

Abstract
Cyanide is a life threatening, bioterrorist agent, preventing cellular respiration by inhibiting cytochrome c oxidase, resulting in cardiopulmonary failure, hypoxic brain injury, and death within minutes. However, even after treatment with various antidotes to protect cytochrome oxidase, cyanide intoxication in humans can induce a delayed-onset neurological syndrome that includes symptoms of Parkinsonism. Additional mechanisms are thought to underlie cyanide-induced neuronal damage, including generation of reactive oxygen species (ROS). This may account for the fact that antioxidants prevent some aspects of cyanide-induced neuronal damage. Here, as a potential preemptive countermeasure against a bioterrorist attack with cyanide, we tested the CNS protective effect of carnosic acid (CA), a pro-electrophilic compound found in the herb rosemary. CA crosses the blood-brain-barrier to upregulate endogenous antioxidant enzymes via activation of the Nrf2 transcriptional pathway. We demonstrate that CA exerts neuroprotective effects on cyanide-induced brain damage in cultured rodent and human induced pluripotent stem cell (hiPSC)-derived neurons in vitro, and in vivo in various brain areas of a non-Swiss albino (NSA) mouse model of cyanide poisoning that simulates damage observed in the human brain.
Keywords: Carnosic acid, Cyanide poisoning, Electrophilic compounds, Nrf2, Anti-oxidant Response Element, Human iPSC-derived neurons
Acute cyanide poisoning is life threatening, blocking cellular respiration by inhibiting the mitochondrial enzyme cytochrome c oxidase. The result is cardiopulmonary failure and death within minutes (Hamel 2011; Nelson 2006). Cyanide is also a potential bioterroristic agent, e.g., that can be toxic via subacute intake in a contaminated water supply. If recognized quickly, systemic cyanide poisoning is treatable by agents binding to cyanide in order to protect cytochrome c oxidase, and efforts devoted to developing more effective antidotes are ongoing (Hamel 2011; Brenner et al. 2010; Hall et al. 2009). In a 2007 annual report from the American Association of Poison Control Centers, most of the 242 cyanide-poisoning cases survived; there were only 5 fatalities (Bronstein et al. 2008). Complete protection from acute cyanide poisoning depends on rapid administration and effective penetration into various tissues. However, current systemic antidotes, such as hydroxocobalamin, may not adequately cross the blood-brain-barrier (BBB), so the brain may remain vulnerable even in humans surviving acute cyanide poisoning. In fact, in over a dozen human suicide cases of cyanide intoxication, a delayed neurological syndrome has been documented that includes dystonia and parkinsonian signs and symptoms, even in patients who had initially appeared to have had a full recovery after the exposure (Rachinger et al. 2002; Riudavets et al. 2005; Rosenow et al. 1995; Uitti et al. 1985; Valenzuela et al. 1992; Borgohain et al. 1995; Rosenberg et al. 1989; Carella et al. 1988). Typically, these patients begin to manifest symptoms and signs after a few weeks or months, with progressive rigidity accompanied by flexed upper limbs and extended lower limbs. CT and MRI examinations of the brain consistently revealed lesions in the basal ganglia, including the globus pallidus and putamen. Damage in the substantia nigra, cerebellum, and cerebral cortex has also been reported in several human cases (Rosenow et al. 1995; Uitti et al. 1985; Carella et al. 1988). This imaging evidence for damage in the human brain has been confirmed at autopsy in some cases (Uitti et al. 1985; Riudavets et al. 2005). Similar observations have been reported in human patients after subchronic, lower dose, non-lethal cyanide exposure (Di Filippo et al. 2008), as might occur in a bioterroristic attempt at contaminating a city’s water supply.
Multiple mechanisms are thought to underlie cyanide-induced neuronal damage. For example, in addition to binding to and poisoning hemoglobin, cyanide inhibits cytochrome c oxidase, thus blocking electron transport in the mitochondrial respiratory chain and disrupting oxidative phosphorylation (OX-PHOS) (Hamel 2011). Such global cessation of aerobic cell metabolism results in severe consequences for active cellular processes requiring ATP. Additionally, increased levels of reactive oxygen species (ROS) generated in response to cyanide poisoning initiate lipid peroxidation that is toxic to neurons (Johnson et al. 1987). Particularly harmful to neurons is the malfunction of glutamate transport and sodium/potassium ion exchangers, contributing to excitotoxicity due to excessive extracellular glutamate (Persson et al. 1985; Patel et al. 1991). Under these conditions, glutamate overstimulates a variety of receptors, including N-methyl-D-aspartate-type glutamate receptors (NMDARs), to literally excite neurons to death. Additionally, the release of normal Mg2+ blockade of NMDARs because of membrane depolarization, with consequent inrush of cations, further overactivates NMDARs (Nowak et al. 1984). Therefore, neurons are particularly vulnerable to cyanide poisoning, although different brain regions show differential sensitivity to cyanide (Prabhakaran et al. 2002).
