

Summary
The organ of Corti harbors highly specialized sensory hair cells and surrounding supporting cells that are essential for the sense of hearing. Here, we report a single cell gene expression data analysis and visualization strategy that allows for the construction of a quantitative spatial map of the neonatal organ of Corti along its major anatomical axes. The map displays gene expression levels of 192 genes for all organ of Corti cell types ordered along the apex-to-base axis of the cochlea. Statistical interrogation of cell type-specific gene expression patterns along the longitudinal gradient revealed features of apical supporting cells indicative of a propensity for proliferative hair cell regeneration. This includes reduced expression of Notch effectors, receptivity for canonical Wnt signaling, and prominent expression of early cell cycle genes. Cochlear hair cells displayed expression gradients of genes indicative of cellular differentiation and the establishment of the tonotopic axis.
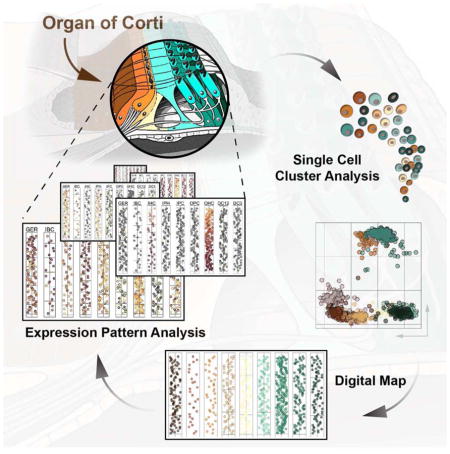
Introduction
The organ of Corti is the mammalian organ of hearing and harbors some of the most rare and unique cell types of the body. Organized in single longitudinal rows of about 800 cells in the adult mouse (Ehret and Frankenreiter, 1977), the organ of Corti cells are arranged in a medial-to-lateral pattern with the more abundant cells of the greater epithelial ridge (GER) defining the medial side (Figure 1A). Laterally situated to the GER are consecutive rows of inner border cells, sensory inner hair cells (IHCs), inner phalangeal cells, inner and outer pillar cells, followed by a mosaic of three rows of sensory outer hair cells (OHCs) and Deiters’ cells. Whereas the integrated function of some individual organ of Corti cell types, particularly of the sensory IHCs and OHCs are well-described (Hudspeth, 2014), the role(s) of the various non-sensory supporting cells are much less understood. The paucity and inaccessibility of organ of Corti cells has made molecular studies challenging. Single cell technology provides an opportunity to overcome such challenges and to study gene expression in the organ of Corti comprehensively.
Figure 1.

Sorting of different organ of Corti cell types. (A) Schematic representation of the mouse organ of Corti at P2 and color code used for different cell types. (B, C) Fluorescent reporter gene expression in a representative mid-basal P2 organ of Corti cryo-section of the Atoh1-nGFP/Fgfr3-iCreERT2/Ai14-tdTomato mouse line. (D) FACS plot and gating strategy for isolation of GFP-expressing cells (gate 1). (E-M) Analogous data representation for three additional mouse lines. Scale bars: B,E,H,K, 10μm. Related to Figure S1.
Here we describe a single cell data analysis and visualization strategy to generate a quantitative gene expression map along the major anatomical axes for all cell types of the organ of Corti. We utilized reporter mice, fluorescence activated cell sorting (FACS) and microfluidic arrays to conduct single cell quantitative (q)RT-PCR measurements for 192 genes representative of individual organ of Corti cell types and major and minor signaling pathways. Iterative k-means clustering and principal component analysis (PCA) was used to classify 808 assayed cells into nine distinct groups. We then developed a method to rebuild the organ’s core anatomical features in silico using spatially derived gene expression information. This strategy resulted in a quantitative, digital, two-dimensional map of the organ of Corti where cell type-specific rows are visualized as one-dimensional trajectories representing apex-to-base orientations. When compared with existing gene expression studies, the map’s nine groups faithfully recapitulated known expression domains that correspond to hair cell and supporting cell subtypes. Moreover, our model revealed distinct expression gradients in specific cell types along the apex-to-base axis of the cochlea. Statistical analyses of gene expression among the different organ of Corti cell types as well as along the apex-to-base axis revealed a domain-specific interplay of reduced Notch activity, elevated canonical Wnt activity and elevated levels of early cell cycle genes that could account for differences in the regenerative potential among supporting cells in the neonatal cochlea. Likewise, we identified several genes that representatively visualize emerging tonotopic patterns in maturing hair cells of the organ of Corti. The general concept introduced in this study is universally applicable and can be utilized to establish comprehensive 2D maps of other complex tissues.
Results
Isolation of Organ of Corti Cells
We used six different mouse reporter alleles that in four combinations target specific hair cell and supporting cell subtypes (Figure 1). Cochlear ducts of postnatal day 2 (P2) mice were divided into apical and basal pieces and enzymatically separated into single cells. We then used FACS to isolate individual cells for subsequent gene expression analysis.
The first mouse line used was Atoh1-nGFP/Fgfr3-CreERT2/Ai14-tdTomato expressing nuclear localized GFP (nGFP) under control of an Atoh1 enhancer element (Lumpkin et al., 2003) in combination with Fgfr3-CreERT2 driver (Young et al., 2010) and Ai14-tdTomato reporter alleles. nGFP was detected in IHCs and OHCs as well as inner border and inner phalangeal cells (Figure 1B,C). Conditional expression of tdTomato was found in pillar cells, Deiters’ cells, and sporadically in OHCs. We collected 192 individual nGFP-positive cells (gate 1, Figures 1D and S1A), which we hypothesized to represent hair cells, inner border and inner phalangeal cells. nGFP-tdTomato double positive cells were not specifically gated as these presumptive OHCs were traceable based on detection of the recombined tdTomato allele (Figure S2A,A′). An additional set of 96 cells was also included that originated from Atoh1-nGFP mice (Figure S1E).
The second mouse line combined the Fgfr3-CreERT2/Ai14-tdTomato alleles with GFP knocked into the Sox2 locus to enrich for pillar and Deiters’ cells co-expressing tdTomato and GFP. Except for Hensen’s cells GFP was strongly expressed in all organ of Corti supporting cell types, including GER cells located adjacent to the inner hair cells (Figure 1E,F). Two distinct populations of double positive cells – characterized by different GFP expression levels – were collected as indicated by gates 3 and 4 (Figures 1G and S1B). In addition, we sorted two populations that expressed GFP exclusively but again at different intensity levels as shown in gate 2 and 7. We hypothesized that cells from these two gates correspond to supporting cell subtypes outside the Fgfr3-domain, namely GER, inner border and inner phalangeal cells. Finally, we sampled 12 cells expressing tdTomato alone that served as a negative control and define cell types from regions outside the organ of Corti (gate 8, Figure 1G and S1B). Applying all five gates we collected in total 480 single cells from this mouse line.
We next used Sox2-GFP/Glast-CreERT2/Ai14-tdTomato triple transgenic mice where tdTomato expression was found in inner border, inner phalangeal and Hensen’s cells, and in a mosaic pattern within the GER (Figure 1H,I). 96 GFP-tdTomato double positive cells were collected in gate 5, presumptively representing GER cells in excess as well as comparable numbers of inner border and inner phalangeal cells (Figures 1J and S1C).
