional level, IP3Rs were first shown to impact contractility and arrhythmias. Atrial myocytes express much higher levels of IP3R2 than ventricular myocytes [136]. They are activated in response to elevated extracellular agonist concentrations, e.g. after ischaemia or during disease. Endothelin-1-induced IP3R activation promotes the inotropy and the occurrence of arrhythmic events in atrial myocytes [141, 142]. While in basal conditions, the cardiac function of IP3R2 knockout mice was similar to one of wild-type mice, the positive inotropic effect and the arrhythmic events induced by endothelin-1 were absent in IP3R2 knockout mice [143]. Hence, the presence of IP3R2 appears to be not essential for the normal functioning of the rodent heart, which is in line with the normal phenotype of the IP3R2 knockout mice generated by Chen and co-workers [143] and by Mikoshiba and co-workers [121].
In the ventricle, far fewer IP3Rs are present but may still contribute to Ca2+ regulation under baseline conditions [144-147]. Moreover, a study by Roderick and co-workers revealed that increased IP3R2-mediated Ca2+ signalling, in response to enhanced IP3 signalling, is responsible for inducing hypertrophic pathways after prolonged endothelin-1 exposure of neonatal or adult rat ventricular cardiomyocytes [148]. Endothelin-1 triggered the expression of atrial natriuretic factor (ANF), a marker for hypertrophy. Endothelin-1-induced hypertrophy was independent of excitation-contraction coupling, but required IP3 signalling and downstream IP3R-mediated Ca2+ release. The latter occurred in the perinuclear region, but not in the cytosol, while Ca2+ transients linked to excitation-contraction coupling occurred throughout the cardiomyocyte. Specifically buffering nuclear Ca2+ by nuclear-targeted calbindin prevented endothelin-1-induced ANF expression. The nuclear Ca2+ signal was mediated by IP3R2 channels, which were enriched in the perinuclear region and led to the activation of calcineurin and downstream NFATc1, which accumulated in the nucleus.
The IP3R/calcineurin/NFATc1 hyperactivity also seems to be operative in response to prolonged β-adrenergic signalling, which occurs during workload-induced cardiac hypertrophy via enhanced excitation-contraction coupling. Interestingly, this model led to increased endothelin-1 signalling. This involved the release of endothelin-1 and autocrine/paracrine-mediated hyperactivation of its receptor (ETAR), thereby triggering downstream IP3 signalling and Ca2+-dependent calcineurin activation. In hypertrophic cardiomyocytes (e.g. derived from spontaneous hypertrophic rats or from aortic-banded mice), IP3R2 not only played a role in the nucleus, where its hyperactivation via increased IP3 signalling downstream of ETAR led to calcineurin/NFATc1 activation and ANF expression, but also became upregulated in the junctional sarcoplasmic reticulum [146]. Here, localization of IP3R2 coincides with RyR2 channels, thereby augmenting Ca2+ transients associated with excitation-contraction coupling or endothelin-1 exposure. As a consequence, the IP3R2-mediated Ca2+ rise during diastole may activate or sensitize RyR2 channels, resulting in spontaneous extra-systolic Ca2+-release events and the occurrence of arrhythmias [147]. This increased “extra-nuclear” expression of IP3R2 was also found in human heart samples derived from patients with heart failure after ischemic dilated cardiomyopathy [146].
The upregulation of IP3R2 channels during cardiac hypertrophy was mediated via a dynamic and Ca2+-dependent regulation of miRNA-133a [149]. In normal physiological conditions, miRNA-133a expression is highly expressed in cardiomyocytes, thereby targeting the 3′ untranslated region of the IP3R2 mRNA. As a consequence miRNA-133a reduces the basal expression of IP3R2 and thereby avoids hypertrophy or arrhythmias resulting from excessive Ca2+ signalling. Interestingly, limiting the expression of miRNA-133a using an antagomir led to hypertrophic signalling (evident from the increased ANF expression), which was dependent on IP3-induced Ca2+ release, since degrading IP3 using IP3 5-phosphatase limited the increase of ANF by miR-133a antagomir.
The role of miR-133a in controlling IP3R2 expression and the initiation of hypertrophic markers was found both ex vivo and in vivo. In isolated, hypertrophic cardiomyocytes from spontaneous hypertensive rats, IP3R2 levels were elevated, while miRNA-133a was downregulated [149]. Overexpression of miRNA-133a in these hypertrophic cardiomyocytes reduced ANF expression to levels similar as control cardiomyocytes. In addition, in vivo application of miRNA-133a antagomir caused IP3R2 upregulation and hypertrophic signalling. Interestingly, increased IP3-induced Ca2+ release was also involved in the decreased miRNA-133a expression in hypertrophic models. Indeed, lowering IP3 signalling by IP3 5-phosphatase transduction blunted the endothelin-1-induced decrease in miRNA-133a and the concomitant increase in IP3R2 protein levels. Collectively, these findings indicate that during pathophysiological conditions associated with increased endothelin-1, increased IP3 signalling can lead to downregulation of miRNA-133a. The latter will lead to upregulation of IP3R2 protein levels, thereby further driving the downregulation of miRNA-133a by boosting IP3-induced IP3R2-mediated Ca2+ signalling. This perpetual feedback cycle will establish a new signalling network that favours the expression of hypertrophic genes like ANF (via hyperactivation of calcineurin/NFATc1) and the occurrence of arrhythmic events.
The mechanism by which IP3-induced Ca2+ release controls miRNA-133a expression seems to involve transcription factors like the serum response factor (SRF), which is negatively regulated by the homeodomain-only protein. SRF induces miRNA-133a expression and subsequent IP3R2 downregulation. However, during hypertrophy, IP3-induced Ca2+ release may increase homeodomain-only protein expression, thereby recruiting class I histone deacetylase (HDAC) and limiting transcriptional activity of SRF.
The importance of IP3 signalling and IP3R2 has also been elegantly addressed by Molkentin and co-workers by the generation of transgenic mice overexpressing an IP3 sponge, which represents a mutated, high-affinity form of the IP3-binding core (to blunt endogenous IP3-induced Ca2+ release by trapping IP3), or overexpressing IP3R2 (to boost IP3-induced Ca2+ release) in cardiomyocytes [150]. Mice overexpressing the IP3 sponge displayed reduced cardiac hypertrophy in response to chronic β-adrenergic stimulation and angiotensin II stimulation. In contrast, IP3R2-overexpressing mice displayed only a mild cardiac hypertrophic phenotype under basal conditions. However, when cardiac hypertrophy was induced (e.g. using transverse aortic constriction, chronic β-adrenergic stimulation or overexpression of Gαq, an upstream phospholipase C activator) mice expressing high IP3R2 levels demonstrated increased hypertrophic responses. Under these conditions, mice expressing low levels of IP3R2 (except for transverse aortic constriction) also displayed enhanced cardiac hypertrophy. Moreover, IP3R2 channels, which already display high sensitivity to IP3, may be further sensitized by increased PKA signalling downstream of β-adrenergic receptor stimulation leading to hyperphosphorylation of IP3R2 at Ser937 [97] (see section 2.4.1). The increased sensitivity of IP3R2-expressing mice to cardiac hypertrophy-inducing conditions could be linked to increased calcineurin and NFAT signalling. Consistent with this, the augmented cardiac hypertrophic response in IP3R2-overexpressing mice were completely blunted when these mice were crossed with calcineurin B-knockout mice, indicating an essential role of calcineurin/NFAT signalling in response to hyperactive IP3R2-mediated Ca2+ signalling. While from the above studies, calcineurin emerged as the downstream target of increased IP3R-mediated Ca2+ signalling, it is important to note that also nuclear CaMKIIδ has been implicated in altered transcription in response to cardiac hypertrophic endothelin-1 signalling [109]. Increased IP3 signalling in response to endothelin-1 triggers a unique nuclear Ca2+ signalling that does not occur during excitation-contraction coupling but activates CaMKII, which together with protein kinase D results in the phosphorylation and nuclear export of class II HDAC5, a transcriptional repressor. In healthy conditions, nuclear HDAC5 forms a complex with the transcription factor MEF2, thereby preventing the transcription of hypertrophic genes. In hypertrophic conditions, HDAC5 is exported from the nucleus, leading to de-repression of MEF2 and the induction of hypertrophic genes. Interestingly, blocking IP3Rs using chemicals like 2-aminoethoxydiphenyl borate or using IP3R2-knockout mice, prevents the nuclear export of HDAC5 and subsequent activation of the hypertrophic transcription program.