Since cyanide poisoning blocks mitochondrial respiration, which produces an increase in ROS in the brain with consequent severe oxidative damage in neurons, one possible strategy for the development of neuroprotective drugs is to use low-molecular-weight compounds that can cross the blood-brain-barrier and counter oxidative damage. In this regard, our group is developing pro-electrophilic drugs (PEDs) to treat neurological disorders due in part to excessive ROS; these drugs become active electrophilic compounds (EPs) when they encounter oxidative insults that chemically convert them to the active form (Satoh and Lipton 2007). By using pro-electrophiles that are themselves innocuous, we increase the chances that a compound will be a clinically-tolerated therapeutic. In contrast, compounds that are electrophilic themselves (often represented by quinone structures) may be toxic, in part, by reacting with and depleting glutathione (GSH) in normal areas of the brain (Satoh et al. 2013). We found that one such pro-electrophilic compound is carnosic acid (CA), a natural substance found in the herb rosemary (Satoh et al. 2008a; Satoh et al. 2008b; Satoh et al. 2011). Previously, we showed that CA readily crosses the blood-brain-barrier and exerts its protective effect after conversion from its catechol form to its quinone form through redox stress-induced oxidation/activation (Satoh et al. 2008a; Satoh et al. 2008b). This conversion makes CA an electrophile that then binds to the protein Keap1 in the cytoplasm of neural cells, probably in astrocytes to a larger extent than neurons, to release the transcription factor Nrf2. Nrf2 is thus ‘activated’ and enters the nucleus to stimulate transcription of an endogenous antioxidant enzyme system known as phase 2 enzymes (Satoh et al. 2008b) (Fig. 1). Here, we show that CA protects a variety of neuronal types from different brain regions from sublethal cyanide poisoning both in vitro and in vivo when administered repeatedly around the time of a subchronic exposure. Importantly, for the first time we are able to test the effects of a potential bioterroristic agent like cyanide in a human context by using human induced pluripotent stem cell (hiPSC)-derived neurons in our in vitro work. Additionally, this approach allows us to test the neuroprotective effect of CA in a human context. The results suggest that CA is a prospective therapeutic agent for preventing brain pathophysiology caused by cyanide poisoning, in which oxidative stress plays an important role.
Fig. 1.

Neuroprotective mechanism of carnosic acid (CA). CA is a pro-electrophilic drug (PED) for treatment of neurological disorders in part due to excessive ROS; these drugs become active electrophilic compounds when they encounter oxidative insults that chemically convert them to the active form (Satoh and Lipton 2007). CA upregulates endogenous antioxidant enzyme systems via reaction with Keap1 to release Nrf2 for translocation to the nucleus where it activates the transcription of neuroprotective Phase 2 antioxidant genes.
Materials and methods
Preparation of rat primary cerebrocortical and mesencephalic cultures
Primary cerebrocortical neurons and astrocytes were isolated from embryonic day 15 (E15) or E16 Sprague-Dawley rats (Harlan) and maintained in culture as described previously (Uehara et al. 2006). In brief, dissected cerebrocortices were treated with trypsin, and dissociated cells were plated on poly-L-lysine coated glass coverslips in D10C medium. Experiments were performed 20–21 days after plating.
Mixed neuronal/glial mesencephalic primary cultures, including the substantia nigra, were prepared from E13 fetuses of C57BL/6J mice. The mesencephalon was dissected in ice-cold EBSS (Gibco-Invitrogen) and subsequently digested in 0.25% trypsin (Gibco-Invitrogen) for 35 minutes at 37 °C in a 7% CO2 humidified incubator. Tissues were washed with preincubation medium containing DMEM, Ham’s F-12 (1:1) (Omega Scientific), 10% fetal bovine serum (FBS, HyClone), 50 IU/ml penicillin (Omega Scientific) and 50 μg/ml streptomycin (Omega Scientific) and dissociated by trituration. Cells were plated on 24-well plates coated with poly-L-lysine (Sigma) in preincubation medium:incubation medium (1:1), the latter containing DMEM Ham’s F-12, 0.5 mM L-glutamine (Gibco-Invitrogen), 1% B27 (Gibco-Invitrogen), 50 IU/ml penicillin and 50 μg/ml streptomycin, and maintained at 37 °C in a 5% CO2 humidified incubator; 24 h after plating, the entire medium was replaced with incubation medium. Subsequently, half of the incubation medium was replaced once every four days. At 14 days in vitro (DIV), cells were treated according to the specific experimental design.
For mesencephalon cultures, glass coverslips in 24-well plates were initially coated with poly-L-lysine (0.1 mg/ml) for 24 h. Coverslips were then coated with laminin for 24 h, following which 500 μl of preincubation medium, consisting of DMEM-F12, 10% FBS, and 1% penicillin/streptomycin (P/S), was allotted to each well and incubated overnight. Mesencephalic tissue was dissected out from E13 fetuses of C57BL/6J mice and placed in dissection medium. Trypsin (500 μl, 0.25%) was added to the tissue and incubated for 35 minutes, at 37 °C and 5% CO2. The preparation was triturated and cells transferred to NPC medium, consisting of 0.25%/0.5 mM Glutamate, 1% P/S, and Neurobasal. The mesencephalic cell suspension (500 μl) in NPC medium, containing 200,000 cells, was pipetted into each well containing preincubation medium, and then incubated for 2 weeks prior to use.
