

Abstract
There is developing interest in the potential association between anesthesia and the onset and progression of Alzheimer's disease. Several anesthetics have thus been demonstrated to induce tau hyperphosphorylation, an effect mostly mediated by anesthesia-induced hypothermia. Here, we tested the hypothesis that acute normothermic administration of dexmedetomidine, an intravenous sedative used in intensive care units, would result in tau hyperphosphorylation in vivo and in vitro. When administered to non-transgenic mice, dexmedetomidine induced tau hyperphosphorylation persisting up to 6h in the hippocampus for the AT8 epitope. Pretreatment with atipamezole, a highly specific α2-adrenergic receptor (α2-AR) antagonist, blocked dexmedetomidine-induced tau hyperphosphorylation. Furthermore, dexmedetomidine dose-dependently increased tau phosphorylation at AT8 in SH-SY5Y cells, impaired mice spatial memory in the Barnes maze, and promoted tau hyperphosphorylation and aggregation in transgenic hTau mice. These findings suggest that dexmedetomidine: i) increases tau phosphorylation, in vivo and in vitro, in the absence of anesthetic-induced hypothermia and through α2-AR activation, ii) promotes tau aggregation in a mouse model of tauopathy, and iii) impacts spatial reference memory.
Keywords: Dexmedetomidine, anesthesia, tau, Alzheimer's disease, mouse, cells
1. Introduction
Tau protein is a microtubule-associated protein abundantly found in neuronal axons, whose main function, the binding and stabilization of microtubules, is partly regulated by phosphorylation (Avila, et al., 2004). In Alzheimer's disease (AD) and other tauopathies, tau can become abnormally hyperphosphorylated, leading to its aggregation and the subsequent formation of intraneuronal neurofibrillary tangles (Grundke-Iqbal, et al., 1986a, Grundke-Iqbal, et al., 1986b). Tau pathology is important as it correlates with dementia in AD as well as with memory loss in normal aging and mild cognitive impairment (Arriagada, et al., 1992, Bretteville and Planel, 2008, Guillozet, et al., 2003, Wilcock and Esiri, 1982).
There is developing interest in the potential association between anesthesia and the onset and progression of AD (Baranov, et al., 2009, Bianchi, et al., 2007, Brunden, et al., 2011, Papon, et al., 2011, Whittington, et al., 2013). In terms of tau protein specifically, we previously demonstrated that both inhalational and intravenous anesthetics induce robust tau hyperphosphorylation and increase tau pathology through anesthesia-induced hypothermia (Planel, et al., 2009, Planel, et al., 2008, Planel, et al., 2007). However, we more recently observed that propofol, a commonly used intravenous sedative-hypnotic agent, increased tau phosphorylation under normothermic conditions (Whittington, et al., 2011). Nevertheless, it remains unclear whether this hyperphosphorylation effect is a unique property of propofol or whether it can be observed with other intravenous agents that are currently used in clinical anesthesia practice.
Dexmedetomidine is a highly selective alpha-2 adrenergic receptor (α2-AR) agonist that produces its sedative response by binding to the α2-ARs in the locus coeruleus (Correa-Sales, et al., 1992). Clinically, the use of dexmedetomidine for sedative purposes in intensive care units has steadily increased over the last several years (Wunsch, et al., 2010). Interestingly, in contrast to benzodiazepines, the use of dexmedetomidine has been associated with decreased delirium (Riker, et al., 2009). Despite this “safer” cognitive profile, we posited the hypothesis that the capacity to induce tau hyperphosphorylation, under normothermic conditions, is not unique to anesthetics that affect γ-aminobutyric acid (GABA) receptors such as propofol, but also a property exhibited by α2-AR agonists such as dexmedetomidine. Thus, we investigated the impact of a hypnotic dose of dexmedetomidine on tau phosphorylation in vivo and in vitro, under normothermic conditions. Furthermore, to determine whether changes in tau phosphorylation levels, following acute dexmedetomidine administration, correlate with changes in memory, short-term and long-term spatial reference memory were assessed in C57BL/6 mice by means of Barnes Maze testing.
We observed that normothermic administration of dexmedetomidine induced robust tau hyperphosphorylation lasting up to 6h for some specific epitopes and impacted spatial memory. While we could not identify a specific kinase or phosphatase responsible for this phenomenon, we demonstrated that it was mediated by α2-AR activation, as treatment with atipamezole blocked dexmedetomidine-induced tau hyperphosphorylation.