All these studies are consistent with a critical role for Ca2+ signalling via IP3R2 in cardiac hypertrophy, being in the nucleus and required for driving transcription of hypertrophic genes and in the junctional sarcoplasmic reticulum being responsible for driving extra-systolic Ca2+ rises and contractions. Moreover, these studies all support the concept of distinct Ca2+ signalling compartments in cardiomyocytes, either in the cytosol during physiological excitation-contraction coupling driven by RyR2 channels or in the nucleus during pathophysiological hypertrophic signalling driven by IP3 and IP3R2 channels [151].
5. The role of IP3R2 in cell death and in senescence
Over the last 20 years, IP3R channels have emerged as key regulators that control cell death and survival in a variety of cellular systems [14, 152-154]. T cells deficient in IP3R1 are resistant to a variety of apoptotic triggers, including chemical stimuli, like corticoids, and biological stimuli, including excessive T-cell receptor stimulation and exposure to Fas ligand [155]. Interestingly, susceptibility to T-cell receptor stimulation could be restored by artificially rising the cytosolic [Ca2+] using the SERCA inhibitor, thapsigargin. Also, a role for IP3R3 has emerged in pro-apoptotic Ca2+ signalling [156], because some studies proposed that this channel may be preferentially located in the mitochondrial ER-associated membranes. As such, IP3R3 channels are thought to be part of the “quasi-synaptic” Ca2+-transport complex between the ER Ca2+ stores and the mitochondria that can involve IP3Rs, GRP75 and VDAC1 [157, 158]. Nevertheless, it is becoming increasingly clear that all IP3R isoforms participate in apoptotic Ca2+ signalling and/or influence the susceptibility of cells towards apoptotic stimuli. This can mean two things: i) not only IP3R3, but also IP3R1 and IP3R2 channels can be part of the ER-mitochondrial junction complexes, and ii) not only direct Ca2+ transfer into the mitochondria, but also other downstream Ca2+-dependent signalling pathways participate in triggering mitochondrial outer membrane permeabilization, the point-of-no-return in apoptosis. Furthermore, it is important to emphasize that a complex interaction exists between IP3Rs and proteins from the B-cell lymphoma (Bcl)-2 family involved in the control of apoptosis, whereby several interaction sites for such proteins have already been identified on the IP3R [159-162].
IP3R-mediated Ca2+ release can lead to calcineurin activation, which dephosphorylates the pro-apoptotic “sensitizer” BH3-only protein, Bad [163, 164]. Phosphorylated Bad is neutralized due to its scaffolding with 14-3-3 proteins and therefore it cannot form a complex with anti-apoptotic Bcl-Xl [165]. Dephosphorylation of Bad by calcineurin, e.g. in response to increases in cytosolic [Ca2+] mediated by IP3Rs [164], results in Bad release from 14-3-3 proteins and its translocation from the cytosol to the mitochondrial membranes. Here, it can bind to and inhibit anti-apoptotic Bcl-Xl proteins [163], thereby displacing Bim/tBid, which then can activate Bax/Bak and induce apoptosis.
These data indicate that dampening the IP3R-mediated Ca2+ rise, either by lowering IP3R levels or altering the IP3R-expression profile, by inhibiting the Ca2+-flux properties of IP3Rs, or by lowering the ER Ca2+ content, which decreases the driving force for Ca2+ release into the cytosol upon IP3R activation, will be cytoprotective [162]. Not surprisingly, different pro-survival signalling mechanisms, which are often oncogenic, appear to have exploited this concept to promote cell survival, including the survival of malignant or altered cells. In many cases, different mechanisms can be simultaneously operative. For instance, oncogenic KRAS mutations appear to switch the expression from IP3R3 into IP3R1 and to lower the ER Ca2+-store content, together suppressing agonist-induced Ca2+ release and mitochondrial Ca2+ accumulation and thus protecting cells against menadione exposure [166]. AKT/PKB phosphorylates all three IP3R isoforms, thereby suppressing their pro-apoptotic Ca2+-release function [84, 156]. This mechanism is also exploited by tumour suppressors like the promyelocytic leukemia protein, which enhance IP3R3 activity by counteracting PKB-mediated IP3R3 phosphorylation [167]. Other survival/anti-apoptotic proteins, like Bcl-2, have been reported to lower ER Ca2+ store-content by sensitizing IP3Rs to basal IP3 levels and to directly suppress IP3R-mediated Ca2+ release, thereby preventing toxic mitochondrial Ca2+ overload [168]. Evidently, these mechanisms will also result in reduced calcineurin activation, thereby limiting Bad dephosphorylation and its subsequent inhibitory effects on the anti-apoptotic Bcl-2 proteins.
While most studies have addressed the role of IP3R1 and IP3R3 channels in apoptosis, there is emerging evidence that IP3R2 channels play a crucial role in mediating proapoptotic Ca2+ signalling. Definitely, IP3R2 with its high sensitivity to IP3 (see section 2.1) may actually be a very critical regulator of cell survival versus cell demise by rendering cells sensitive to basal IP3 signalling. The role of IP3R2 in cell death has been elucidated in different studies and using different approaches.
First of all, there is evidence that cell death triggered by cellular exposure to cytotoxic compounds or agents that induce oxidative stress has been associated with an increase in IP3R2 levels and activity. lncreasing oxidative stress in a neuronal cell line exposed to sub-lethal concentrations of tert-butyl hydroperoxide-mediated oxidative stress led to prominent upregulation of IP3R2 mRNA and protein levels, while IP3R1 and IP3R3-expression levels remained unaltered [38]. Consistent with elevated IP3R expression levels, Ca2+ release from the nucleoplasm in response to a cell-permeable IP3 ester was strongly potentiated in tert-butyl hydroperoxide-treated cells. Also, the nephrotoxic compound uranyl acetate has been shown to increase IP3R2 mRNA and protein levels in human epithelial kidney cells, thereby increasing the basal cytosolic [Ca2+] and apoptosis levels [169]. Similar findings have been reported in HeLa cells exposed to fast H2S donors, although in this case IP3R1 expression levels were also increased [170]. Interestingly, IP3Rs may also be directly affected by reactive oxygen species (ROS) [171]. In intact DT40 cells, superoxide anions caused Ca2+ release from the ER, likely via a mechanism that sensitizes IP3Rs to basal levels of IP3 signalling. In these DT40 cells, the presence of IP3R2 and IP3R1 isoforms, but not of IP3R3, was required for superoxide anion-induced [Ca2+] rise in the cytosol.
The role of IP3R2 channels in apoptotic Ca2+ signalling was also identified in B-cell cancer cells, in particular in a subset of “primed to death” diffuse large B-cell lymphoma cell lines [172]. Cells expressing high IP3R2 levels seem “addicted” to the presence and recruitment of anti-apoptotic Bcl-2 proteins at the ER and especially in the IP3R protein complex [173]. By interacting via its BH4 domain with the modulatory and transducing domain of the IP3Rs, Bcl-2 inhibits IP3-induced Ca2+ release [174-176]. The binding site for the BH4 domain of Bcl-2 (Figure 1) has been identified [175] and is conserved between the three IP3R isoforms [177]. Importantly, a peptide tool designed to disrupt IP3R/Bcl-2-complexes by targeting Bcl-2’s BH4 domain (see [175-178]) was very effective in inducing intracellular Ca2+ overload and provoking cell death in DL-BCL cells that express high levels of IP3R2, like SU-DHL-4 cells [172]. In contrast, cells that expressed very low levels of IP3R2 were virtually resistant to this peptide tool. The apoptotic resistance of these cells to this peptide was not due to a general defect in the initiation or execution of apoptosis, since staurosporine or BH3-mimetic drugs were very effective in these cells. We hypothesize that anti-apoptotic Bcl-2 is required at the ER to associate with the IP3R2 to prevent its hyperactivity in response to the ongoing IP3 signalling downstream of the B-cell receptor [173]. It remains to be elucidated whether these findings translate into primary B-cell cancer cells. In any case, disrupting IP3R/Bcl-2 complexes results in excessive Ca2+-signalling patterns and apoptotic cell death in primary peripheral mononuclear blood cells (mainly B cells) isolated from chronic lymphocytic leukaemia patients [179]. Remarkably, a gene expression profile analysis using the GeneSapiens microarray database revealed an upregulation of IP3R2 at the mRNA level in chronic lymphocytic leukaemia samples [173].