The fourth mouse line featured GFP knocked into the Lgr5 locus. As previously reported (Chai et al., 2011; Shi et al., 2012), GFP expression at P2 occurred in inner border cells, inner phalangeal cells, inner pillar cells, in the 3rd row of Deiters’ cells, as well as in GER cells in close proximity to the inner hair cell row (Figure 1K,L). 96 Lgr5-GFP positive cells were sorted from gate 6 (Figures 1M and S1D).
In total we collected 960 single cells, representative for all hair cell and supporting cell types of the organ of Corti. For quality control purposes and downstream analyses each cell is associated to a string of metadata that is of non-transcriptional information including multi-well plate position, date of experiment, mouse line, FACS gate and whether it came from the cochlear apex or base.
Identification of Organ of Corti Cell Types Based on Transcriptional Profiles
Each cell was subjected to a series of multi-parallel qRT-PCR reactions (Table S1). A quarter of the 192 assayed genes represented previously known markers associated with cellular identities (Table S2). The remaining primer pairs monitored the activity of signaling pathway related genes that have been implicated in early inner ear development or were detectable in previously conducted bulk RNA analyses of neonatal cochlea-derived cells (Sajan et al., 2007; Sinkkonen et al., 2011). Data was normalized as previously reported (Durruthy-Durruthy et al., 2014) and subjected to stringent control evaluation methods, leaving a total of 877 single cells for subsequent analyses (Figure S2B-H and Table S1).
We hypothesized that a grouping strategy, applied in successively ranked steps would allow us to classify all 877 cells and identify the major organ of Corti cell types (Figure 2A,B). For this purpose we employed k-means clustering (MacQueen, 1967), an unsupervised learning algorithm that associates similar objects of a given data set (here cells) to an a priori defined number of clusters. To visualize the multivariate data and recognize patterns, we performed principal component analysis (PCA, (Jolliffe, 2002)) and projected the data onto a compressed two-dimensional coordinate system.
Figure 2.

Sequential clustering of organ of Corti cell types into cell type specific groups. (A) Legend and color key for panels C-K. (B) Subcluster tree of identified populations of all sorted cells across ten levels (I – X); grey boxes indicate intermediate groups and black boxes indicate final groups. (C) Principal component analysis (PCA) and k-means clustering of all 877 cells. Shown are projections onto lower-dimensional data space (principal component scores (PC1 and PC2)) visualizing different color-coded parameters. Each data point represents a single cell. Upper left: FACS-Gates 1 – 8. Upper right: Variable factor map indicates six representative genes that are most contributive for data separation. This representation shows the correlation between the first two components and the original variables (genes), which are called component loadings. Analogous to correlation coefficients their negative/positive values and direction can be understood as a measure of how much of variation in a variable is explained by the component. Second row: For each population expression levels of one representative marker is projected onto data, here Jag2 and Jag1. Gene expression levels range from grey (absent), green (low expression) to red (high expression). Bar graphs indicate k-means cluster-related relative contribution for each FACS gate (in %). Highest gate contribution for each group is indicated. Large square plot: Cells are colored according to computed k-means clusters (black = hair cells, grey = non-hair cells). (D-K) Analogous data representation for subsequently clustered subpopulations. (L) Table summarizing total cell number determined for each identified population. Related to Figures S2, S3 and S6.
We applied this strategy to all 877 cells in an unbiased approach, disregarding each cell’s associated metadata (i.e. mouse line, FACS gate). We hypothesized that the cells could be initially subdivided into major cell identities, comprising the presumptive hair cells and supporting cell lineages. PCA recognized two main populations that coincided with afore-defined k-means clusters (Figures 2B,C and S3A). High levels of well-described hair cell markers including Pou4f3, Cdkn2d (p19-INK4d), and Jag2 (Chen et al., 2003; Erkman et al., 1996; Lanford et al., 1999) were associated with one of the two clusters. In contrast, the cells of the other group lacked expression of hair cell markers and predominantly expressed supporting cell associated genes like Jag1, Cdkn1b (p27(Kip1)) and Sox9 (Chen and Segil, 1999; Lanford et al., 1999; Lowenheim et al., 1999; Mak et al., 2009). To further discriminate within each of the identified groups of presumptive hair cells and supporting cells we iteratively applied our integrative approach to dissect the dataset by successive bifurcations into smaller subpopulations (Figure 2B).
The hair cell population segregated into two subclusters (Figures 2D and S3B), representing 205 OHCs characterized by expression of Cdh1 (E-Cadherin), Fgfr3, and Slc26a5 (Prestin) (Anttonen et al., 2012; Simonneau et al., 2003; Tateya et al., 2013; Zheng et al., 2000), and 40 IHCs expressing known identifiers Fgf8, Fgf10 and S100a1 (Buckiova and Syka, 2009; Pirvola et al., 2002).
The non-hair cell group of 632 cells was bisected by k-means into two groups that were notably characterized by either absence or presence of Fgfr3 expression (Figures 2E and S3C). 78% of the Fgfr3-negative population originated from the Atoh1-nGFP/Fgfr3-CreERT2/Ai14-tdTomato line, Sox2-GFP/Glast-CreERT2/Ai14-tdTomato line and Lgr5-GFP line (gates 1, 5 and 6, respectively). According to our sorting strategy, we expected these gates to contain inner border, inner phalangeal and GER cells. Consistent with this assumption those cells were characterized by high Gjb2 and Gjb6 (Connexin 26 and 30) expression (Zhang et al., 2005). On the other hand 83% of the Fgfr3-positive population was derived from gate 3 (Sox2-GFP/Fgfr3-CreERT2/Ai14-tdTomato), characterized by high levels of known pillar and Deiters’ cells markers such as Fgfr3 and Prox1 (Anttonen et al., 2012; Tateya et al., 2013). Reciprocal Etv4 (Mansour et al., 2013) and Dkk3 (Huang et al., 2011) expression divided the Fgfr3-positive population into pillar cells and presumptive Deiters’ cells (Figures 2F and S3D). Lgr5 (Chai et al., 2011) and Ngfr (Shim et al., 2005; White et al., 2006) expression distinguished 99 inner pillar cells from 87 outer pillar cells that were positive for Dkk3 and Aqp4 (Figures 2G and S3E). The organ of Corti is equipped with three rows of Deiters’ cells whereby in neonates the third row is distinguishable from rows 1 and 2 by Lgr5 expression (Figure 1K,L (Chai et al., 2011)). Based on the opposing expression patterns of Lgr5 and Kcnj10 (Kir4.1) we were able to identify 82 third row Deiters’ cells (DC3) and 96 cells associated with the first and second row of Deiters’ cells (Figures 2H and S3F). We refrained from further subdividing the first two rows of Deiters’ cells due to the lack of known distinctive markers.
The Fgfr3-negative cell population constituted two subgroups (Figures 2I and S3G). Cells associated with the smaller group stem mainly from gate 6, gate 7 and the negative control gate 8. The fact that all negative control cells constituted this population (Figure S3J), prompted us to consider these cells as off target (Figure S3K).