Generation of neurons from human iPSCs
(a) Differentiation protocol for cortical neurons
Human iPSCs were generated from normal human fibroblasts (Hs27, ATCC CRL-1634) by using an integration free reprogramming method (Okita et al. 2011), which uses three episomal vectors that collectively encode OCT3/4, SOX2, KLF4, L-MYC, LYN28, p53-shRNA. Details of the hiPSC characterization are described elsewhere (Talantova et al. 2013). hiPSCs were maintained as we described before (Lin et al. 2009). Cortical neuron differentiation of hiPSCs was performed by modification of a previously method (Chambers et al. 2009). Briefly, for 1 week, hiPSC colonies were exposed to small-molecule inhibitors of bone morphogenic protein (Dorsomorphin, Wnt/Activin/Nodal (A83-01). Then, colonies were exposed to small-molecule inhibitors as neurospheres for ~2–3 weeks in the presence of basic FGF (20 ng/mL). Mature neurospheres were triturated and dissociated with Accutase (Invitrogen) and adhered as a monolayer of cells on polyornithine/laminin-coated dishes. Clusters of neural stem cells (NSCs) exhibiting rosette morphology were manually isolated and expanded for several passages in DMEM/F12 supplemented with basic FGF (20 ng/mL), N2 and B27 (Invitrogen) and later seeded onto polyornithine/laminin-coated coverslips for terminal differentiation. NSCs were terminally differentiated for at least 6 weeks in the presence of medium supplemented with N2 and B27, BDNF (10 ng/mL) and GDNF (10 ng/mL) before experiments were conducted. Cells were immunostained for neuronal markers (Activin/Nodal, A83-0, NeuN) and/or cortical markers (TBR1, CTIP2) and further characterized as described to ascertain their identity and maturity.
(b) Differentiation protocol for dopaminergic neurons
Differentiation of hiPSCs into A9-type dopaminergic neurons (the type of neurons initially injured in Parkinson’s disease) was performed as recently described (Ryan et al. 2013). Immediately preceding differentiation, the colonies were dissociated into a single cell suspension using Accutase. To purify hiPSCs and remove fibroblast feeders, cultured cells were dissociated and placed in gelatin-coated dishes. After adherence of the fibroblasts, supernatant containing purified hiPSCs was collected and re-plated at 4×104 cells/cm2 on Matrigel (BD)-coated tissue culture dishes for differentiation. Floor-plate induction was carried out using medium containing knockout serum replacement (KSR), LDN193189 (100 nM), SB431542 (10 μM), Sonic Hedgehog (SHH) C25II (100 ng/ml), Purmorphamine (2 μM), fibroblast growth factor 8 (FGF8; 100 ng/ml), and CHIR99021 (3 μM). On day 5 of differentiation, KSR medium was incrementally shifted to N2 medium (25%, 50%, 75%) every 2 days. On day 11, the medium was changed to Neurobasal/B27/Glutamax supplemented with CHIR. On day 13, CHIR was replaced with Brain Derived Neurotrophic Factor (BDNF; 20 ng/ml), ascorbic acid (0.2 mM), Glial Derived Neurotrophic Factor (GDNF; 20 ng/ml), transforming growth factor beta 3 (TGFβ3; 1 ng/ml), dibutyryl cAMP (db cAMP; 0.5 mM), and DAPT (10 μM) for 9 days. On day 20, cells were dissociated using Accutase and re-plated under high cell density (4×105 cells/cm2) in terminal differentiation medium (NB/B27 + BDNF, ascorbic acid, GDNF, db cAMP, TGFβ3 and DAPT), designated Dopaminergic Neuron Medium, on dishes precoated with polyornithine (15 μg/ml):laminin (1 μg/ml):fibronectin (2 μg/ml).
Analysis of cell death by TUNEL in cultured neurons
Neuronal cultures on coverslips were treated with CA dissolved in DMSO as a 10 mM stock (1000X) and incubated for 24 h prior to insult with KCN. The cultures were then exposed to 500 μM KCN for 24 h. Immediately after exposure, cells were washed with PBS, and then fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature (RT). After fixation, cells were washed with PBS three times followed by permeabilization with 3% Triton X-100 for 30 min at RT. After permeabilization, cells were washed with PBS three times, and then stained with a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) In Situ Cell Death Detection Kit, TMR red (Roche). Coverslips were washed three times with PBS and mounted on slides with DAPI fluoromount. Images were taken using a deconvolution microscope in 5 random fields per preselected brain area for each brain and quantified by a masked observer.
Mouse model of cyanide poisoning and rescue by carnosic acid (CA)
Two- to three-month-old male NSA mice were purchased from Harlan Laboratories. All procedures involving live animals were approved by the Sanford-Burnham Medical Research Institute IACUC. Mice were split into four groups that received intraperitoneal (i.p.) injections of either vehicle (PBS, pH 7.4) or KCN dissolved in PBS, with or without CA. Mice were pretreated for one week with oral CA in food pellets (0.05% wt/wt CA) or the same diet without added CA. Thereafter, mice were kept on the same diet while injected i.p. twice daily with 5–6 mg KCN/kg body weight in a volume equivalent to 10 μl solution/g body weight for 8–12 days. The model was chosen based on the prior published model of Gary Isom (Mills et al. 1999), and the doses were chosen to simulate levels attained after human ingestion of cyanide, for example, in the water supply after a bioterroristic attack using modeling data supplied by the NIH CounterACT Program. In this series of experiments, CA was administered before subacute KCN exposure, as it was felt in a ‘real-world’ scenario in the face of a threatened bioterroristic attack, pretreatment with an innocuous agent like CA was a reasonable approach. One to two weeks after KCN exposure (with or without CA treatment), mice were analyzed for behavioral changes. Mice were then deeply anesthetized and sacrificed by transcardial perfusion, first with PBS and then with 4% PFA. Following completion of behavioral testing, mice were placed under deep anesthesia with isoflurane and sacrificed by transcardial perfusion, first with saline followed by 4% PFA. Brains were sectioned and used for neuropathological analysis of cortex, hippocampus, and striatum.