IP3R2 channels are not only implicated in apoptosis but also play a role in cellular senescence. Stable cell cycle arrest is a key feature of cellular senescence, which is activated in response to cellular stress. Factors include oncogenic stress following loss of PTEN function, DNA damage or telomere attrition, oxidative stress and replicative stress [180]. The arrest in proliferation depends on the major tumour suppressor pathways involving p53/p21 and p16/Rb [180, 181]. At the physiological level, cellular senescence contributes to ageing at the level of the organism [182]. However, cellular senescence can also function as an important “health keeper” fighting pathophysiological conditions associated with oncogenic stress [183, 184]. As such, cellular senescence, in addition to apoptosis, is one of the pathways that counteract cancer cell initiation and tumour development [185, 186]. For instance, in pre-malignant hepatocytes, senescence led to the secretion of chemo- and cytokines, resulting in their clearance by CD4+ T cells [187]. Loss of immune surveillance caused the progression of the pre-malignant hepatocytes into hepatocellular carcinomas. Recently, Wiel et al. [188] performed an elegant shRNA-based screen to identify which “loss-of-function” genes can cause escape from oncogene-induced senescence in immortalized human mammary epithelial cells (HEC). Interestingly, the gene coding for IP3R2 was identified as a prominent modulator of this form of senescence. These findings correlated with an analysis performed by the authors using the Oncomine database, which indicated that many malignant tumours displayed a decrease in IP3R2 mRNA levels. IP3R2 shRNAs alleviated the growth arrest in HEC exposed to oncogenic stress. Prolonged incubation of these cells with cell-permeable IP3 repressed cell growth and induced pre-mature senescence. Oncogenic stress-induced senescence led to an increase in the Ca2+ accumulation in the mitochondria, a process that did not occur in the IP3R2 shRNA-treated cells, and also boosted IP3-induced mitochondrial Ca2+ uptake. This mitochondrial Ca2+ uptake was proposed to be responsible for the decrease in mitochondrial potential observed during oncogene-induced senescence, because shRNA against the IP3R2 or against the mitochondrial Ca2+ uniporter prevented this decline in mitochondrial potential. Interestingly, chemical induction of mitochondrial depolarization blocked cell growth and induced pre-mature senescence. The role of IP3R2 and of the subsequent mitochondrial Ca2+ accumulation was linked to an increase in ROS production, since anti-oxidants promoted oncogene-induced senescence escape. Finally, these concepts may not be limited to oncogene-induced senescence, but may also be applicable in models of replicative senescence. IP3R2 knockdown counteracted the increase in mitochondrial Ca2+ and the decline in mitochondrial potential observed during replicative senescence, thereby delaying the occurrence of senescence in these models.
6. Conclusions
IP3R2 is characterized by a number of important and specific properties, including, but not limited to, its high sensitivity to IP3 and ATP. Other properties such as its regulation by protein kinases, its interaction with adenylate cyclase to couple to cAMP production, its ability to recruit associated proteins and its low mobility in the ER, remain underexplored. At the functional level, it is clear that IP3R2 is not only important for regulating secretion, but also is implicated in health and disease, including prominent roles in cardiac function and tumour growth. The available evidence indicates that tumour cells either downregulate IP3R2 expression or dampen its activity via Bcl-2, since IP3R2 can promote senescence and/or apoptosis. It is now anticipated that further research will elucidate additional important functions of IP3R2 in other tissues and organs and further that developing tools specifically targeting or impacting IP3R2 will allow modulating its function in disease states.
Highlights.
* The understanding of IP3R2 has long lagged behind that of the other IP3R isoforms
* IP3R2 is an intracellular Ca2+-release channel with important and unique properties
* IP3R2 performs crucial physiological functions in various cell types
* IP3R2 is implicated in health and disease, including cardiac hypertrophy and cancer
* IP3R2 forms an important target for further research
Acknowledgments
Work performed on the topic in the laboratory of J.B.P. and G.B was supported by various grants of the Research Foundation-Flanders and of the Research Council of the KU Leuven as well as by the Interuniversity Attraction Pole Programs of the Belgian Science Policy. T.V. is the recipient of a fellowship of the “Vlaamse Liga tegen Kanker”. D.Y. is supported by grants from the NIH (RO1-DE019245 and R01-DE041756).
The authors are indebted to past and present members of their laboratory, and especially to Professors H. De Smedt and L. Missiaen, for stimulating discussions. The authors wish also to acknowledge Professors C.W. Distelhorst, K. Mikoshiba, D. Bernard and P. Gailly for interesting discussions.
Abbreviations
- AC6
Adenylate cyclase type 6
- AKT/PKB
Protein kinase B
- ANF
Atrial natriuretic factor
- Bcl
B-cell lymphoma
- BH
Bcl-2 homology domain
- CaMKII
Ca2+/calmodulin dependent protein kinase II
- DT40 TKO
DT40 triple knockout
- EMT
Epithelial-mesenchymal transition
- ER
Endoplasmic reticulum
- ETAR
Endothelin receptor type A
- HDAC
Histone deacetylase
- HEC
Immortalized human mammary epithelial cells
- IP3
Inositol 1,4,5-trisphosphate
- IP3R(1, 2, 3)
Inositol 1,4,5-trisphosphate receptor (type 1, type 2, type 3)
- NFAT
Nuclear factor of activated T cells
- PKA
Protein kinase A
- PKC
Protein kinase C
- Po
Open probability
- ROS
Reactive oxygen species
- RyR
Ryanodine receptor
- SERCA
Sarco/endoplasmic reticulum Ca2+ ATPase
- SRF
Serum response factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Parys JB, De Smedt H. Inositol 1,4,5-trisphosphate and its receptors. Adv Exp Med Biol. 2012;740:255–279. doi: 10.1007/978-94-007-2888-2_11. [DOI] [PubMed] [Google Scholar]
- [3].Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- [4].Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–38. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- [5].Südhof TC, Newton CL, Archer BT, 3rd, Ushkaryov YA, Mignery GA. Structure of a novel InsP3 receptor. Embo J. 1991;10:3199–3206. doi: 10.1002/j.1460-2075.1991.tb04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blondel O, Takeda J, Janssen H, Seino S, Bell GI. Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3R-3, expressed in pancreatic islets, kidney, gastrointestinal tract, and other tissues. J Biol Chem. 1993;268:11356–11363. [PubMed] [Google Scholar]
- [7].Bosanac I, Michikawa T, Mikoshiba K, Ikura M. Structural insights into the regulatory mechanism of IP3 receptor. Biochim Biophys Acta. 2004;1742:89–102. doi: 10.1016/j.bbamcr.2004.09.016. [DOI] [PubMed] [Google Scholar]
- [8].Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium. 1999;26:237–251. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- [9].Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 1999;25:247–264. doi: 10.1054/ceca.1999.0021. [DOI] [PubMed] [Google Scholar]
- [10].Newton CL, Mignery GA, Südhof TC. Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J Biol Chem. 1994;269:28613–28619. [PubMed] [Google Scholar]
- [11].De Smedt H, Missiaen L, Parys JB, Bootman MD, Mertens L, Van Den Bosch L, Casteels R. Determination of relative amounts of inositol trisphosphate receptor mRNA isoforms by ratio polymerase chain reaction. J Biol Chem. 1994;269:21691–21698. [PubMed] [Google Scholar]
- [12].Wojcikiewicz RJ, I Type. II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J Biol Chem. 1995;270:11678–11683. doi: 10.1074/jbc.270.19.11678. [DOI] [PubMed] [Google Scholar]