2. Methods
2.1 Mice Treatment and Anesthesia Administration
The Columbia University Animal Care and Use Committee approved the experimental protocol, and appropriate measures were taken to minimize pain and discomfort in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals as well as the EU Directive 2010/63/EU for animal experiments. Eight to 10-week-old C57BL/6 male mice, purchased from a commercial vendor (Taconic, Germantown, NY), were used in this study. We also used 3-month old hTau mice, a well-characterized model of tau pathology overexpressing human tau on a murine tau knockout background and developing tau pathology (Andorfer, et al., 2005, Andorfer, et al., 2003). All mice were housed at 22°C in a temperature-controlled room and kept on a 12h/12h light/dark cycle. Food and water were made available ad libitum, and all mice underwent an acclimatization period of at least 24h prior to their use in the experiments.
On the day of the study, a 30 μg/ml solution of dexmedetomidine hydrochloride (Tocris Bioscience, Ellisville, MO) in 0.9% saline was prepared. In experiments examining the acute effect of dexmedetomidine on tau phosphorylation in vivo, the mice were treated with either dexmedetomidine 300 μg/kg or 0.9% saline as a control (100 μL per 10 g of mouse body weight) via intraperitoneal (i.p.) injection. This dose of dexmedetomidine was based on doses used in previous studies, which demonstrated transient loss of righting reflex without any significant adverse effects (Hunter, et al., 1997, Vulliemoz, et al., 1998).
In the studies in which the effects of dexmedetomidine were pharmacologically antagonized with atipamezole, a highly selective α2-AR antagonist, 100 (atipamezole low) and 200 (atipamezole high) μg/mL solutions of atipamezole hydrochloride (Tocris Bioscience, Ellisville, MO) in 0.9% saline were prepared. The mice were then treated with atipamezole 1 mg/kg, 2 mg/kg, or an equal volume of saline (control for atipamezole) i.p. 15 min prior to receiving dexmedetomidine 300 μg/kg or saline i.p. (control for dexmedetomidine). These atipamezole doses were based on previously published studies in mice wherein several of the physiological effects of dexmedetomidine, including its sedative effects, were reversed without any adverse sequelae (Hocker, et al., 2008, Nelson, et al., 2003).
After receiving their treatment injections, all control (unanesthetized) mice were returned to their respective home cage, which was maintained at room temperature. The mice that received dexmedetomidine were initially anesthetized in their home cage at room temperature, and, once sedated, transferred to a Thermocare ICS therapy warmer unit (Thermocare, Incline Village, NV), which was set to maintain a mouse body temperature of approximately 37°C throughout the study. All anesthetized mice were maintained in their home cage while in the incubator, in order to avoid the stress of a foreign environment following the initial emergence from anesthesia. For those mice sacrificed 2 or 6h following dexmedetomidine treatment, upon emergence from anesthesia, the cage was removed from the incubator and placed on a warming pad located adjacent to the control mice. Rectal temperatures were monitored using an electronic thermometer (Thermalert TH-5, Physitemp, Clifton, NJ), and these temperatures, at the time of brain tissue collection, were compared to ensure that normothermia was present in each group. The control mice were not placed in the warmer unit, as this could have potentially introduced the stress of a foreign environment, which could have resulted in an artifactual increase in tau phosphorylation (Rissman, 2009).
2.2 Preparation of Mouse Brain Protein Samples
The mice were killed by cervical dislocation without anesthesia at 30 min, 2h, 6h, or, in the case of the Barnes Maze studies, 1 week following dexmedetomidine or saline administration. Each brain was immediately harvested and dissected in ice-cold Tris-EDTA buffer (100 mM Tris HCl, 1 mM EDTA, pH 7.4), then the hippocampal and cortical tissues were immediately frozen in liquid nitrogen and stored at -80°C as previously described (Planel, et al., 2009, Planel, et al., 2007, Whittington, et al., 2011). Tissue homogenates were also prepared using our previously described method (Planel, et al., 2008). Briefly, hippocampal and cortical tissues were homogenized in 5× v/w RIPA buffer containing 50 mM Tris-HCl (Sigma-Aldrich, St. Louis, MO), pH 7.4, 1 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 0.1% SDS (Sigma), 0.5% sodium deoxycholate, 1% Nonidet P-40 (Sigma-Aldrich), phosphatase inhibitors (Cocktail 1 and 2, Sigma-Aldrich, 1:100 dilution), and protease inhibitors (Cocktail set III, Calbiochem, EMD Biosciences Inc., La Jolla, CA, 1:200 dilution). All homogenates were incubated on ice for 30 min, sonicated for 30s in pulse mode, and then centrifuged at 11,000 g for 10 min at 4°C. The total protein content was determined in the supernatant using the bicinchoninate (BCA) method (Smith, et al., 1985).