Contemplating all previously identified cell populations we expected the remaining Fgfr3-negative cells, which were all Sox2-positive, to represent either GER cells, inner border cells or inner phalangeal cells. As expected, 103 presumptive GER cells isolated mainly from Sox2-GFP/Glast-CreERT2/Ai14-tdTomato (gate 5) and Lgr5-GFP (gate 6) mouse lines, were identified based on high Fgfr1 and Dkk3 expression levels (Figures 2J and S3H) (Hayashi et al., 2010; Huang et al., 2011). In comparison, inner border cells and inner phalangeal cells were not only sorted from the Sox2-GFP/Glast-CreERT2/Ai14-tdTomato and Lgr5-GFP lines, but also 49% of the cells originated from the Atoh1-nGFP/Fgfr3-CreERT2/Ai14-tdTomato line gated for GFP expression (gate 1). We were able to separate 96 presumptive inner border and inner phalangeal cells from the GER population based on characteristic Lfng (Kiernan et al., 2005; Zhang et al., 2000) and Cdkn1a expression (Figures 2J and S3H). Different levels of Otol1 (Deans et al., 2010) expression further distinguished 27 inner border cells from 69 inner phalangeal cells, which were characterized by Gjb1 (Lopez-Bigas et al., 2002), Gjb3, as well as Clrn1 expression (Figure 2K and S3I).
In summary, we partitioned all 877 cells into 9 subpopulations of distinct presumptive organ of Corti cell types consisting of two sensory hair cell populations (OHC, IHC) and seven non-sensory cell groups (GER, IB, IPH, OP, IP, 1st/2nd row of DC, 3rd row of DC) (Figure 2L); 69 off target cells were excluded. The groups of cells are not characterized by single marker gene expression, but rather by the aggregate of all genes assayed. Likewise, our analysis does not simply apply binary codes (i.e. whether a transcript is detectable or not), but uses quantitative gene expression data. Compared with published expression studies, which were often not conducted at exactly the same age (P2) as our study, we generally confirmed previous reports (Table S2). We contend that the identified 9 organ of Corti derived groups represent a quantitative and high-resolution blueprint of the neonatal organ and we utilized this platform to reconstruct some of the fundamental spatial features of the organ of Corti in silico.
In Silico Reconstruction of the Organ of Corti
The morphology of the cochlea resembles a snail shell-like structure in which organ of Corti cells spiral in rows along the longitudinal apex-to-base axis. Our experimental design allowed for the identification of apical- and basal-derived cells as we encoded their origin during the procedural steps of dissection and FACS (Figures 3A and S1). We hypothesized that instead of utilizing all genes for principal component calculation (Figure 3B,B′) employing only the specific subsets that include differentially expressed genes between apex and base will resolve spatially encrypted patterns in lower-dimensional space (Figure 3C,C′). For each of the nine determined cell groups, we calculated the differences between averaged expression levels for every gene (ΔLog2Ex) from apical versus basal associated cells (Figures 3B-D and S4A show GER-assigned cells as an example). Each subpopulation was then reanalyzed by PCA, taking only genes into account with ΔLog2Ex values greater than ±1, corresponding to an at least 2-fold expression difference between both anatomical sides (Figure 3C). Upon projection onto PC1 and PC2, cells segregated according to their apical and basal character, although they did not form two distinct groups (Figure 3C′). This result agrees with our expectations because all cells originated from a contiguous structure and most differentially expressed genes occur in gradients along the apex-to-base axis of the cochlear duct (Lelli et al., 2009; Son et al., 2012). Calculating the centroid locations for apical and basal-derived cells within the 2D coordinate system allowed us to determine a vector along which an apex-to-base gradient might be represented most accurately. Computations were performed for all ten possible 2-component subspace combinations considering PC1 through PC5 (Figure 3C″, S4A). Centroid distance was then used as measure of maximum separation of the data with respect to apical/basal distinction. PC1 was contributing most to the presumed spatial separation throughout all cell populations, only the second contributor varied (Figure S4A,B). The method’s robustness was validated by assessing the change of each cell’s relative rank position from apex to base for all nine populations after varying PC combinations or ΔLog2Ex threshold values (Figure S4C,D). For the purpose of data visualization we aligned all nine cell populations in their respective coordinate space such that the apex-to-base axis (defined by the centroid-centroid vector) was presented vertically (Figure 3D). This geometric modification resulted in the data being rotated into an apical-to-basal orientation. In an effort to resemble the organ of Corti’s distinctive row format, we compressed the dataset along the abscissa, rank normalized the distribution of cells, and projected each subpopulation in one-dimensional space. Finally, we assembled the nine cell populations side-by-side in an order that reflects the medial-to-lateral anatomical axis (Figure 3E). Prediction of the apex-to-base accuracy was confirmed by projecting data of cochlear duct cells purposely isolated from the middle turn or from the whole organ, showing that their location in the map was in agreement with the region they were dissected from (Figure S5). The resulting digitally reconstructed 2D single cell map of the organ of Corti can be interrogated to display expression patterns for each of the 192 genes (Figure S6).
Figure 3.

Spatial trajectory analysis of organ of Corti longitudinal rows. (A) Schematic illustration and color code. Tissue was dissected into apical and basal halves prior to cell sorting. (B, B′) PCA of GER cells (Figure 2J) utilizing all genes regardless of apex-to-base expression differences. Cells are projected onto PC1/PC2 and color-coded in blue and orange based on anatomical origin. No obvious segregation of apical versus basal derived cells is apparent. (C, C′) PCA of GER cells using only genes that differ by at least 2-fold expression between apical and basal-derived cells. Upon projection onto PC1/PC2, a differential distribution of apical and basal cells is apparent, though no isolated clusters are formed. (C″) Apical and basal centroids were calculated for each population and centroid distance was determined. This distance measurement was repeated across all ten combinatorial 2-dimension representations for PC1 to PC5, and the representation with largest centroid distance was selected for subsequent analysis (see Figure S4A,B). (D) Data space was rotated such that the centroid-centroid-vector was orientated parallel to PC1, apex facing up. To accomplish row-format appearance of the data, it was constricted along the x-axis and visualized in a rank normalized format. (E) Result of afore-described procedure for each of the 9 subpopulations arranged in medial-to-lateral order (GER – DC3). Cells are projected onto 2D subspace and colored based on apical (blue) or basal (orange) origin. (B′, C′, C″, D, E) Cells colored in light grey were derived from a few selected control experiments where we explicitly isolated cells from the middle turn or from the whole cochlear duct. This control was done to validate the accuracy of the methodology and is illustrated in detail in Figure S5. Related to Figures S1, S4 and S5.
In Silico Predicted Map Resembles Organ of Corti In Situ Composition
The accuracy of the organ of Corti map was assessed by comparing patterns of fluorescent reporter expression of the mouse lines used in this study with measured transcript expression levels. GFP fluorescence from the Atoh1-nGFP mouse line was detected at two distinct intensity levels using flow cytometry (Figure 1D, S1A,E). Confocal imaging confirmed high nGFP expression in IHCs and OHCs, whereas inner border and inner phalangeal cells displayed lower fluorescence intensity (Figure 4A). The mean expression of Atoh1 mRNA across all cells in each row-associated population confirmed statistically higher levels within the two hair cell populations compared to each of the supporting cell populations (Figure 4A′ and 4G). Neither inner phalangeal cells nor inner border cells showed significantly elevated Atoh1 mRNA expression levels compared to the remaining supporting cell populations (Figure 4G). To demonstrate clustering accuracy, we projected Atoh1 mRNA levels on all 808 cells, unambiguously illustrating specificity of high Atoh1 mRNA expression in IHCs and OHCs (Figure 4A″). Next we projected only gate 1 sorted cells on the map to highlight that all gate 1 sorted Atoh1-nGFP cells were allocated to IHCs and OHCs as well as inner border and inner phalangeal cells. Likewise, high GFP mRNA levels of gate 1 sorted cells were detected in hair cells, low levels in inner border and inner phalangeal cells (Figure 4A″). Our interpretation of these observations is that the GFP fluorescence intensity levels as measured by FACS and microscopy reflect enhancer activity levels of the transgenic Atoh1-nGFP allele, rather than quantitative Atoh1 mRNA levels as measured by single cell qRT-PCR. This analysis effectively exemplifies the high level of accuracy of the in silico map.