- [13].De Smedt H, Missiaen L, Parys JB, Henning RH, Sienaert I, Vanlingen S, Gijsens A, Himpens B, Casteels R. Isoform diversity of the inositol trisphosphate receptor in cell types of mouse origin. Biochem J. 1997;322:575–583. doi: 10.1042/bj3220575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014;1843:2164–2183. doi: 10.1016/j.bbamcr.2014.03.007. [DOI] [PubMed] [Google Scholar]
- [15].Sugiyama T, Yamamoto-Hino M, Miyawaki A, Furuichi T, Mikoshiba K, Hasegawa M. Subtypes of inositol 1,4,5-trisphosphate receptor in human hematopoietic cell lines: dynamic aspects of their cell-type specific expression. FEBS Lett. 1994;349:191–196. doi: 10.1016/0014-5793(94)00662-8. [DOI] [PubMed] [Google Scholar]
- [16].Lee B, Jonas JC, Weir GC, Laychock SG. Glucose regulates expression of inositol 1,4,5-trisphosphate receptor isoforms in isolated rat pancreatic islets. Endocrinology. 1999;140:2173–2182. doi: 10.1210/endo.140.5.6738. [DOI] [PubMed] [Google Scholar]
- [17].Mountian I, Manolopoulos VG, De Smedt H, Parys JB, Missiaen L, Wuytack F. Expression patterns of sarco/endoplasmic reticulum Ca2+-ATPase and inositol 1,4,5-trisphosphate receptor isoforms in vascular endothelial cells. Cell Calcium. 1999;25:371–380. doi: 10.1054/ceca.1999.0034. [DOI] [PubMed] [Google Scholar]
- [18].Siefjediers A, Hardt M, Prinz G, Diener M. Characterization of inositol 1,4,5-trisphosphate (IP3) receptor subtypes at rat colonic epithelium. Cell Calcium. 2007;41:303–315. doi: 10.1016/j.ceca.2006.07.009. [DOI] [PubMed] [Google Scholar]
- [19].Mountian II, Baba-Aissa F, Jonas JC, Humbert De S, Wuytack F, Parys JB. Expression of Ca2+ transport genes in platelets and endothelial cells in hypertension. Hypertension. 2001;37:135–141. doi: 10.1161/01.hyp.37.1.135. [DOI] [PubMed] [Google Scholar]
- [20].Steffl M, Schweiger M, Amselgruber WM. Oestrous cycle-regulated expression of inositol 1,4,5-trisphosphate receptor type 2 in the pig ovary. Acta Histochem. 2004;106:137–144. doi: 10.1016/j.acthis.2003.10.006. [DOI] [PubMed] [Google Scholar]
- [21].Jurkovicova D, Kopacek J, Stefanik P, Kubovcakova L, Zahradnikova A, Jr., Zahradnikova A, Pastorekova S, Krizanova O. Hypoxia modulates gene expression of IP3 receptors in rodent cerebellum. Pflügers Arch. 2007;454:415–425. doi: 10.1007/s00424-007-0214-6. [DOI] [PubMed] [Google Scholar]
- [22].Vermassen E, Parys JB, Mauger JP. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell. 2004;96:3–17. doi: 10.1016/j.biolcel.2003.11.004. [DOI] [PubMed] [Google Scholar]
- [23].Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta. 2009;1793:959–970. doi: 10.1016/j.bbamcr.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yule DI, Betzenhauser MJ, Joseph SK. Linking structure to function: recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium. 2010;47:469–479. doi: 10.1016/j.ceca.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2005;38:261–272. doi: 10.1016/j.ceca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- [26].Laflamme K, Domingue O, Guillemette BI, Guillemette G. Immunohistochemical localization of type 2 inositol 1,4,5-trisphosphate receptor to the nucleus of different mammalian cells. J Cell Biochem. 2002;85:219–228. [PubMed] [Google Scholar]
- [27].Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, Ehrlich BE, Nathanson MH. Nuclear and cytosolic calcium are regulated independently. Proc Natl Acad Sci U S A. 2003;100:2975–2980. doi: 10.1073/pnas.0536590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Duncan RS, Hwang SY, Koulen P. Differential inositol 1,4,5-trisphosphate receptor signaling in a neuronal cell line. Int J Biochem Cell Biol. 2007;39:1852–1862. doi: 10.1016/j.biocel.2007.05.003. [DOI] [PubMed] [Google Scholar]
- [29].Hirata K, Pusl T, O’Neill AF, Dranoff JA, Nathanson MH. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology. 2002;122:1088–1100. doi: 10.1053/gast.2002.32363. [DOI] [PubMed] [Google Scholar]
- [30].Gerasimenko O, Gerasimenko J. New aspects of nuclear calcium signalling. J Cell Sci. 2004;117:3087–3094. doi: 10.1242/jcs.01295. [DOI] [PubMed] [Google Scholar]
- [31].Bootman MD, Fearnley C, Smyrnias I, MacDonald F, Roderick HL. An update on nuclear calcium signalling. J Cell Sci. 2009;122:2337–2350. doi: 10.1242/jcs.028100. [DOI] [PubMed] [Google Scholar]
- [32].Bengtson CP, Bading H. Nuclear calcium signaling. Adv Exp Med Biol. 2012;970:377–405. doi: 10.1007/978-3-7091-0932-8_17. [DOI] [PubMed] [Google Scholar]
- [33].Echevarria W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat Cell Biol. 2003;5:440–446. doi: 10.1038/ncb980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zima AV, Bare DJ, Mignery GA, Blatter LA. IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J Physiol. 2007;584:601–611. doi: 10.1113/jphysiol.2007.140731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morikawa K, Ohbayashi T, Nakagawa M, Konishi Y, Makino Y, Yamada M, Miyawaki A, Furuichi T, Mikoshiba K, Tamura T. Transcription initiation sites and promoter structure of the mouse type 2 inositol 1,4,5-trisphosphate receptor gene. Gene. 1997;196:181–185. doi: 10.1016/s0378-1119(97)00224-2. [DOI] [PubMed] [Google Scholar]
- [36].Sankar N, deTombe PP, Mignery GA. Calcineurin-NFATc regulates type 2 inositol 1,4,5-trisphosphate receptor (InsP3R2) expression during cardiac remodeling. J Biol Chem. 2014;289:6188–6198. doi: 10.1074/jbc.M113.495242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang W, Nurbaeva MK, Schmid E, Russo A, Almilaji A, Szteyn K, Yan J, Faggio C, Shumilina E, Lang F. Akt2- and ETS1-dependent IP3 receptor 2 expression in dendritic cell migration. Cell Physiol Biochem. 2014;33:222–236. doi: 10.1159/000356664. [DOI] [PubMed] [Google Scholar]
- [38].Kaja S, Duncan RS, Longoria S, Hilgenberg JD, Payne AJ, Desai NM, Parikh RA, Burroughs SL, Gregg EV, Goad DL, Koulen P. Novel mechanism of increased Ca2+ release following oxidative stress in neuronal cells involves type 2 inositol-1,4,5-trisphosphate receptors. Neuroscience. 2011;175:281–291. doi: 10.1016/j.neuroscience.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Futatsugi A, Kuwajima G, Mikoshiba K. Muscle-specific mRNA isoform encodes a protein composed mainly of the N-terminal 175 residues of type 2 Ins(1,4,5)P3 receptor. Biochem J. 1998;334:559–563. doi: 10.1042/bj3340559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chan J, Yamazaki H, Ishiyama N, Seo MD, Mal TK, Michikawa T, Mikoshiba K, Ikura M. Structural studies of inositol 1,4,5-trisphosphate receptor: coupling ligand binding to channel gating. J Biol Chem. 2010;285:36092–36099. doi: 10.1074/jbc.M110.140160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Iwai M, Tateishi Y, Hattori M, Mizutani A, Nakamura T, Futatsugi A, Inoue T, Furuichi T, Michikawa T, Mikoshiba K. Molecular cloning of mouse type 2 and type 3 inositol 1,4,5-trisphosphate receptors and identification of a novel type 2 receptor splice variant. J Biol Chem. 2005;280:10305–10317. doi: 10.1074/jbc.M413824200. [DOI] [PubMed] [Google Scholar]
- [42].Wojcikiewicz RJ, Furuichi T, Nakade S, Mikoshiba K, Nahorski SR. Muscarinic receptor activation down-regulates the type I inositol 1,4,5-trisphosphate receptor by accelerating its degradation. J Biol Chem. 1994;269:7963–7969. [PubMed] [Google Scholar]
- [43].Sipma H, Deelman L, De Smedt H, Missiaen L, Parys JB, Vanlingen S, Henning RH, Casteels R. Agonist-induced down-regulation of type 1 and type 3 inositol 1,4,5-trisphosphate receptors in A7r5 and DDT1 MF-2 smooth muscle cells. Cell Calcium. 1998;23:11–21. doi: 10.1016/s0143-4160(98)90070-7. [DOI] [PubMed] [Google Scholar]