2.3 Preparation of insoluble and soluble tau fraction
The insoluble tau fraction was prepared according to our protocol previously used in mouse models of tauopathies (Julien, et al., 2012). This procedure uses 1% sarkosyl and is similar to ones previously used to isolate tau aggregates from the brains of Alzheimer's disease patients (Greenberg and Davies, 1990). Briefly, the RIPA supernatant from hTau mice brains was adjusted to 1% sarkosyl (N-lauroylsarcosine), incubated for 30 min at room temperature with constant shaking, and centrifuged at 100,000 × g for 1 hour at 20°C. The pellet containing sarkosyl-insoluble aggregated (insoluble fraction) was resuspended and diluted in Sample buffer (NuPAGE LDS) containing 5% of 2-β-mercapto-ethanol, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 10 μl/ml of Proteases Inhibitors Cocktail (P8340, Sigma-Aldrich), boiled for 5 min, and kept at -20°C. The soluble, aggregate-free fraction was obtained by boiling an aliquot of RIPA supernatant for 5 min and removing protein aggregates by centrifugation at 20,000 g for 20 min at 4°C and resuspension in Sample Buffer (Planel, et al., 2009).
2.4 SH-SY5Y Cell Culture Studies and Cell Lysate Preparation
Dexmedetomidine-induced tau phosphorylation was also examined in vitro using SH-SY5Y human neuroblastoma cells stably transfected to constitutively express human tau with 3 microtubule binding domains (Delobel, et al., 2002, Hamdane, et al., 2003, Mailliot, et al., 2000). These Tau-SH-SY5Y cells were a kind gift of Dr. Luc Buée (Unité 422, INSERM, Lille, France), and our previous studies have demonstrated that they are suitable for examining the impact of anesthetics on tau phosphorylation in an environment devoid of anesthesia-induced systemic physiological changes (Whittington, et al., 2011). The Tau-SH-SY5Y cells were grown initially in 25-cm2 flasks containing Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum, 2 mM L-glutamine, 1 mM nonessential amino acids as well as 50 units/ml penicillin/streptomycin (DMEM and cell culture additives were purchased from Invitrogen, Carlsbad, CA), and were maintained in a 5% CO2-95% O2 humidified incubator at 37°C (Tatebayashi, et al., 2006). In preparation for dexmedetomidine or vehicle exposure, the cells were transferred into 6-well plates and used at approximately 70-80% confluency.
In the 4h dexmedetomidine treatment experiments, each well was incubated with either dexmedetomidine (0.1 or 10 μM) in growth medium (DMEM with 2 mM L-glutamine, non-essential amino acids, and 10% fetal bovine serum) or growth medium alone (control) at 37°C. In the 24h experiments, each well was incubated with either dexmedetomidine (1, 10, or 100 μM) in growth medium (DMEM with 2 mM L-glutamine, non-essential amino acids, and 10% fetal bovine serum) or growth medium alone (control) for 24h at 37°C. The dexmedetomidine concentrations used in this study were based on those previously utilized in neuronal cell culture studies (Sanders, et al., 2010) as well as concentrations previously demonstrated to be neuroprotective in an organotypic hippocampal slice culture model of traumatic brain injury (Schoeler, et al., 2012). At the end of the exposure period, the media were removed by aspiration, and each well was rinsed twice with 1 ml of phosphate buffered saline warmed to 37°C. The cells were harvested by scraping in 200 μl of ice-cold RIPA buffer containing phosphatase (Cocktail 1 and 2, Sigma-Aldrich, 1:100 dilution) and protease inhibitors (Cocktail set III, EMD Biosciences Inc., La Jolla, CA, 1:200 dilution). All samples were stored at -80°C until they were used in the immunoblotting analyses. The preparation of the Tau-SH-SY5Y cell lysates for SDS-PAGE and Western blot analysis was performed as we have previously described (Whittington, et al., 2011). Poly(ADP-ribose) polymerase (PARP) cleavage was measured in the 24h cell culture experiments using a polyclonal PARP antibody (Catalog #9542, New England Biolabs, Whitby, Ontario, Canada), to examine whether apoptosis played a role in mediating any dexmedetomidine-induced phosphorylation changes in vitro.