Figure 4.

Comparative validation of the organ of Corti map. (A-E) For each mouse line expression of GFP and tdTomato is shown in organ of Corti cross-sections. (A′-E′) Bar graphs represent mean gene expression levels per population. (A″-E″) Three in situ maps of the organ of Corti are shown in which individual cells are represented by dots that are color-coded based on marker gene mRNA expression (top), FACS gate origin (middle), and reporter allele mRNA expression (bottom). (F) Confocal images of whole mount preparations of P2 cochleae from Fgfr3-CreERT2/Ai14-tdTomato mice immunolabeled with antibody to hair cell marker myosin 7A. (G) Statistical analyses of mean expression level differences pairwise among all nine cell type groups. Only p-values <0.0001 are shown and indicated by colored lines (red = higher mean expression value, green = lower mean expression value). Scale bars: A-F, 10μm. Related to Figures S6 and S7.
Sox2-GFP was used as a marker to fluorescently label all organ of Corti supporting cell types in two mouse lines used (Figure 1E,H). Sorting for GFP positive cells (Figures 1G,J and S1B,C), we identified 4 different target populations of high and lower GFP fluorescence intensities (Gates 2–5). High GFP intensity was detectable in all supporting cell populations whereas the hair cell populations showed less intense fluorescence (Figure 4B). This was confirmed by statistically different mean Sox2 mRNA expression between hair cells and supporting cells (Figure 4B′ and 4G). Projecting Sox2 mRNA levels onto the organ of Corti map visualized high expression in the supporting cell populations and an apex-to-base expression gradient within both hair cell populations (Figure 4B″). The gate contribution distinctly confirmed that cells sorted from the Sox2-GFP/Fgfr3-CreERT2/Ai14-tdTomato line in gate 3 displaying high GFP fluorescence intensity were allocated to the pillar and Deiters’ cell populations (Figure 4B″). Gate 4-derived cells were exclusively assigned to the OHC population with a bias towards the apex, whereas cells sorted from the Sox2-GFP/Glast-CreERT2/Ai14-tdTomato line in gate 5 were confirmed as GER, inner border and inner phalangeal cells. Assuming that gate 2 sorted cells would correspond to Fgfr3-negative supporting cell types, they were correctly linked with GER, inner border and inner phalangeal cells. GFP mRNA was detected in all supporting cells from gate 2 to 5.
Lgr5-GFP expression was analyzed to further confirm the map’s cell-subtype specific resolution (Figure 4C). We stringently sorted for GFP high-level cells only (Figures 1M and S1D, gate 6), targeting third row of Deiters’ cells in particular but also inner pillar, inner phalangeal, inner border and GER cells (Figure 1K,L). High GFP expression in the target populations was microscopically confirmed (Figure 4C) and quantitatively assessed by corresponding statistical differences of mean expression values of Lgr5 mRNA among all examined cell groups (Figure 4C′ and 4G). Visualization of Lgr5 mRNA expression levels at single cell resolution demonstrates the specificity of the predicted 2D projection, as well as the correct mapping of the gate 6-sorted cells within the dataset (Figure 4C″).
Glast-CreERT2/Ai14-tdTomato recombination was restricted to GER, inner border, inner phalangeal, and Hensen’s cells (Figure 1H and 4D). To isolate GER, inner border and inner phalangeal cell populations in the Sox2-GFP/Glast-CreERT2/Ai14-tdTomato line, we sorted for double positive cells accordingly (Gate 5, Figures 1J and S1C). Statistically significant expression of Glast mRNA was found in GER, IBC, and IPH (Figure 4D′ and 4G). The three Deiters’ cell rows also expressed Glast mRNA, albeit at moderate levels, however Cre-mediated recombination was not detected in Deiters’ cells as visualized in the gate 5 distribution map and by lack of tdTomato mRNA expression (Figure 4D″). Combining the Glast- and the Sox2 alleles allowed us to extract the target populations at high purity, as validated by the computed in situ data projection.
Finally, we used Fgfr3-CreERT2/Ai14-tdTomato in conjunction with the Sox2-GFP allele to enrich for double positive pillar and Deiters’ cells (Figures 1E-G and S1B). Strong tdTomato fluorescence was detected in both cell types (Figure 4E). This qualitative finding was quantitatively confirmed by calculating mean Fgfr3 mRNA expression for each cell population and by statistical analysis (Figure 4E′ and 4G). The organ of Corti map mirrored the Fgfr3 mRNA expression domain in pillar and Deiters’ cell populations and revealed an apical-to-basal gradient within the OHC population (Figure 4E″). Projecting gates 3 and 4 sorted cells onto the map showed that both supporting cell populations were associated with gate 3, whereas preferentially apical OHCs originated in gate 4 only. The specific primer set that reports recombination at the Ai14-tdTomato locus also allowed us to measure tdTomato mRNA expression levels (Figure S2A,A′) and detected expression in gates 3 and 4 sorted cells. Moreover, projecting tdTomato expression levels onto gate 1 sorted Fgfr3-CreERT2/Ai14-tdTomato/Atoh1-nGFP cells confirmed that recombination in this population was restricted to OHCs as well (Figure S7A). Imaging of organ of Corti whole mount preparations confirmed higher rates of recombination in apical versus basal OHCs, potentially a reflection of the higher levels of Fgfr3 mRNA expression in the apex at P2 (Figure 4F). FACS, qRT-PCR and microscopy data concurred with each other such that tdTomato intensity was detected at equivalent levels for both hair cell and supporting cell populations. Expression levels of tdTomato correspond to Rosa26-rather than Fgfr3-driven transcription (Figure S2A-A′); hence expression in OHCs follows a quasi-binary pattern governed by recombination activity (Figure 4E-E″ and F).
In conclusion, the expression analysis of reporter gene-based marker activity validated the spatial accuracy of the organ of Corti map. Figure S7B illustrates the expression patterns of 16 marker genes previously identified to be associated with specific cell populations, further illustrating a high level of prediction accuracy. The established organ of Corti map reveals mRNA expression patterns of individual genes, similar to conventional mRNA in situ hybridization assays. The novelty lies in the fact that expression pattern evaluation can be carried out in parallel for many genes, at single cell resolution, and in readily quantitative format that is digitalized.