- [44].Saleem H, Tovey SC, Molinski TF, Taylor CW. Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br J Pharmacol. 2014;171:3298–3312. doi: 10.1111/bph.12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Taylor CW, da Fonseca PC, Morris EP. IP3 receptors: the search for structure. Trends Biochem Sci. 2004;29:210–219. doi: 10.1016/j.tibs.2004.02.010. [DOI] [PubMed] [Google Scholar]
- [46].Iwai M, Michikawa T, Bosanac I, Ikura M, Mikoshiba K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2007;282:12755–12764. doi: 10.1074/jbc.M609833200. [DOI] [PubMed] [Google Scholar]
- [47].Vanlingen S, Sipma H, De Smet P, Callewaert G, Missiaen L, De Smedt H, Parys JB. Ca2+ and calmodulin differentially modulate myo-inositol 1,4, 5-trisphosphate (IP3)-binding to the recombinant ligand-binding domains of the various IP3 receptor isoforms. Biochem J. 2000;346:275–280. [PMC free article] [PubMed] [Google Scholar]
- [48].Miyakawa T, Maeda A, Yamazawa T, Hirose K, Kurosaki T, Iino M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999;18:1303–1308. doi: 10.1093/emboj/18.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Morel JL, Fritz N, Lavie JL, Mironneau J. Crucial role of type 2 inositol 1,4,5-trisphosphate receptors for acetylcholine-induced Ca2+ oscillations in vascular myocytes. Arterioscler Thromb Vasc Biol. 2003;23:1567–1575. doi: 10.1161/01.ATV.0000089013.82552.5D. [DOI] [PubMed] [Google Scholar]
- [50].Fritz N, Mironneau J, Macrez N, Morel JL. Acetylcholine-induced Ca2+ oscillations are modulated by a Ca2+ regulation of InsP3R2 in rat portal vein myocytes. Pflügers Arch. 2008;456:277–283. doi: 10.1007/s00424-007-0379-z. [DOI] [PubMed] [Google Scholar]
- [51].Ramos-Franco J, Fill M, Mignery GA. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys J. 1998;75:834–839. doi: 10.1016/S0006-3495(98)77572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tu H, Wang Z, Nosyreva E, De Smedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophys J. 2005;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wojcikiewicz RJ, Luo SG. Differences among type I, II, and III inositol-1,4,5-trisphosphate receptors in ligand-binding affinity influence the sensitivity of calcium stores to inositol-1,4,5-trisphosphate. Mol Pharmacol. 1998;53:656–662. doi: 10.1124/mol.53.4.656. [DOI] [PubMed] [Google Scholar]
- [54].Nerou EP, Riley AM, Potter BV, Taylor CW. Selective recognition of inositol phosphates by subtypes of the inositol trisphosphate receptor. Biochem J. 2001;355:59–69. doi: 10.1042/0264-6021:3550059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J Gen Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- [57].Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- [58].Parys JB, Sernett SW, DeLisle S, Snyder PM, Welsh MJ, Campbell KP. Isolation, characterization, and localization of the inositol 1,4,5-trisphosphate receptor protein in Xenopus laevis oocytes. J Biol Chem. 1992;267:18776–18782. [PubMed] [Google Scholar]
- [59].Ramos-Franco J, Bare D, Caenepeel S, Nani A, Fill M, Mignery G. Single-channel function of recombinant type 2 inositol 1,4, 5-trisphosphate receptor. Biophys J. 2000;79:1388–1399. doi: 10.1016/S0006-3495(00)76391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tu H, Wang Z, Bezprozvanny I. Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J. 2005;88:1056–1069. doi: 10.1529/biophysj.104.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Smith JB, Smith L, Higgins BL. Temperature and nucleotide dependence of calcium release by myo-inositol 1,4,5-trisphosphate in cultured vascular smooth muscle cells. J Biol Chem. 1985;260:14413–14416. [PubMed] [Google Scholar]
- [62].Hirata M, Kukita M, Sasaguri T, Suematsu E, Hashimoto T, Koga T. Increase in Ca2+ permeability of intracellular Ca2+ store membrane of saponin-treated guinea pig peritoneal macrophages by inositol 1,4,5-trisphosphate. J Biochem. 1985;97:1575–1582. doi: 10.1093/oxfordjournals.jbchem.a135214. [DOI] [PubMed] [Google Scholar]
- [63].Ehrlich BE, Watras J. Inositol 1,4,5-trisphosphate activates a channel from smooth muscle sarcoplasmic reticulum. Nature. 1988;336:583–586. doi: 10.1038/336583a0. [DOI] [PubMed] [Google Scholar]
- [64].Ferris CD, Huganir RL, Snyder SH. Calcium flux mediated by purified inositol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles is allosterically regulated by adenine nucleotides. Proc Natl Acad Sci U S A. 1990;87:2147–2151. doi: 10.1073/pnas.87.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Maeda N, Kawasaki T, Nakade S, Yokota N, Taguchi T, Kasai M, Mikoshiba K. Structural and functional characterization of inositol 1,4,5-trisphosphate receptor channel from mouse cerebellum. J Biol Chem. 1991;266:1109–1116. [PubMed] [Google Scholar]
- [66].Iino M. Effects of adenine nucleotides on inositol 1,4,5-trisphosphate-induced calcium release in vascular smooth muscle cells. J Gen Physiol. 1991;98:681–698. doi: 10.1085/jgp.98.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bezprozvanny I, Ehrlich BE. ATP modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron. 1993;10:1175–1184. doi: 10.1016/0896-6273(93)90065-y. [DOI] [PubMed] [Google Scholar]
- [68].Missiaen L, Parys JB, De Smedt H, Sienaert I, Sipma H, Vanlingen S, Maes K, Casteels R. Effect of adenine nucleotides on myo-inositol-1,4,5-trisphosphate-induced calcium release. Biochem J. 1997;325:661–666. doi: 10.1042/bj3250661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Betzenhauser MJ, Wagner LE, 2nd, Iwai M, Michikawa T, Mikoshiba K, Yule DI. ATP modulation of Ca2+ release by type-2 and type-3 inositol (1, 4, 5)-triphosphate receptors. Differing ATP sensitivities and molecular determinants of action. J Biol Chem. 2008;283:21579–21587. doi: 10.1074/jbc.M801680200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Betzenhauser MJ, Wagner LE, 2nd, Park HS, Yule DI. ATP regulation of type-1 inositol 1,4,5-trisphosphate receptor activity does not require walker A-type ATP-binding motifs. J Biol Chem. 2009;284:16156–16163. doi: 10.1074/jbc.M109.006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Maes K, Missiaen L, De Smet P, Vanlingen S, Callewaert G, Parys JB, De Smedt H. Differential modulation of inositol 1,4,5-trisphosphate receptor type 1 and type 3 by ATP. Cell Calcium. 2000;27:257–267. doi: 10.1054/ceca.2000.0121. [DOI] [PubMed] [Google Scholar]
- [72].Mak DO, McBride S, Foskett JK. Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 InsP3 receptors. J Gen Physiol. 2001;117:435–446. doi: 10.1085/jgp.117.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mak DO, McBride S, Foskett JK. ATP regulation of recombinant type 3 inositol 1,4,5-trisphosphate receptor gating. J Gen Physiol. 2001;117:447–456. doi: 10.1085/jgp.117.5.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Maeda N, Kawasaki T, Nakade S, Yokota N, Taguchi T, Kasai M, Mikoshiba K. Structural and functional characterization of inositol 1,4,5-trisphosphate receptor channel from mouse cerebellum. J Biol Chem. 1991;266:1109–1116. [PubMed] [Google Scholar]
- [76].Maes K, Missiaen L, Parys JB, De Smet P, Sienaert I, Waelkens E, Callewaert G, De Smedt H. Mapping of the ATP-binding sites on inositol 1,4,5-trisphosphate receptor type 1 and type 3 homotetramers by controlled proteolysis and photoaffinity labeling. J Biol Chem. 2001;276:3492–3497. doi: 10.1074/jbc.M006082200. [DOI] [PubMed] [Google Scholar]
- [77].Maes K, Missiaen L, Parys JB, Sienaert I, Bultynck G, Zizi M, De Smet P, Casteels R, De Smedt H. Adenine-nucleotide binding sites on the inositol 1,4,5-trisphosphate receptor bind caffeine, but not adenophostin A or cyclic ADP-ribose. Cell Calcium. 1999;25:143–152. doi: 10.1054/ceca.1998.0011. [DOI] [PubMed] [Google Scholar]
- [78].Mak DO, McBride S, Foskett JK. ATP regulation of type 1 inositol 1,4,5-trisphosphate receptor channel gating by allosteric tuning of Ca2+ activation. J Biol Chem. 1999;274:22231–22237. doi: 10.1074/jbc.274.32.22231. [DOI] [PubMed] [Google Scholar]