Neuropathological analysis of mouse brains
Neuropathological analysis was performed using methods previously published by our group (Talantova et al. 2013; Meng et al. 2011; Cho et al. 2011). To determine the effects of cyanide exposure and CA treatment, brains were fixed in 4% PFA, cut into 40 μm-thick vibratome sections and immunolabeled with mouse monoclonal antibodies against the neuronal-specific proteins, MAP2 (1:100; Millipore), NeuN (1:500; Millipore), and synaptophysin (1:500; Millipore). Primary antibodies were detected with horse anti-mouse FITC-conjugated antibody (1:75; Vector) and mounted under glass coverslips using anti-fading media (Vector). All sections were processed by standardized conditions. Staining was analyzed by quantitative confocal immunofluorescence using blind-coded sections, serially imaged on a laser-scanning confocal microscope, and quantified using NIH Image 1.43 software. A total of three sections for each brain and four fields for each section were analyzed in each brain area. For MAP2 and synaptophysin staining, results are expressed as percent area (% area) of the neuropil occupied by immunoreactive dendrites or terminals. NeuN-positive nuclei were counted stereologically using Stereo Investigator software (MicroBrightField, Wiliston, VT).
Animal behavior tests
For the neurobehavioral tests, we monitored mice receiving the 5 mg/kg cyanide dosing since it was found to be sublethal in the vast majority of cases. The Open Field Test was conducted to assess spontaneous exploratory and anxiety-related behavior in a 30 min trial. During the test, animals were placed in a chamber measuring 16 × 16 inches that provided a novel environment and were allowed to roam freely for the full duration of the test. Tracking software was used to divide the chamber into an inner square measuring 8 × 8 inches and an outer region encompassing the remaining portion of the chamber. The distance traveled within the inner and outer regions was recorded over the full 30 min period by sensing infrared beam breaks (each infrared sensing beam was 1 inch apart in both the x and y directions). Environmental odors were removed after each trial by cleaning the chamber so as to avoid any influence on the animal subject in the subsequent trial. Data were analyzed using AnyMaze computer software (San Diego Instruments).
The Grip (Motor) Test was performed on a flat metal grating in a round bucket. Each animal was placed in the center of the flat metal grating and then the metal grating was turned upside down on top of the bucket so that the animal could not escape through the sides and could only hold onto the grating or fall into the bucket. As soon as the metal grating was turned onto the top of the bucket, the observer began timing and looked at the animal’s hind legs for the proper gripping behavior criteria to stop the test. The test was terminated when any of the following three criteria were met: The animal’s hind legs came off the grating and were suspended in the air for at least two seconds (this two-second time period was used as the criterion because some animals would pull their hind legs off momentarily in order to reposition them on the grating); the animal fell off the grating completely into the bucket; or the animal held onto the grating for the full three minute period. Once one of these three criteria was met, the observer would stop the test and record the time. The test was repeated three times for each animal with at least five minutes of rest between trials.
Kaplan-Meier survival curves
Animal fatality was monitored daily during KCN exposure and documented using GraphPad Prism to generate the Kaplan Meier Curve. Fatalities were recorded with the dosing regimen of 5 mg/kg KCN (see Results).
Statistics
Data are presented as mean + SEM. Comparisons between two conditions were performed by a two-tailed Student’s t test. Multiple comparisons were performed by a one-way ANOVA with a Tukey’s post hoc test. Significance was set at a p value of < 0.05. Statistical calculations were performed using GraphPad PRISM software.
Results
Carnosic acid (CA) antagonizes cyanide-induced cytotoxicity in neuronal cultures
We first established an in vitro model of cyanide poisoning using rat primary cerebrocortical cell cultures and found in a preliminary dose-ranging study that exposure to 300 μM KCN for 24 h significantly increased neuronal apoptosis. We then treated cultures for 24 h in 10 μM CA, a concentration previously determined to be neuroprotective in dose-ranging studies (Satoh et al. 2008a; Satoh et al. 2008b), prior to KCN exposure and found significant protection from apoptosis (Fig. 2a). Next, in order to expand our investigations into a human context, we repeated these experiments using hiPSCs differentiated into cerebrocortical neurons. Similar to cultured rat cortical cells, KCN exposure dramatically increased apoptotic cell death above that of vehicle-treated control, whereas CA treatment protected neurons, reducing the number of apoptotic cells to a level not significantly different from controls (Fig. 2b).
Fig. 2.
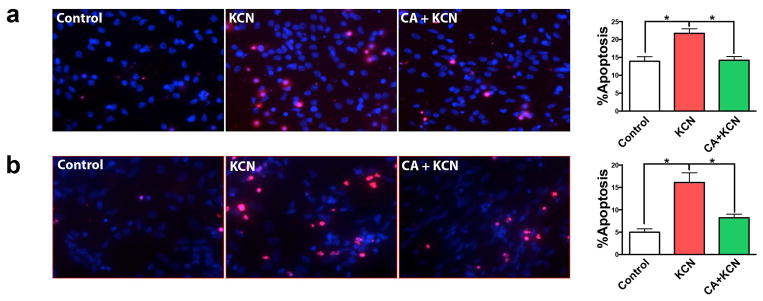
Protective effect of CA on KCN-induced apoptotic cell death in rat and human cortical neuronal cultures. (a) Representative images of primary rat cortical neurons: vehicle-treated control and KCN-exposed with or without treatment with CA. Apoptotic cell death was identified by TUNEL staining (red); all cell nuclei were stained with DAPI (blue). Histogram shows the percentage of apoptotic neurons in each group. (b) KCN-induced apoptotic death and rescue by CA was investigated in human iPSC-derived cortical neurons. All data are expressed as mean + SEM (n = 20; *p < 0.001 by one-way ANOVA with Tukey’s post test).