Hierarchical Clustering Confirms Subpopulation Assignments
Successive PCA and k-means clustering resulted in 9 subpopulations associated with different organ of Corti cell types. To exclude a potential bias introduced by this approach, we employed hierarchical clustering as an additional unsupervised statistical approach and visualized the data in conventional heatmap format. Eleven different hierarchical clusters were found (Figure 5A). Hierarchical clusters 1 and 2 corresponded to inner and outer hair cell populations, respectively. Previously identified inner pillar and outer pillar cells were separated in hierarchical clusters 3 and 4 respectively, whereas cluster 5 contained Deiters’ cell rows 1 through 3. Cluster 6 matched inner border and inner phalangeal cells and cluster 7 contained the GER cell population. Hierarchical clusters 8, 9, 10 and 11 corresponded to the off target cell populations (Figure S3J,K), which did not contribute to the organ of Corti map. The 7 organ of Corti groups identified by hierarchical clustering matched the previously determined 9 cell populations to a very high degree (94.64±5.83%SD), but did not resolve differences between inner border and inner phalangeal cells and between the different rows of Deiters’ cells. In addition, clustering was not guided by confounding factors such as the date of experiment, different FACS gates or different mouse lines as indicated by the unbiased spread of cells across and within the hierarchical clusters (Figure 5A,B).
Figure 5.

Hierarchical clustering of all 877 single cells. (A) Shown is the result of hierarchical clustering in heatmap format. Seven hierarchical clusters correspond to previously identified sub-groups of cells. Off-target cells are partitioned into four groups (8 – 11). (B) Different levels of metadata information are color-coded and shown is the distribution of this metadata across cells and subclusters to visualize that these parameters had no confounding effects on clustering.
Using the Organ of Corti Map as a Statistical Tool
In an effort to identify genes that are expressed in gradients along the apex-to-base axis or differentially between subpopulations, we examined the in silico map with statistical methods. Fgfr3 mRNA expression in OHCs is shown in Figure 6A-C″ and is used to illustrate our strategy. Along the longitudinal axis we partitioned the population into three segments, representing apex, middle, and base with proportionally the same number of cells per segment. Next we present the data in an apex-to-base progression curve by fitting each cell’s associated expression value as a function of its relative position along the longitudinal axis. The progress line of the calculated mean expression for each of the three segmented regions resembled the fitted curve (Figure 6A). Data rendering was repeated for all 9 organ of Corti subpopulations and visualizes a gradual decrease of Fgfr3 mRNA expression from apex to base for OHCs (Figure 6B,B′). Next, to identify genes with expression levels that significantly change between at least 2 segments, we employed a binary comparison approach where we contrasted mean expression levels between apex- and middle-segment, middle- and base-segment, apex- and base-segment, as well as between subpopulations (Figure 6C). This strategy enabled us to identify genes expressed in statistically significant gradients along the longitudinal axis for each organ of Corti cell type, for example for OHC we found 62 transcripts (Figure 6C′, Table S3). It also allowed us to inquire whether there is significantly different expression of a specific gene between select subpopulations in segment-specific fashion (Figure 6C″).
Figure 6.

Identification of statistically significant differences to reveal differential gene expression in organ of Corti supporting cells. The procedure is illustrated using Fgfr3 expression as an example. (A) Along the apex-to-base axis the OHC population was divided into three compartments. The plot shows expression data as well as fitted data from apex to base across the three compartments. Compartments’ mean expression values are concurrent with fitted data. (B, B′) Application of this strategy to all 9 cell groups translates the organ of Corti map into a statistical tool, (C-C″) allowing for 2-fold comparisons resolving statistical differences between different compartments along the longitudinal axis of one population (comparisons 1 – 3), or within one compartment between different cell groups (comparisons 4 – 6). (D) Asymmetric gene expression increasing in statistically different manner (comparisons 1–3, *p≤0.05) from the base towards the apex for Kcnj10, Gjb1, Itpr2, Inhba and Pdgfra. (E) Fgf10 and Hmga2 show inversely oriented gradients. (F-H)) Notch effectors (Hes5, Hey1 and Lfng), Wnt-(Lgr5, Fzd6 and Dkk3), and cell cycle genes (Ccnd1 (Cyclin D1) and Cdk6) are differentially expressed (comparison 4, *p≤0.05) between apical inner pillar cells and apical outer pillar cells.
Inversely Correlated Signaling Pathway Patterns in Neonatal Supporting Cells
Regenerative capacities and developmental maturation of supporting cells in the developing organ of Corti are coordinated in opposing fashion. In agreement with this proposed pattern significantly higher mRNA levels of the potassium channel Kcnj10 (Kir4.1) and gap junction gene Gjb1 (Connexin 32) were detected in the basal compartments compared to the respective apical compartments of different supporting cell subtypes (Figure 6D). Expression of Kcnj10 is an indication of supporting cell maturation, for which Gjb1 expression at P2 appears to be also a robust indicator (Hibino et al., 1997; Lopez-Bigas et al., 2002). Similar patterns were detected for the EGF receptor Itpr2 in all organ of Corti supporting cells except for the 3rd row of Deiters’ cells, as well as for alpha-type platelet-derived growth factor receptor (Pdgfra), and Inhba, indicative of increased Activin/Inhibin production in the more mature base in all organ of Corti cell types except for the GER. The gradients indicate that these genes could play roles in cellular maturation or that they are involved in suppression of possible rudimentary regenerative capability.
In contrast to the apex-to-base increasing gradients, Fgf10 expression followed an inverse pattern with highest levels in the more immature apex and decreasing expression towards the base in inner and outer pillar cells, the 3rd row of Deiters’ cells and in inner phalangeal cells (Figure 6E). Interestingly pillar, Deiters’ and GER cells expressed Hmga2 in a gradient significantly decreasing toward the base. This nuclear protein controls changes in transcriptional patterns that contribute to pro-proliferative state and self-renewal in fetal cells and stem cells and has been shown to decline during aging (Nishino et al., 2008), which could be a distinct indicator of the maturation gradient in the P2 neonatal organ of Corti.
In summary, utilizing statistical tools we found differentially expressed candidate genes connected to FGF, EGF, PDGF and TGFβ signaling along the organ of Corti longitudinal axes supporting the hypothesis that the concerted action of multiple signaling pathways coordinately modulate maturation and possibly loss of regenerative ability. This spurred our interest to examine the data set for specific pathway-related transcripts that could reveal more information about the proposed regenerative potential of one particular neonatal supporting cell type, the inner pillar cell.
Molecular Mechanisms Priming the Regenerative Potential of Inner Pillar Cells
Neonatal inner pillar cells can reenter the cell cycle, divide and differentiate into new hair cells in vivo and in vitro (Cox et al., 2014; Li et al., 2015; Liu et al., 2012; White et al., 2006). This potential is particularly evident in the cochlear apex. Neonatal outer pillar cells do not display such increased regenerative potential. To identify genes that may be responsible for the differential regenerative capabilities, we searched for markers with significantly differing expression between the apical inner and outer pillar cell populations. Particularly focusing on the apex we found three Notch effector genes, Hes5, Hey1, and Lfng (Figure 6F), to be significantly lower expressed in apical inner pillar cells compared to the corresponding compartment in outer pillar cells. Active Notch signaling represses hair cell regeneration in the organ of Corti whereas pharmacological and genetic reduction of Notch signaling promotes the generation of new hair cells (Korrapati et al., 2013; Li et al., 2015; Mizutari et al., 2013). Our data suggests that reduced Notch signaling in apical inner pillar cells represents a cellular state that is inversely correlated with the enhanced regenerative potential of those cells.