- [79].Wagner LE, Yule DI. Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J Physiol. 2012;590:3245–3259. doi: 10.1113/jphysiol.2012.228320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Park HS, Betzenhauser MJ, Won JH, Chen J, Yule DI. The type 2 inositol (1,4,5)-trisphosphate (InsP3) receptor determines the sensitivity of InsP3-induced Ca2+ release to ATP in pancreatic acinar cells. J Biol Chem. 2008;283:26081–26088. doi: 10.1074/jbc.M804184200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Park HS, Betzenhauser MJ, Zhang Y, Yule DI. Regulation of Ca2+ release through inositol 1,4,5-trisphosphate receptors by adenine nucleotides in parotid acinar cells. Am J Physiol Gastrointest Liver Physiol. 2012;302:G97–G104. doi: 10.1152/ajpgi.00328.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Alzayady KJ, Wagner LE, 2nd, Chandrasekhar R, Monteagudo A, Godiska R, Tall GG, Joseph SK, Yule DI. Functional inositol 1,4,5-trisphosphate receptors assembled from concatenated homo- and heteromeric subunits. J Biol Chem. 2013;288:29772–29784. doi: 10.1074/jbc.M113.502203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Khan MT, Wagner L, Yule DI, Bhanumathy CD, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol.Chem. 2006;281:3731–3737. doi: 10.1074/jbc.M509262200. [DOI] [PubMed] [Google Scholar]
- [84].Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Söderberg O, Bootman MD, Roderick HL. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A. 2008;105:2427–2432. doi: 10.1073/pnas.0711324105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Betzenhauser MJ, Yule DI. Regulation of inositol 1,4,5-trisphosphate receptors by phosphorylation and adenine nucleotides. Curr Top Membr. 2010;66C:273–298. doi: 10.1016/S1063-5823(10)66012-7. [DOI] [PubMed] [Google Scholar]
- [86].Walaas SI, Nairn AC, Greengard P. PCPP-260, a Purkinje cell-specific cyclic AMP-regulated membrane phosphoprotein of Mr 260,000. J Neurosci. 1986;6:954–961. doi: 10.1523/JNEUROSCI.06-04-00954.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mignery GA, Newton CL, Archer BT, 3rd, Südhof TC. Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1990;265:12679–12685. [PubMed] [Google Scholar]
- [88].Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SH. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochem Biophys Res Commun. 1991;175:192–198. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- [89].Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase A and protein phosphatase 1α. J Neurosci. 2003;23:403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wagner LE, 2nd, Li WH, Yule DI. Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases. A mutational analysis of the functionally important sites in the S2+ and S2− splice variants. J Biol Chem. 2003;278:45811–45817. doi: 10.1074/jbc.M306270200. [DOI] [PubMed] [Google Scholar]
- [91].Wagner LE, 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol. 2008;586:3577–3596. doi: 10.1113/jphysiol.2008.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wagner LE, 2nd, Li WH, Joseph SK, Yule DI. Functional consequences of phosphomimetic mutations at key cAMP-dependent protein kinase phosphorylation sites in the type 1 inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2004;279:46242–46252. doi: 10.1074/jbc.M405849200. [DOI] [PubMed] [Google Scholar]
- [93].Bruce JI, Shuttleworth TJ, Giovannucci DR, Yule DI. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J Biol Chem. 2002;277:1340–1348. doi: 10.1074/jbc.M106609200. [DOI] [PubMed] [Google Scholar]
- [94].Regimbald-Dumas Y, Arguin G, Fregeau MO, Guillemette G. cAMP-dependent protein kinase enhances inositol 1,4,5-trisphosphate-induced Ca2+ release in AR4-2J cells. J Cell Biochem. 2007;101:609–618. doi: 10.1002/jcb.21221. [DOI] [PubMed] [Google Scholar]
- [95].Hajnoczky G, Gao E, Nomura T, Hoek JB, Thomas AP. Multiple mechanisms by which protein kinase A potentiates inositol 1,4,5-trisphosphate-induced Ca2+ mobilization in permeabilized hepatocytes. Biochem J. 1993;293:413–422. doi: 10.1042/bj2930413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wojcikiewicz RJ, Luo SG. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J Biol Chem. 1998;273:5670–5677. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- [97].Betzenhauser MJ, Fike JL, Wagner LE, 2nd, Yule DI. Protein kinase A increases type-2 inositol 1,4,5-trisphosphate receptor activity by phosphorylation of serine 937. J Biol Chem. 2009;284:25116–25125. doi: 10.1074/jbc.M109.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bruce JI, Straub SV, Yule DI. Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium. 2003;34:431–444. doi: 10.1016/s0143-4160(03)00150-7. [DOI] [PubMed] [Google Scholar]
- [100].Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919–939. doi: 10.1006/jmcc.2002.2038. [DOI] [PubMed] [Google Scholar]
- [103].Ferris CD, Huganir RL, Bredt DS, Cameron AM, Snyder SH. Inositol trisphosphate receptor: phosphorylation by protein kinase C and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc Natl Acad Sci U S A. 1991;88:2232–2235. doi: 10.1073/pnas.88.6.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Matifat F, Hague F, Brulé G, Collin T. Regulation of InsP3-mediated Ca2+ release by CaMKII in Xenopus oocytes. Pflügers Arch. 2001;441:796–801. doi: 10.1007/s004240000479. [DOI] [PubMed] [Google Scholar]
- [105].Zhu DM, Tekle E, Chock PB, Huang CY. Reversible phosphorylation as a controlling factor for sustaining calcium oscillations in HeLa cells: involvement of calmodulin-dependent kinase II and a calyculin A-inhibitable phosphatase. Biochemistry. 1996;35:7214–7223. doi: 10.1021/bi952471h. [DOI] [PubMed] [Google Scholar]
- [106].Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA. Cardiac type 2 inositol 1,4,5-trisphosphate receptor. Interaction and modulation by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:15912–15920. doi: 10.1074/jbc.M414212200. [DOI] [PubMed] [Google Scholar]
- [107].Maxwell JT, Natesan S, Mignery GA. Modulation of inositol 1,4,5-trisphosphate receptor type 2 channel activity by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated phosphorylation. J Biol Chem. 2012;287:39419–39428. doi: 10.1074/jbc.M112.374058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Bootman MD, Roderick HL. Why, where, and when do cardiac myocytes express inositol 1,4,5-trisphosphate receptors? Am J Physiol Heart Circ Physiol. 2008;294:H579–581. doi: 10.1152/ajpheart.01378.2007. [DOI] [PubMed] [Google Scholar]
- [109].Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Vermassen E, Fissore RA, Nadif Kasri N, Vanderheyden V, Callewaert G, Missiaen L, Parys JB, De Smedt H. Regulation of the phosphorylation of the inositol 1,4,5-trisphosphate receptor by protein kinase C. Biochem Biophys Res Commun. 2004;319:888–893. doi: 10.1016/j.bbrc.2004.05.071. [DOI] [PubMed] [Google Scholar]
- [111].Matter N, Ritz MF, Freyermuth S, Rogue P, Malviya AN. Stimulation of nuclear protein kinase C leads to phosphorylation of nuclear inositol 1,4,5-trisphosphate receptor and accelerated calcium release by inositol 1,4,5-trisphosphate from isolated rat liver nuclei. J Biol Chem. 1993;268:732–736. [PubMed] [Google Scholar]
- [112].Arguin G, Regimbald-Dumas Y, Fregeau MO, Caron AZ, Guillemette G. Protein kinase C phosphorylates the inositol 1,4,5-trisphosphate receptor type 2 and decreases the mobilization of Ca2+ in pancreatoma AR4-2J cells. J Endocrinol. 2007;192:659–668. doi: 10.1677/JOE-06-0179. [DOI] [PubMed] [Google Scholar]