Since humans surviving cyanide exposure may exhibit parkinsonian symptoms and damage in the striatum, we next tested neurons that project to the striatum and are most vulnerable in Parkinson’s disease. Accordingly, we investigated the effect of KCN exposure on neurons obtained from the rat mesencephalon and on human iPS cells differentiated into A9-type dopaminergic neurons. Rat mesencephalic neurons were highly sensitive to KCN, as KCN exposure induced approximately twice the level of apoptotic cell death found in cortical neurons, and CA reduced KCN-induced cell death (Fig. 3a). Similarly, CA treatment decreased the percentage of KCN-induced cell death in hiPSC-derived dopaminergic neurons (Fig. 3b). Next, we tested the effect of CA in protecting neurons in an in vivo model of cyanide poisoning.
Fig. 3.
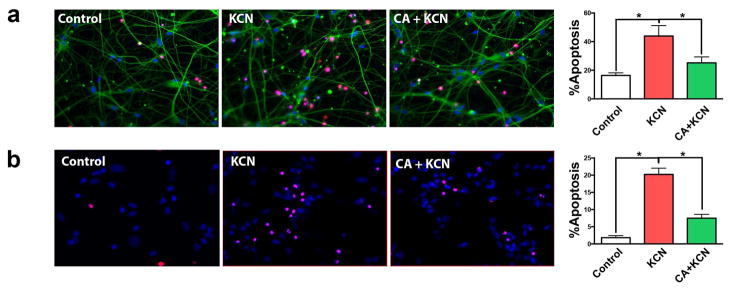
Protective effect of CA on KCN-induced apoptotic cell death in mouse and human dopaminergic neuronal cultures. (a) Representative images show apoptotic cells (red, TUNEL staining) in primary mouse mesencephalic neurons for vehicle-treated control, 300 μM KCN exposed with and without CA treatment. Cell nuclei were stained by DAPI (blue) and neurites were stained with MAP2 (green). Histogram shows the percentage of apoptotic cells under the corresponding treatment conditions. (b) Similar tests were conducted using human iPSC-derived DA neurons, and corresponding representative images and quantification histograms are displayed. All data are expressed as mean + SEM (n = 10; *p < 0.05 by one-way ANOVA with Tukey’s post test).
CA reduces neurotoxicity and improves neurobehavioral outcome in an in vivo mouse model of cyanide poisoning
(a) NeuroBehavioral improvement after treatment with CA
Previous work had shown that parenteral injections of KCN produced a delayed-onset type of damage in various brain areas (Mills et al. 1999). Here, we developed a model of cyanide poisoning that might simulate a ‘real world scenario’ in a bioterroristic attack resulting in CNS damage in animals not succumbing to a subacute insult (see Materials and methods). In preliminary experiments with non-Swiss albino (NSA) mice, we found that ≥ 6 mg/kg KCN, administered i.p. twice daily for 8 to 12 days, produced excessive neurological morbidity and mortality; hence, subsequent studies were performed using a dose of 5 mg/kg. With these results in hand, the experimental paradigm for testing the protective effect of CA is shown in Figure 4a. Treatment with CA was followed by KCN exposure. We then performed neurobehavioral tests that are sensitive to damage in brain regions known to be affected by cyanide poisoning and associated with motor dysfunction and anxiety. Subsequently, we sacrificed the mice for histological evaluation.
Fig. 4.

In vivo mouse model of cyanide poisoning. (a) Experimental paradigm for KCN administration and CA treatment. Non-Swiss albino mice were fed normal chow or chow containing 0.05% (wt/wt) CA for 7 days prior to exposure to KCN via i.p. injection twice daily for 8 days. Mice remained on the same chow throughout the experiment. Mice underwent behavioral testing for two weeks after the final injection, whereupon brains were harvested for neuropathological analysis. (b) Kaplan-Meier curve shows the survival of mice under various exposure/treatment conditions (n = 83 total animals). Control received vehicle only. (c) Neuromuscular strength was assessed by a Grip Test under the various exposure/treatment conditions (total n = 81, *p < 0.01). (d) Anxiety level was measured by the Open Field Test for mice under the various exposure/treatment conditions (total n = 29; *p < 0.05 by one-way ANOVA). Data for panels c and d are expressed as mean + SEM.
We found that a dose of 5 mg/kg resulted in significant mortality in mice receiving the control diet; however, as shown in Figure 4b, this mortality could be largely prevented by treatment with CA (0.05% wt/wt in food pellets), a dose previously shown to produce micromolar brain levels that were neuroprotective (Satoh et al. 2008a; Satoh et al. 2008b) and similar to those used in the in vitro experiments described above. In surviving animals, we found in this model that behavioral testing 1–2 weeks post cyanide exposure resulted in a reduced time that the mice were able to hold onto a metal screen with their hind legs in the grip/motor strength test by 50% (n = 9, p < 0.0001) (Fig. 4c). Importantly, CA treatment protected against this deficit by 90% (n = 24; p < 0.01). KCN exposure also caused an increase in anxiety behavior as measured by total time in the center of the apparatus on the Open Field Test; this deficit was corrected by CA treatment (Fig. 4d).