Beside inhibition of Notch signaling the activation of Wnt signaling has been shown to induce hair cell generation in the postnatal organ of Corti (Chai et al., 2012; Jacques et al., 2014; Shi et al., 2013). Recent evidence suggests that Notch signaling suppresses canonical Wnt signaling in Lgr5-expressing neonatal cochlear supporting cells and furthermore, that Notch-inhibition via interaction with the canonical Wnt pathway results in proliferation and generation of new hair cells from inner pillar cells (Li et al., 2015). In agreement with this hypothesis we found inversely correlating canonical Wnt and Notch candidate gene expression patterns in inner and outer pillar cells. Wnt receptor Fzd6 and the Wnt-reporter Lgr5 were significantly higher expressed in inner pillar cells compared to outer pillar cells (Figure 6G). In contrast the Wnt-antagonist Dkk3 was not detected in the majority of inner pillar cells compared to outer pillar cells where it was expressed in an apex-to-base gradient. Our findings suggest a primed state of canonical Wnt in inner pillar cells with lower levels of Notch effectors compared to outer pillar cells that do not display regenerative potential.
In further support of a Wnt dependent regenerative priming of inner pillar cells we found significantly higher levels of early cell cycle transcripts Cyclin D1 and Cdk6 (Coller, 2007) when compared to outer pillar cells (Figure 6H). In addition, Cyclin D1 in inner pillar cells was significantly higher expressed in the apex compared to the base. No statistically significant increase of expression for late cell cycle markers including Cyclin B1, Cyclin A2 or Pcna was detected. Consolidation of these quantitative gene expression analyses suggest that reduced Notch activity correlated with elevated Wnt signaling and elevated levels of early cell cycle genes synergistically define a primed regenerative state that specifically endows apical, neonatal inner pillar cells with the ability to re-enter the cell cycle.
Developmental Gradients and Emerging Tonotopy in Hair Cells
Maturation of hair cells happens in a gradient from apex-to-base at multiple levels ranging from changes in hair bundle morphology to functional maturation of the mechanoelectrical transduction machinery (Lelli et al., 2009; Waguespack et al., 2007). Markers expected in mature hair cells, like Otof, Ptprq and Clrn1 were expressed in gradients significantly increasing from the apex towards the base (Figure 7A). In addition, Calb1, Calb2, and Ocm, encoding mobile calcium buffers calbindin, calretinin, and oncomodulin, respectively, as well as the voltage-gated calcium channel CaV1.3 (Cacna1d) exhibited gradients with similar orientation (Figure 7B,C). Conversely, genes related to assembly of the hair bundle apparatus such as beta actin (Actb), Myosin 15 and protocadherin 15 (Pcdh15) showed highest expression levels in the apex (Figure 7D) where bundle growth and assembly appears to require significant mRNA levels of these genes at this neonatal time (Kaltenbach et al., 1994). Interestingly, we identified a group of genes, extensively studied during early otic development (Hidalgo-Sanchez et al., 2000; Ozaki et al., 2004; Radde-Gallwitz et al., 2004), to be expressed in maturing hair cells (Figure 7E). Coinciding with gradual maturation, expression of these genes decreased from apex to base.
Figure 7.

Gene expression gradients in organ of Corti hair cells. (A) Genes expected to be expressed in mature hair cells including Otof (Otoferlin), Clrn1 (Clarin1) and Ptprq show increasing expression levels from apex to base. (B) Calcium binding proteins Calb1 (Calbindin), Calb2 (Calretinin) and Ocm (Oncomodulin) are expressed in gradients with the same orientation, (C) as shown for calcium channel Cacna1d (CaV1.3) as well. (D) Hair bundle genes like Actb (beta-Actin), Myo15 (Myosin15) and Pcdh15 (Protocadherin 15) are significantly higher expressed in the apex compared to the base. The same pattern is present for four transcription factor genes (Isl1, Dach1, Eya2 and Pax2).
Discussion
Known for more than one-and-a-half century (Retzius, 1884), organ of Corti cells have resisted extensive molecular interrogation because they are not only rare but also encased in highly dense bone. For molecular and cellular biology of the inner ear, single cell gene expression analysis offers an unprecedentedly powerful tool that has the potential to annihilate existing obstacles. Here we present a strategy for data analysis to generate a 2D map of the mammalian organ of Corti at single cell resolution that allows researchers to assess expression patterns for many genes in parallel in a quantitative and digital framework. This approach is not specific to the inner ear and can theoretically be applied to many organ systems that consist of multiple cell types and well described spatial organization.
When compared with known expression patterns of cochlear-specific genes, the generated 2D map of the neonatal mouse organ of Corti displayed remarkable robustness and accuracy. Along the medial-to-lateral axis, we were able to conclusively differentiate between virtually all major cell types of the organ of Corti as shown by k-means clustering and principal component analysis, and confirmed this result with hierarchical clustering. Additional separation of cells such as in the GER population, the individual rows of OHCs and the 1st and 2nd row of Deiters’ cells was not possible due to the lack of definite markers. Conceivably, RNA-Seq would be needed to potentially identify distinguishing marker genes that differentiate between these rows. For all other cell types, displaying the cells in row format is equivalent to the native configuration as in the organ. For computing the apex-to-base axis of the map, we developed a strategy that establishes spatial-related trajectories. The basis of this algorithm features the usage of lower-dimensional variable space images that derive from asymmetric gene expression information between two classes of cells, here the cochlear apex and base. Similar but also distinctively different to recently introduced concepts (Durruthy-Durruthy et al., 2014; Trapnell et al., 2014), which exploit temporally defined gene expression differences to establish trajectories in time, we were able to order cells along an axis that represents space. For this strategy no pre-defined markers were required and the generated maps were highly consistent with known gene expression information along the longitudinal cochlear axis (Lelli et al., 2009; Son et al., 2012). Segmentation allowed us to examine significantly different gene expression between compartments of specific organ of Corti cell types. Applying the same strategy to resolve the medial-to-lateral axis was initially considered, but micro-dissection of the organ of Corti reliably into medial and lateral compartments was not feasible for technical reasons.
We used the map to explore whether we can identify genes that provide mechanistic insight into two important biological topics, sensory hair cell regeneration and cochlear tonotopy. We identified a candidate mechanism that explains regenerative predisposition in inner pillar cells, which appear to be primed for a canonical Wnt-mediated cell cycle re-entry that is suppressed but not to the degree of complete inhibition by well-balanced low level Notch activation. This primed state, probably a relic of delayed developmental maturation in the apex, is transient and disappears when supporting cells mature. Higher expression of genes associated with a more advanced cellular differentiation state was found in the basal segment, which supports this assumption. We also identified gradients in genes that are involved in maturation of hair cells, an indication of establishment of cochlear tonotopy. Rodent hair cells mature in precisely controlled fashion where hair bundle morphology and acquisition of mature physiological features are initially more advanced at the base and over the course of days reach the cochlear apex (Lelli et al., 2009; Waguespack et al., 2007). Conceivably, one can also use the population-specific gene expression data to identify correlated and anti-correlated genes or gene classes that characterize any of the nine organ of Corti cell groups. An example for such correlation analysis is shown in Figure S8. We note that the number of preselected genes for this study (192) limits the overall power of this type of analysis. Whole transcriptome data derived from RNA-Seq studies on the other hand would provide a viable opportunity to investigate gene-to-gene relationships in a rather complete fashion. Correlation coefficients for all genes in organ of Corti cell type-specific manner is provided in Table S4 for the interested reader.