- [113].Tovey SC, Dedos SG, Taylor EJ, Church JE, Taylor CW. Selective coupling of type 6 adenylyl cyclase with type 2 IP3 receptors mediates direct sensitization of IP3 receptors by cAMP. J Cell Biol. 2008;183:297–311. doi: 10.1083/jcb.200803172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Tovey SC, Dedos SG, Rahman T, Taylor EJ, Pantazaka E, Taylor CW. Regulation of inositol 1,4,5-trisphosphate receptors by cAMP independent of cAMP-dependent protein kinase. J Biol Chem. 2010;285:12979–12989. doi: 10.1074/jbc.M109.096016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wilson BS, Pfeiffer JR, Smith AJ, Oliver JM, Oberdorf JA, Wojcikiewicz RJ. Calcium-dependent clustering of inositol 1,4,5-trisphosphate receptors. Mol Biol Cell. 1998;9:1465–1478. doi: 10.1091/mbc.9.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Diambra L, Marchant JS. Localization and socialization: experimental insights into the functional architecture of IP3 receptors. Chaos. 2009;19:037103. doi: 10.1063/1.3147425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Sheppard CA, Simpson PB, Sharp AH, Nucifora FC, Ross CA, Lange GD, Russell JT. Comparison of type 2 inositol 1,4,5-trisphosphate receptor distribution and subcellular Ca2+ release sites that support Ca2+ waves in cultured astrocytes. J Neurochem. 1997;68:2317–2327. doi: 10.1046/j.1471-4159.1997.68062317.x. [DOI] [PubMed] [Google Scholar]
- [118].Pantazaka E, Taylor CW. Differential distribution, clustering, and lateral diffusion of subtypes of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2011;286:23378–23387. doi: 10.1074/jbc.M111.236372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Yule DI, Ernst SA, Ohnishi H, Wojcikiewicz RJ. Evidence that zymogen granules are not a physiologically relevant calcium pool. Defining the distribution of inositol 1,4,5-trisphosphate receptors in pancreatic acinar cells. J Biol Chem. 1997;272:9093–9098. doi: 10.1074/jbc.272.14.9093. [DOI] [PubMed] [Google Scholar]
- [120].Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH, Wuytack F, Racymaekers L, Muallem S. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J Biol Chem. 1997;272:15765–15770. doi: 10.1074/jbc.272.25.15765. [DOI] [PubMed] [Google Scholar]
- [121].Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi-Iwanaga H, Noda T, Aruga J, Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science. 2005;309:2232–2234. doi: 10.1126/science.1114110. [DOI] [PubMed] [Google Scholar]
- [122].Zhang X, Wen J, Bidasee KR, Besch HR, Jr., Wojcikiewicz RJ, Lee B, Rubin RP. Ryanodine and inositol trisphosphate receptors are differentially distributed and expressed in rat parotid gland. Biochem J. 1999;340:519–527. [PMC free article] [PubMed] [Google Scholar]
- [123].Yamamoto-Hino M, Miyawaki A, Segawa A, Adachi E, Yamashina S, Fujimoto T, Sugiyama T, Furuichi T, Hasegawa M, Mikoshiba K. Apical vesicles bearing inositol 1,4,5-trisphosphate receptors in the Ca2+ initiation site of ductal epithelium of submandibular gland. J Cell Biol. 1998;141:135–142. doi: 10.1083/jcb.141.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Inaba T, Hisatsune C, Sasaki Y, Ogawa Y, Ebisui E, Ogawa N, Matsui M, Takeuchi T, Mikoshiba K, Tsubota K. Mice lacking inositol 1,4,5-trisphosphate receptors exhibit dry eye. PLoS One. 2014;9:e99205. doi: 10.1371/journal.pone.0099205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Fukuda N, Shirasu M, Sato K, Ebisui E, Touhara K, Mikoshiba K. Decreased olfactory mucus secretion and nasal abnormality in mice lacking type 2 and type 3 IP3 receptors. Eur J Neurosci. 2008;27:2665–2675. doi: 10.1111/j.1460-9568.2008.06240.x. [DOI] [PubMed] [Google Scholar]
- [126].Klar J, Hisatsune C, Baig SM, Tariq M, Johansson AC, Rasool M, Malik NA, Ameur A, Sugiura K, Feuk L, Mikoshiba K, Dahl N. Abolished InsP3R2 function inhibits sweat secretion in both humans and mice. J Clin Invest. 2014;124:4773–4780. doi: 10.1172/JCI70720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Shibao K, Hirata K, Robert ME, Nathanson MH. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology. 2003;125:1175–1187. doi: 10.1016/s0016-5085(03)01201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Li M, Miyawaki A, Yamamoto-Hino M, Yasutomi D, Furuichi T, Hasegawa M, Mikoshiba K. Differential cellular expression of three types of inositol 1,4,5-trisphosphate receptor in rat gastrointestinal epithelium. Biomed Res. 1996;17:45–51. [Google Scholar]
- [129].Williams JA, Yule DI. Stimulus-secretion coupling in pancreatic acinar cells. In: Johnson ELR, editor. Physiology of the Gastrointestinal Tract. 5th Edition Academic Press; San Diego: 2012. pp. 1361–1398. [Google Scholar]
- [130].Melvin JE, Yule D, Shuttleworth T, Begenisich T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol. 2005;67:445–469. doi: 10.1146/annurev.physiol.67.041703.084745. [DOI] [PubMed] [Google Scholar]
- [131].Kasai H, Augustine GJ. Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature. 1990;348:735–738. doi: 10.1038/348735a0. [DOI] [PubMed] [Google Scholar]
- [132].Kasai H, Li YX, Miyashita Y. Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell. 1993;74:669–677. doi: 10.1016/0092-8674(93)90514-q. [DOI] [PubMed] [Google Scholar]
- [133].Thorn P, Lawrie AM, Smith PM, Gallacher DV, Petersen OH. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell. 1993;74:661–668. doi: 10.1016/0092-8674(93)90513-p. [DOI] [PubMed] [Google Scholar]
- [134].Kruglov EA, Gautam S, Guerra MT, Nathanson MH. Type 2 inositol 1,4,5-trisphosphate receptor modulates bile salt export pump activity in rat hepatocytes. Hepatology. 2011;54:1790–1799. doi: 10.1002/hep.24548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hernandez E, Leite MF, Guerra MT, Kruglov EA, Bruna-Romero O, Rodrigues MA, Gomes DA, Giordano FJ, Dranoff JA, Nathanson MH. The spatial distribution of inositol 1,4,5-trisphosphate receptor isoforms shapes Ca2+ waves. J Biol Chem. 2007;282:10057–10067. doi: 10.1074/jbc.M700746200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000;10:939–942. doi: 10.1016/s0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- [137].Uchida K, Aramaki M, Nakazawa M, Yamagishi C, Makino S, Fukuda K, Nakamura T, Takahashi T, Mikoshiba K, Yamagishi H. Gene knock-outs of inositol 1,4,5-trisphosphate receptors types 1 and 2 result in perturbation of cardiogenesis. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- [139].Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res. 2003;92:1305–1313. doi: 10.1161/01.RES.0000077045.84609.9F. [DOI] [PubMed] [Google Scholar]
- [140].Nakazawa M, Uchida K, Aramaki M, Kodo K, Yamagishi C, Takahashi T, Mikoshiba K, Yamagishi H. Inositol 1,4,5-trisphosphate receptors are essential for the development of the second heart field. J Mol Cell Cardiol. 2011;51:58–66. doi: 10.1016/j.yjmcc.2011.02.014. [DOI] [PubMed] [Google Scholar]
- [141].Mackenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, Li WH, Lipp P. The role of inositol 1,4,5-trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol. 2002;541:395–409. doi: 10.1113/jphysiol.2001.013411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zima AV, Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation-contraction coupling and arrhythmias. J Physiol. 2004;555:607–615. doi: 10.1113/jphysiol.2003.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- [144].Proven A, Roderick HL, Conway SJ, Berridge MJ, Horton JK, Capper SJ, Bootman MD. Inositol 1,4,5-trisphosphate supports the arrhythmogenic action of endothelin-1 on ventricular cardiac myocytes. J Cell Sci. 2006;119:3363–3375. doi: 10.1242/jcs.03073. [DOI] [PubMed] [Google Scholar]
- [145].Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H596–604. doi: 10.1152/ajpheart.01155.2007. [DOI] [PubMed] [Google Scholar]
- [146].Harzheim D, Movassagh M, Foo RS, Ritter O, Tashfeen A, Conway SJ, Bootman MD, Roderick HL. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:11406–11411. doi: 10.1073/pnas.0905485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Harzheim D, Talasila A, Movassagh M, Foo RS, Figg N, Bootman MD, Roderick HL. Elevated InsP3R expression underlies enhanced calcium fluxes and spontaneous extra-systolic calcium release events in hypertrophic cardiac myocytes. Channels (Austin) 2010;4:67–71. doi: 10.4161/chan.4.1.10531. [DOI] [PubMed] [Google Scholar]