(b) Histological evidence for neuroprotection by CA
To evaluate KCN-induced damage in neocortex, hippocampus, and striatum, we analyzed sections of mouse brain for cell death by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). We observed significant KCN-induced damage in the frontal cortex (p < 0.01), hippocampus (p < 0.03) and striatum (p < 0.01), as quantified by the apoptotic score (Fig. 5).
Fig. 5.
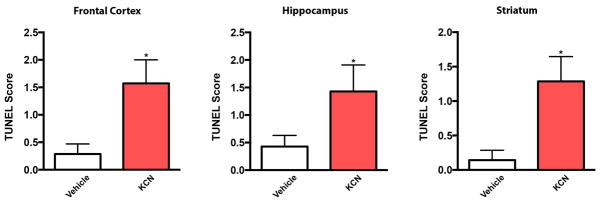
KCN exposure causes neuronal apoptosis. Histograms show the TUNEL Score (number of TUNEL-positive neurons per 0.1 sq. mm) for mouse frontal cortex, hippocampus, and striatum in vehicle- and KCN-exposed tissue. Data are expressed as mean + SEM (n = 7 for each group; *p < 0.01 for cortex and striatum, and *p < 0.03 for hippocampus by Student’s t test).
We further examined neuronal integrity and the ability of CA to protect against KCN exposure using NeuN, microtubule-associated protein 2 (MAP2), and synaptophysin antibodies to quantify surviving neurons, their processes, and synapses, respectively (see Materials and methods). We used NeuN staining to quantify the total number of neurons in the cortex, hippocampus, and striatum of KCN- vs. vehicle-exposed mice in the presence or absence of CA treatment (Fig. 6). We observed a significant reduction in the total number of neurons in the KCN-exposed group. Importantly, treatment with CA offered protection from this loss of neurons. Specifically, in the neocortex the number of NeuN positive cells in the KCN-exposed group was significantly lower (p < 0.002, n = 8) compared to vehicle control (n = 14) or the CA-only control group (n = 7). The KCN group receiving CA exhibited significant preservation of neurons (p = 0.01, n = 8) compared to the KCN-exposed group. Similarly, total neuronal cell counts in the hippocampus revealed a significant decrement in NeuN-labeled neurons in the KCN-exposed group (p < 0.0004, n = 8) compared to the vehicle control (n = 14) or the CA-only control group (n = 7). Additionally, CA significantly protected from KCN-induced neuronal cell loss (p < 0.007, n = 8). Finally, KCN also induced significant neuronal loss in the striatum (p ≤ 0.0007, n = 8) compared to vehicle control (n = 14) or the CA-only control group (n = 7). Again, CA provided significant neuroprotection (p ≤ 0.0007, n = 8) from KCN.
Fig. 6.

Protective effect of CA on KCN-induced loss of neurons. Representative NeuN staining of neocortex (Nctx), hippocampus (Hippo), and striatum (Str) of mouse brain exposed to vehicle control, CA-only, KCN, or CA+KCN. Low and high magnification images are shown on top and bottom, respectively (scale bars, 250 μm at low power and 40 μm at high power). Histograms below show stereological cell counts of the affected brain areas in neocortex, hippocampus, and striatum under different treatment conditions. All data are expressed as mean + SEM (total n = 37; *p < 0.01 by one-way ANOVA with Tukey’s post test).
By quantifying MAP2 staining, we observed a significant reduction in neuropil due to KCN (p < 0.003, n = 8) compared to the groups treated with vehicle control (n = 14) or CA-only (n = 7; Fig. 7). Mice pretreated with CA prior to KCN exposure (n = 8) exhibited significant protection of neuropil compared to the KCN-exposed group (p < 0.04). Similarly in the hippocampus, there was a significant reduction of neuropil in the KCN-exposed group (p < 0.0001, n = 8) compared to the vehicle control (n = 14) or CA-only group (n = 7), while the CA+KCN group was significantly protected (p ≤ 0.0007, n = 8). Finally, in the hippocampus, synaptophysin staining showed that cyanide exposure decreased synaptic density (n = 8), which was restored to normal levels by CA treatment (p < 0.001, n = 15) (Fig. 7). Taken together, these data indicate that treatment with CA can protect from neuronal damage in vivo in this mouse model of cyanide poisoning.
Fig. 7.

Protective effect of CA from KCN-induced damage to neuropil and synapses. Representative images of MAP2 and synaptophysin staining of the brain following exposure of mice to vehicle, CA-only, KCN, or CA+KCN. Low and high magnification images are shown on top and bottom, respectively (scale bars, 250 μm at low power and 40 μm at high power). Histograms below show quantitative analysis of the neuropil in neocortex (Nctx) and hippocampus (Hipp) by MAP2 staining, and synaptic density in the hippocampus via synaptophysin staining. All data are expressed as mean + SEM (total n = 37; *p < 0.04 by one-way ANOVA with Tukey’s post test).
Discussion
Currently, no treatment is available to protect the brain from cyanide poisoning, e.g., in the event of sublethal ingestion of cyanide in a contaminated water supply or in food sources. Additionally, many patients with acute cyanide inhalation survive when medical intervention with systemic antidotes, such as hydroxocobalamin, is immediately available. However, in both circumstances, the delayed-onset of dystonia and parkinsonian-like symptoms can severely impact the quality of life and productivity of affected individuals. In the present study, we investigated the use of CA, a pro-electrophilic compound found in the herb Rosemary, for protection against cyanide-associated brain injury. Cyanide-induced neuronal damage is mediated by several processes that result in the production of oxidative stress in the brain. To counteract this effect, we used CA, which crosses the blood-brain-barrier to exert its effects by upregulating endogenous phase 2 antioxidant enzymes via the Nrf2 transcriptional pathway (Satoh et al. 2008a; Satoh et al. 2008b; Satoh and Lipton 2007; Satoh et al. 2013).