In summary, we present a method to reconstruct spatial core features of a complex organ. We used the organ of Corti as an example to illustrate the power and usefulness of this approach, yet its field of applications can be applied to other organs. Development and functional maturation of cells in multicellular organisms is entwined with dynamic changes of gene expression along temporal as well as spatial trajectories. Practical ways to interrogate these dynamics adequately and in high-enough throughput are lacking and the present study addresses this deficit.
Experimental Procedures
Animal Breeding
Four different mouse lines were generated from six different alleles: Atoh1-nGFP (Lumpkin et al., 2003), Sox2-GFP (Jax#: 017592), Lgr5-EGFP-IRES-CreERT2 (Jax#: 008875), Fgfr3-CreERT2 (Young et al., 2010), Glast-CreERT2 (Jax#: 012586), Ai14-tdTomato (Jax#: 007908). Pups were injected with tamoxifen (0.2mg/g) at P0 and dissected at P2. Procedures involving animals were performed in accordance with institutional and governmental regulations and were approved by the Stanford University Institutional Animal Care and Use Committee.
Cell Isolation and Sorting
Cochlear ducts were microdissected as one piece (full 1.8 turn) or divided into either two parts (apex/base, each about 0.9 turns long) or three parts (apex/middle/base, each about 0.6 turns long). Apportioned tissues were enzymatically and mechanically dissociated and passed through a strainer for removal of clumps. Gating strategy was designed to maximize capture of one individual live cell per well (Figure S1). For details see (Durruthy-Durruthy et al., 2014).
RNA Processing and Single-Cell qRT-PCR
RNA was processed and analyzed by qRT-PCR as described previously (Durruthy-Durruthy et al., 2014). For assay composition see Table S2.
Confocal Microscopy
Specimens were fixed with paraformaldehyde (4%) for 2 hrs and analyzed as cryosections or whole mount preparations using DAPI as a nuclear stain and antibody to myosinVIIa (Axxora) to label hair cells. Imaging was performed with a confocal laser-scanning microscope (LSM700, Zeiss).
Data Analysis
Analysis was conducted with R software and Excel (Microsoft). See Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Unbiased clustering of single organ of Corti cells based on gene expression data
Clusters correspond to nine major medial-to-lateral aligned organ of Corti cell types
Spatial reconstruction of apex-to-base localization of each organ of Corti cell
Statistical analysis reveals changes in gene expression along the tonotopic axis
Acknowledgments
We thank the staff at the Stanford Shared FACS Facility for their help and support. Members of the Heller laboratory and Dr. Daniel Ellwanger for comments on the manuscript. This work was supported by NIH grants DC006167 and DC012250 to S.H., by P30 core support (DC010363), by the Hearing Health Foundation’s Hearing Restoration Project, and by the Stanford Initiative to Cure Hearing Loss. J.W. is supported in part by a fellowship (WA3420/1) from the Deutsche Forschungsgemeinschaft.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anttonen T, Kirjavainen A, Belevich I, Laos M, Richardson WD, Jokitalo E, Brakebusch C, Pirvola U. Cdc42-dependent structural development of auditory supporting cells is required for wound healing at adulthood. Scientific reports. 2012;2:978. doi: 10.1038/srep00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckiova D, Syka J. Calbindin and S100 protein expression in the developing inner ear in mice. J Comp Neurol. 2009;513:469–482. doi: 10.1002/cne.21967. [DOI] [PubMed] [Google Scholar]
- Chai R, Kuo B, Wang T, Liaw EJ, Xia A, Jan TA, Liu Z, Taketo MM, Oghalai JS, Nusse R, et al. Wnt signaling induces proliferation of sensory precursors in the postnatal mouse cochlea. Proc Natl Acad Sci U S A. 2012;109:8167–8172. doi: 10.1073/pnas.1202774109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai R, Xia A, Wang T, Jan TA, Hayashi T, Bermingham-McDonogh O, Cheng AG. Dynamic expression of Lgr5, a Wnt target gene, in the developing and mature mouse cochlea. J Assoc Res Otolaryngol. 2011;12:455–469. doi: 10.1007/s10162-011-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development. 1999;126:1581–1590. doi: 10.1242/dev.126.8.1581. [DOI] [PubMed] [Google Scholar]
- Chen P, Zindy F, Abdala C, Liu F, Li X, Roussel MF, Segil N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat Cell Biol. 2003;5:422–426. doi: 10.1038/ncb976. [DOI] [PubMed] [Google Scholar]
- Coller HA. What’s taking so long? S-phase entry from quiescence versus proliferation. Nature reviews Molecular cell biology. 2007;8:667–670. doi: 10.1038/nrm2223. [DOI] [PubMed] [Google Scholar]
- Cox BC, Chai R, Lenoir A, Liu Z, Zhang L, Nguyen DH, Chalasani K, Steigelman KA, Fang J, Rubel EW, et al. Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development. 2014;141:816–829. doi: 10.1242/dev.103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Peterson JM, Wong GW. Mammalian Otolin: a multimeric glycoprotein specific to the inner ear that interacts with otoconial matrix protein Otoconin-90 and Cerebellin-1. PLoS One. 2010;5:e12765. doi: 10.1371/journal.pone.0012765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, Heller S. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell. 2014;157:964–978. doi: 10.1016/j.cell.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret G, Frankenreiter M. Quantitative analysis of cochlear structures in the house mouse in relation to mechanisms of acoustical information processing. J Comp Physiol. 1977;122:65–85. [Google Scholar]
- Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Ray CA, Younkins C, Bermingham-McDonogh O. Expression patterns of FGF receptors in the developing mammalian cochlea. Dev Dyn. 2010;239:1019–1026. doi: 10.1002/dvdy.22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Horio Y, Inanobe A, Doi K, Ito M, Yamada M, Gotow T, Uchiyama Y, Kawamura M, Kubo T, et al. An ATP-dependent inwardly rectifying potassium channel, KAB-2 (Kir4. 1), in cochlear stria vascularis of inner ear: its specific subcellular localization and correlation with the formation of endocochlear potential. J Neurosci. 1997;17:4711–4721. doi: 10.1523/JNEUROSCI.17-12-04711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo-Sanchez M, Alvarado-Mallart R, Alvarez IS. Pax2, Otx2, Gbx2 and Fgf8 expression in early otic vesicle development. Mech Dev. 2000;95:225–229. doi: 10.1016/s0925-4773(00)00332-4. [DOI] [PubMed] [Google Scholar]
- Huang M, Sage C, Tang Y, Lee SG, Petrillo M, Hinds PW, Chen ZY. Overlapping and distinct pRb pathways in the mammalian auditory and vestibular organs. Cell Cycle. 2011;10:337–351. doi: 10.4161/cc.10.2.14640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth AJ. Integrating the active process of hair cells with cochlear function. Nat Rev Neurosci. 2014;15:600–614. doi: 10.1038/nrn3786. [DOI] [PubMed] [Google Scholar]
- Jacques BE, Montgomery WHt, Uribe PM, Yatteau A, Asuncion JD, Resendiz G, Matsui JI, Dabdoub A. The role of Wnt/beta-catenin signaling in proliferation and regeneration of the developing basilar papilla and lateral line. Dev Neurobiol. 2014;74:438–456. doi: 10.1002/dneu.22134. [DOI] [PubMed] [Google Scholar]