- [148].Higazi DR, Fearnley CJ, Drawnel FM, Talasila A, Corps EM, Ritter O, McDonald F, Mikoshiba K, Bootman MD, Roderick HL. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol Cell. 2009;33:472–482. doi: 10.1016/j.molcel.2009.02.005. [DOI] [PubMed] [Google Scholar]
- [149].Drawnel FM, Wachten D, Molkentin JD, Maillet M, Aronsen JM, Swift F, Sjaastad I, Liu N, Catalucci D, Mikoshiba K, Hisatsune C, Okkenhaug H, Andrews SR, Bootman MD, Roderick HL. Mutual antagonism between IP3RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J Cell Biol. 2012;199:783–798. doi: 10.1083/jcb.201111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, Lorenz JN, Blatter LA, Bers DM, Molkentin JD. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res. 2010;107:659–666. doi: 10.1161/CIRCRESAHA.110.220038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Molkentin JD. Dichotomy of Ca2+ in the heart: contraction versus intracellular signaling. J Clin Invest. 2006;116:623–626. doi: 10.1172/JCI27824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Joseph SK, Hajnoczky G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis. 2007;12:951–968. doi: 10.1007/s10495-007-0719-7. [DOI] [PubMed] [Google Scholar]
- [153].Harr MW, Distelhorst CW. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol. 2010;2:a005579. doi: 10.1101/cshperspect.a005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta. 2011;1813:1003–1013. doi: 10.1016/j.bbamcr.2010.11.023. [DOI] [PubMed] [Google Scholar]
- [155].Jayaraman T, Marks AR. T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol Cell Biol. 1997;17:3005–3012. doi: 10.1128/mcb.17.6.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 2012;3:e304. doi: 10.1038/cddis.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Csordás G, Thomas AP, Hajnóczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Parys JB. The IP3 receptor as a hub for Bcl-2 family proteins in cell death control and beyond. Sci Signal. 2014;7:e4. doi: 10.1126/scisignal.2005093. [DOI] [PubMed] [Google Scholar]
- [160].Distelhorst CW, Bootman MD. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: role in Ca2+ signaling and disease. Cell Calcium. 2011;50:234–241. doi: 10.1016/j.ceca.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Monaco G, Vervliet T, Akl H, Bultynck G. The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol Life Sci. 2013;70:1171–1183. doi: 10.1007/s00018-012-1118-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Akl H, Bultynck G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta. 2013;1835:180–193. doi: 10.1016/j.bbcan.2012.12.001. [DOI] [PubMed] [Google Scholar]
- [163].Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- [164].Jayaraman T, Marks AR. Calcineurin is downstream of the inositol 1,4,5-trisphosphate receptor in the apoptotic and cell growth pathways. J Biol Chem. 2000;275:6417–6420. doi: 10.1074/jbc.275.9.6417. [DOI] [PubMed] [Google Scholar]
- [165].Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- [166].Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J Cell Sci. 2014;127:1607–1619. doi: 10.1242/jcs.141408. [DOI] [PubMed] [Google Scholar]
- [167].Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330:1247–1251. doi: 10.1126/science.1189157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Kopacek J, Ondrias K, Sedlakova B, Tomaskova J, Zahradnikova L, Sedlak J, Sulova Z, Zahradnikova A, Pastorek J, Krizanova O. Type 2 IP3 receptors are involved in uranyl acetate induced apoptosis in HEK 293 cells. Toxicology. 2009;262:73–79. doi: 10.1016/j.tox.2009.05.006. [DOI] [PubMed] [Google Scholar]
- [170].Lencesova L, Hudecova S, Csaderova L, Markova J, Soltysova A, Pastorek M, Sedlak J, Wood ME, Whiteman M, Ondrias K, Krizanova O. Sulphide signalling potentiates apoptosis through the up-regulation of IP3 receptor types 1 and 2. Acta Physiol (Oxf) 2013;208:350–361. doi: 10.1111/apha.12105. [DOI] [PubMed] [Google Scholar]
- [171].Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, Yule DI, Joseph SK, Hajnoczky G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem. 2014;289:8170–8181. doi: 10.1074/jbc.M113.504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, Molgo J, Distelhorst CW, Missiaen L, Mikoshiba K, Parys JB, De Smedt H, Bultynck G. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013;4:e632. doi: 10.1038/cddis.2013.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Akl H, Vervloessem T, Kiviluoto S, Bittremieux M, Parys JB, De Smedt H, Bultynck G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: mitochondria versus endoplasmic reticulum. Biochim Biophys Acta. 2014;1843:2240–2252. doi: 10.1016/j.bbamcr.2014.04.017. [DOI] [PubMed] [Google Scholar]
- [174].Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD, Mignery GA, Parys JB, De Smedt H, Distelhorst CW. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Molecular Cell. 2008;31:255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A. 2009;106:14397–14402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, Parys JB, Leybaert L, Bultynck G. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19:295–309. doi: 10.1038/cdd.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Rong YP, Barr P, Yee VC, Distelhorst CW. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim Biophys Acta. 2009;1793:971–978. doi: 10.1016/j.bbamcr.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, Rong YP, Molitoris JK, Lam M, Ryder C, Matsuyama S, Distelhorst CW. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood. 2011;117:2924–2934. doi: 10.1182/blood-2010-09-307405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Xu S, Cai Y, Wei Y. mTOR signaling from cellular senescence to organismal aging. Aging Dis. 2014;5:263–273. doi: 10.14336/AD.2014.0500263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- [183].Burgess DJ. Senescence: tumorigenesis under surveillance. Nat Rev Cancer. 2012;12:6. doi: 10.1038/nrc3188. [DOI] [PubMed] [Google Scholar]
- [184].Serrano M. Cancer: final act of senescence. Nature. 2011;479:481–482. doi: 10.1038/479481a. [DOI] [PubMed] [Google Scholar]
- [185].Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- [186].Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, Beach D, Serrano M. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- [187].Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- [188].Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D, Bernard D. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun. 2014;5:3792. doi: 10.1038/ncomms4792. [DOI] [PubMed] [Google Scholar]
- [189].Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type 2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272:23961–23969. doi: 10.1074/jbc.272.38.23961. [DOI] [PubMed] [Google Scholar]
- [190].Sharp AH, Nucifora FC, Jr., Blondel O, Sheppard CA, Zhang C, Snyder SH, Russell JT, Ryugo DK, Ross CA. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J Comp Neurol. 1999;406:207–220. [PubMed] [Google Scholar]
- [191].Monkawa T, Hayashi M, Miyawaki A, Sugiyama T, Yamamoto-Hino M, Hasegawa M, Furuichi T, Mikoshiba K, Saruta T. Localization of inositol 1,4,5-trisphosphate receptors in the rat kidney. Kidney Int. 1998;53:296–301. doi: 10.1046/j.1523-1755.1998.00763.x. [DOI] [PubMed] [Google Scholar]
- [192].Parys JB, De Smedt H, Missiaen L, Bootman MD, Sienaert I, Casteels R. Rat basophilic leukemia cells as model system for inositol 1,4,5-trisphosphate receptor IV, a receptor of the type II family: functional comparison and immunological detection. Cell Calcium. 1995;17:239–249. doi: 10.1016/0143-4160(95)90070-5. [DOI] [PubMed] [Google Scholar]