Previously, we demonstrated neuroprotection by CA in a variety of in vitro and in vivo models of neurological damage. We and others also reported that CA enters the brain to attain neuroprotective levels, as monitored by mass spectrometry of HPLC samples, after oral, parenteral, or transnasal administration (Satoh et al. 2008b; Vaka et al. 2011; Rezaie et al. 2012). These prior findings suggested that CA may have clinical application for various forms of neuronal damage induced predominantly by oxidative stress, and thus motivated our use of CA to prevent cyanide-related brain damage.
After repeated i.p. injection of cyanide in mice, the group of Dr. Gary Isom (Mills et al. 1999) had previously demonstrated delayed-onset loss of neurons in some brain areas also found to be damaged after human cyanide poisoning. Our improved mouse model even more closely simulates brain damage found in surviving human cyanide victims, including delayed loss of neuronal cell bodies, neuropil, and synapses in striatum, neocortex, or hippocampus. Importantly, we found that pretreatment with oral CA significantly protected from cyanide-induced brain damage in our mouse model. Since we have previously shown that CA can also be delivered to the brain after parenteral or transnasal administration, the drug is also potentially amenable to rapid deployment for the treatment of humans exposed to cyanide in the field during a bioterrorist attack, e.g., in conjunction with systemic cyanide-scavenging agents. Moreover, we have confirmed the neuroprotective effect of CA in ‘a human context’ using hiPSC-derived neurons in vitro. Taken together, these data are consistent with the notion that CA may be useful for protecting the human brain from cyanide exposure.
In summary, our studies demonstrate both in vitro in hiPSC-derived neurons and in vivo in a robust mouse model that CA is an effective drug to reduce brain injury occurring after sublethal cyanide poisoning. Since CA is a natural compound appearing on the FDA’s ‘generally regarded as safe’ (GRAS) list, easily crosses the blood-brain-barrier, activates various antioxidant genes, and produces a long-lasting, amplified action, we propose that the development of CA for treatment of cyanide-induced brain injury represents a feasible therapeutic approach.
Acknowledgments
This work was supported in part by NIH grants R21 NS080799, R01 NS086890, R01 ES017462, P01 HD29587, the La Jolla Interdisciplinary Neuroscience Center Core Grant P30 NS076411, and by the Michael J. Fox Foundation. We thank Ravi Agarwal for graphic assistance.
Abbreviations
- NMDAR
N-methyl-D-aspartate-type glutamate receptors
- ROS
reactive oxygen species
- CA
carnosic acid
- hiPSC
human induced pluripotent stem cell
- BBB
blood-brain-barrier
- OX-PHOS
oxidative phosphorylation
- PEDs
pro-electrophilic drugs
- EPs
electrophilic compounds
- GSH
glutathione
- NSA
non-Swiss albino
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- GRAS
generally regarded as safe
- DIV
days in vitro
- NSCs
neural stem cells
- KSR
knockout serum replacement
- BDNF
Brain Derived Neurotrophic Factor
- GDNF
Glial Derived Neurotrophic Factor
- TGFβ3
transforming growth factor beta 3
- PFA
paraformaldehyde
Footnotes
Conflict-of-interest disclosure
The authors declare no competing interests.
References
- Borgohain R, Singh AK, Radhakrishna H, Rao VC, Mohandas S. Delayed onset generalised dystonia after cyanide poisoning. Clin Neurol Neurosurg. 1995;97:213–215. doi: 10.1016/0303-8467(95)00029-j. [DOI] [PubMed] [Google Scholar]
- Brenner M, Mahon SB, Lee J, et al. Comparison of cobinamide to hydroxocobalamin in reversing cyanide physiologic effects in rabbits using diffuse optical spectroscopy monitoring. J Biomed Opt. 2010;15:017001. doi: 10.1117/1.3290816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein AC, Spyker DA, Cantilena LR, Jr, Green JL, Rumack BH, Heard SE. 2007 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 25th Annual Report. Clin Toxicol (Phila) 2008;46:927–1057. doi: 10.1080/15563650802559632. [DOI] [PubMed] [Google Scholar]
- Carella F, Grassi MP, Savoiardo M, Contri P, Rapuzzi B, Mangoni A. Dystonic-Parkinsonian syndrome after cyanide poisoning: clinical and MRI findings. J Neurol Neurosurg Psychiatry. 1988;51:1345–1348. doi: 10.1136/jnnp.51.10.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EG, Zaremba JD, McKercher SR, et al. MEF2C enhances dopaminergic neuron differentiation of human embryonic stem cells in a parkinsonian rat model. PLoS ONE. 2011;6:e24027. doi: 10.1371/journal.pone.0024027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Filippo M, Tambasco N, Muzi G, Balucani C, Saggese E, Parnetti L, Calabresi P, Rossi A. Parkinsonism and cognitive impairment following chronic exposure to potassium cyanide. Mov Disord. 2008;23:468–470. doi: 10.1002/mds.21871. [DOI] [PubMed] [Google Scholar]
- Hall AH, Saiers J, Baud F. Which cyanide antidote? Crit Rev Toxicol. 2009;39:541–552. doi: 10.1080/10408440802304944. [DOI] [PubMed] [Google Scholar]