- Jolliffe IT. Principal component analysis. 2. New York: Springer; 2002. [Google Scholar]
- Kaltenbach JA, Falzarano PR, Simpson TH. Postnatal development of the hamster cochlea. II. Growth and differentiation of stereocilia bundles. J Comp Neurol. 1994;350:187–198. doi: 10.1002/cne.903500204. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- Korrapati S, Roux I, Glowatzki E, Doetzlhofer A. Notch signaling limits supporting cell plasticity in the hair cell-damaged early postnatal murine cochlea. PLoS One. 2013;8:e73276. doi: 10.1371/journal.pone.0073276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, Kelley MW. Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet. 1999;21:289–292. doi: 10.1038/6804. [DOI] [PubMed] [Google Scholar]
- Lelli A, Asai Y, Forge A, Holt JR, Geleoc GS. Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol. 2009;101:2961–2973. doi: 10.1152/jn.00136.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wu J, Yang J, Sun S, Chai R, Chen ZY, Li H. Notch inhibition induces mitotically generated hair cells in mammalian cochleae via activating the Wnt pathway. Proc Natl Acad Sci U S A. 2015;112:166–171. doi: 10.1073/pnas.1415901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Walters BJ, Owen T, Brimble MA, Steigelman KA, Zhang L, Mellado Lagarde MM, Valentine MB, Yu Y, Cox BC, et al. Regulation of p27Kip1 by Sox2 maintains quiescence of inner pillar cells in the murine auditory sensory epithelium. J Neurosci. 2012;32:10530–10540. doi: 10.1523/JNEUROSCI.0686-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bigas N, Arbones ML, Estivill X, Simonneau L. Expression profiles of the connexin genes, Gjb1 and Gjb3, in the developing mouse cochlea. Mech Dev. 2002;119(Suppl 1):S111–115. doi: 10.1016/s0925-4773(03)00102-3. [DOI] [PubMed] [Google Scholar]
- Lowenheim H, Furness DN, Kil J, Zinn C, Gultig K, Fero ML, Frost D, Gummer AW, Roberts JM, Rubel EW, et al. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of corti. Proc Natl Acad Sci U S A. 1999;96:4084–4088. doi: 10.1073/pnas.96.7.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, et al. Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns. 2003;3:389–395. doi: 10.1016/s1567-133x(03)00089-9. [DOI] [PubMed] [Google Scholar]
- MacQueen J. Some methods for classification and analysis of multivariate observations. In: Le Cam LM, Neyman J, editors. Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability Volume I, Statistics. University of California Press; 1967. [Google Scholar]
- Mak AC, Szeto IY, Fritzsch B, Cheah KS. Differential and overlapping expression pattern of SOX2 and SOX9 in inner ear development. Gene Expr Patterns. 2009;9:444–453. doi: 10.1016/j.gep.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SL, Li C, Urness LD. Genetic rescue of Muenke syndrome model hearing loss reveals prolonged FGF-dependent plasticity in cochlear supporting cell fates. Genes Dev. 2013;27:2320–2331. doi: 10.1101/gad.228957.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutari K, Fujioka M, Hosoya M, Bramhall N, Okano HJ, Okano H, Edge AS. Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron. 2013;77:58–69. doi: 10.1016/j.neuron.2012.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki H, Nakamura K, Funahashi J, Ikeda K, Yamada G, Tokano H, Okamura HO, Kitamura K, Muto S, Kotaki H, et al. Six1 controls patterning of the mouse otic vesicle. Development. 2004;131:551–562. doi: 10.1242/dev.00943. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Radde-Gallwitz K, Pan L, Gan L, Lin X, Segil N, Chen P. Expression of Islet1 marks the sensory and neuronal lineages in the mammalian inner ear. J Comp Neurol. 2004;477:412–421. doi: 10.1002/cne.20257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retzius G. Das Gehörorgan der Reptilien, der Vögel und der Säugethiere. 1884. [Reprint of the Original from 1884] (eBooks on Demand (EOD) Network) [Google Scholar]
- Sajan SA, Warchol ME, Lovett M. Toward a systems biology of mouse inner ear organogenesis: gene expression pathways, patterns and network analysis. Genetics. 2007;177:631–653. doi: 10.1534/genetics.107.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Hu L, Edge AS. Generation of hair cells in neonatal mice by beta-catenin overexpression in Lgr5-positive cochlear progenitors. Proc Natl Acad Sci U S A. 2013;110:13851–13856. doi: 10.1073/pnas.1219952110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Kempfle JS, Edge AS. Wnt-responsive lgr5-expressing stem cells are hair cell progenitors in the cochlea. J Neurosci. 2012;32:9639–9648. doi: 10.1523/JNEUROSCI.1064-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim K, Minowada G, Coling DE, Martin GR. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev Cell. 2005;8:553–564. doi: 10.1016/j.devcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Simonneau L, Gallego M, Pujol R. Comparative expression patterns of T-, N-, E-cadherins, beta-catenin, and polysialic acid neural cell adhesion molecule in rat cochlea during development: implications for the nature of Kolliker’s organ. J Comp Neurol. 2003;459:113–126. doi: 10.1002/cne.10604. [DOI] [PubMed] [Google Scholar]
- Sinkkonen ST, Chai R, Jan TA, Hartman BH, Laske RD, Gahlen F, Sinkkonen W, Cheng AG, Oshima K, Heller S. Intrinsic regenerative potential of murine cochlear supporting cells. Scientific reports. 2011;1:26. doi: 10.1038/srep00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son EJ, Wu L, Yoon H, Kim S, Choi JY, Bok J. Developmental gene expression profiling along the tonotopic axis of the mouse cochlea. PLoS One. 2012;7:e40735. doi: 10.1371/journal.pone.0040735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateya T, Imayoshi I, Tateya I, Hamaguchi K, Torii H, Ito J, Kageyama R. Hedgehog signaling regulates prosensory cell properties during the basal-to-apical wave of hair cell differentiation in the mammalian cochlea. Development. 2013;140:3848–3857. doi: 10.1242/dev.095398. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waguespack J, Salles FT, Kachar B, Ricci AJ. Stepwise morphological and functional maturation of mechanotransduction in rat outer hair cells. J Neurosci. 2007;27:13890–13902. doi: 10.1523/JNEUROSCI.2159-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PM, Doetzlhofer A, Lee YS, Groves AK, Segil N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature. 2006;441:984–987. doi: 10.1038/nature04849. [DOI] [PubMed] [Google Scholar]
- Young KM, Mitsumori T, Pringle N, Grist M, Kessaris N, Richardson WD. An Fgfr3-iCreER(T2) transgenic mouse line for studies of neural stem cells and astrocytes. Glia. 2010;58:943–953. doi: 10.1002/glia.20976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Martin GV, Kelley MW, Gridley T. A mutation in the Lunatic fringe gene suppresses the effects of a Jagged2 mutation on inner hair cell development in the cochlea. Curr Biol. 2000;10:659–662. doi: 10.1016/s0960-9822(00)00522-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci U S A. 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.