- Hamel J. A review of acute cyanide poisoning with a treatment update. Crit Care Nurse. 2011;31:72–81. doi: 10.4037/ccn2011799. quiz 82. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Conroy WG, Burris KD, Isom GE. Peroxidation of brain lipids following cyanide intoxication in mice. Toxicology. 1987;46:21–28. doi: 10.1016/0300-483x(87)90134-x. [DOI] [PubMed] [Google Scholar]
- Lin T, Ambasudhan R, Yuan X, et al. A chemical platform for improved induction of human iPSCs. Nat Methods. 2009;6:805–808. doi: 10.1038/nmeth.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Yao D, Shi Y, et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener. 2011;6:34. doi: 10.1186/1750-1326-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EM, Gunasekar PG, Li L, Borowitz JL, Isom GE. Differential susceptibility of brain areas to cyanide involves different modes of cell death. Toxicol Appl Pharmacol. 1999;156:6–16. doi: 10.1006/taap.1999.8630. [DOI] [PubMed] [Google Scholar]
- Nelson L. Acute cyanide toxicity: mechanisms and manifestations. J Emerg Nurs. 2006;32:S8–11. doi: 10.1016/j.jen.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbert A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, et al. A more efficient method to generate intergration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Patel MN, Ardelt BK, Yim GK, Isom GE. Cyanide induces Ca(2+)-dependent and -independent release of glutamate from mouse brain slices. Neurosci Lett. 1991;131:42–44. doi: 10.1016/0304-3940(91)90332-n. [DOI] [PubMed] [Google Scholar]
- Persson SA, Cassel G, Sellstrom A. Acute cyanide intoxication and central transmitter systems. Fundam Appl Toxicol. 1985;5:S150–159. doi: 10.1016/0272-0590(85)90124-1. [DOI] [PubMed] [Google Scholar]
- Prabhakaran K, Li L, Borowitz JL, Isom GE. Cyanide induces different modes of death in cortical and mesencephalon cells. J Pharmacol Exp Ther. 2002;303:510–519. doi: 10.1124/jpet.102.039453. [DOI] [PubMed] [Google Scholar]
- Rachinger J, Fellner FA, Stieglbauer K, Trenkler J. MR changes after acute cyanide intoxication. AJNR Am J Neuroradiol. 2002;23:1398–1401. [PMC free article] [PubMed] [Google Scholar]
- Rezaie T, McKercher SR, Kosaka K, et al. Protective effect of carnosic acid, a pro-electrophilic compound, in models of oxidative stress and light-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53:7847–7854. doi: 10.1167/iovs.12-10793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riudavets MA, Aronica-Pollak P, Troncoso JC. Pseudolaminar necrosis in cyanide intoxication: a neuropathology case report. Am J Forensic Med Pathol. 2005;26:189–191. [PubMed] [Google Scholar]
- Rosenberg NL, Myers JA, Martin WR. Cyanide-induced parkinsonism: clinical, MRI, and 6-fluorodopa PET studies. Neurology. 1989;39:142–144. doi: 10.1212/wnl.39.1.142. [DOI] [PubMed] [Google Scholar]
- Rosenow F, Herholz K, Lanfermann H, Weuthen G, Ebner R, Kessler J, Ghaemi M, Heiss WD. Neurological sequelae of cyanide intoxication--the patterns of clinical, magnetic resonance imaging, and positron emission tomography findings. Ann Neurol. 1995;38:825–828. doi: 10.1002/ana.410380518. [DOI] [PubMed] [Google Scholar]
- Ryan SD, Dolatabadi N, Chan SF, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Izumi M, Inukai Y, et al. Carnosic acid protects neuronal HT22 Cells through activation of the antioxidant-responsive element in free carboxylic acid- and catechol hydroxyl moieties-dependent manners. Neurosci Lett. 2008a;434:260–265. doi: 10.1016/j.neulet.2008.01.079. [DOI] [PubMed] [Google Scholar]
- Satoh T, Kosaka K, Itoh K, et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2008b;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Lipton SA. Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 2007;30:37–45. doi: 10.1016/j.tins.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Satoh T, McKercher SR, Lipton SA. Nrf2;ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med. 2013;65:645–657. doi: 10.1016/j.freeradbiomed.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Rezaie T, Seki M, et al. Dual neuroprotective pathways of a pro-electrophilic compound via HSF-1-activated heat-shock proteins and Nrf2-activated phase 2 antioxidant response enzymes. J Neurochem. 2011;119:569–578. doi: 10.1111/j.1471-4159.2011.07449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Nat Acad Sci USA. 2013;110:E2518–2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Uitti RJ, Rajput AH, Ashenhurst EM, Rozdilsky B. Cyanide-induced parkinsonism: a clinicopathologic report. Neurology. 1985;35:921–925. doi: 10.1212/wnl.35.6.921. [DOI] [PubMed] [Google Scholar]
- Vaka SR, Murthy SN, Repka MA, Nagy T. Upregulation of endogenous neurotrophin levels in the brain by intranasal administration of carnosic acid. J Pharm Sci. 2011;100:3139–3145. doi: 10.1002/jps.22528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela R, Court J, Godoy J. Delayed cyanide induced dystonia. J Neurol Neurosurg Psychiatry. 1992;55:198–199. doi: 10.1136/jnnp.55.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]