2.5 SDS-PAGE and Western Blot Analysis
The protein expressions of phosphorylated tau, total tau as well as the tau kinases and phosphatases were determined using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) coupled with Western blot analysis, as we have previously described (Whittington, et al., 2011). In brief, brain homogenate or cell lysate aliquots containing 30 μg protein were separated on a SDS-10% polyacrylamide gel then transferred onto nitrocellulose membranes (GE Healthcare Life Sciences, Piscataway, NJ). Non-specific binding sites were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1h at room temperature then incubated overnight at 4°C with antibodies directed against total tau, phosphorylated tau, a specific tau kinase, or phosphatase. Following washing, the membranes were incubated for 1h at room temperature with a horseradish peroxidase-linked secondary anti-mouse antibody (Cell Signaling Tecphnology, Boston, MA, 1:2500 dilution), and the immunoreactive band signal intensity was visualized by enhanced chemiluminescence (ECL Plus, GE Healthcare Life Sciences, Piscataway, NJ). The immunoreactive bands were scanned using an ImageQuant LAS 4000 imaging system (GE Healthcare Life Sciences, Piscataway, NJ), and densitometric quantification was performed on these scans with ImageQuant® TL 7.0 software (GE Healthcare Life Sciences, Piscataway, NJ). All immunoblots were normalized for gel loading with β-actin using an antibody purchased from Sigma-Aldrich (1:10,000 Catalog# A2228 Sigma-Aldrich, St. Louis, MO).
2.6 Detection of Total and Phosphorylated Tau
Antibodies utilized in this study were directed at tau phosphorylated at the following epitopes: AT8 (pSer202/pThr205, 1:250 dilution, Pierce Biotechnology) (Goedert, et al., 1995), PHF-1 (pSer396/Ser404, 1:1000) (Otvos, et al., 1994), CP13 (pSer202, 1:1000) (Weaver, et al., 2000), pSer199 (1:2000, Invitrogen, Carlsbad, CA), pSer422 (1:1000, Invitrogen), Tau-1 (Szendrei, et al., 1993) (recognizes tau dephosphorylated Ser195/198/199/202, 1:1000, Millipore, Billerica, MA), 12E8 (Ser262/Ser356, 1:1000) (Seubert, et al., 1995). These phosphorylated tau antibodies were selected for examination, as their epitopes have been associated with pre-tangle formation (p Ser199, CP13) and paired helical filament and NFT formation (AT8, PHF-1) (Augustinack, et al., 2002, Bussiere, et al., 1999, Goedert, et al., 1994, Goedert, et al., 1993, Maurage, et al., 2003), while 12E8 can regulate tau binding to microtubules (Seubert, et al., 1995). Moreover, our previous study examining propofol-induced tau phosphorylation demonstrated that, when compared to CP13 and PHF-1, the AT8 phosphoepitope was particularly sensitive to direct phosphorylation by propofol (Whittington, et al., 2011); therefore, we sought to determine whether the same phosphorylation pattern would be observed with dexmedetomidine. Total tau was detected using one of the following antibodies that detects all 6 six isoforms of tau: Tau46 (monoclonal, 1:2500, Cell Signaling Technology, Danvers, MA) or Tau A0024 (polyclonal, 1:10,000, Dako Cytomation, Carpinteria, CA). Phosphorylated tau levels were normalized to total tau level.
2.7 Detection of Kinases and Phosphatases
The changes in the expression and activation of tau kinases were examined using the following antibodies: GSK-3β (1:1000, BD Transduction Lab, Franklin Lakes, NJ) as well as the following antibodies purchased from Cell Signaling Technology, Inc. (Danvers, MA): phospho-GSK-3β (Ser9, 1:1000), p35/p25 (1:1000), phospho-Akt (Ser473, 1:1000), Akt (1:1000), SAPK/JNK (1:1000), phospho-SAPK-JNK (Thr183/Tyr185, 1:1000), p44/42 MAPK (Erk 1/2, 1:1000), phospho-p44/42 MAPK (Erk 1/2, Thr202/Tyr204, 1:1000), p38 MAPK (P38, 1:1000). Cdk5 (C-8), phospho-Ca2+/calmodulin-dependent protein kinases II (CaMKII, Thr286, 1:1000) and total CaMKII (1:1000) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-p38 (clone 6E5.2, p-P38, Thr180/Tyr182, 1/1000) was purchased from Millipore, and GSK-3 α/β PY279/PY216 (Tyr279/Tyr216, 1:1000) from Life Technologies (Grand Island, NY).
The changes in the expression of the tau phosphatases were determined using antibodies directed at PP2A-C (1:1000, Cell Signaling), PP5 (1:1000, Cell Signaling), PP2A-Bα subunit (2G9), 1:1000, Cell Signaling), demethylated PP2A-C subunit (4B7, 1:1000, Santa Cruz), PP2A-A subunit (1:1000, Cell Signaling), pan-calcineurin A (PP2B, 1:1000, Cell Signaling), and the PP1 catalytic subunit (E-9, 1:1000, Santa Cruz).
2.8 Barnes Maze Testing
Barnes maze testing (Barnes, 1979) was selected for the assessment of spatial reference memory in the C57BL/6 mice following dexmedetomidine administration as, unlike the Morris Water Maze, the Barnes maze is neither associated with the stress of forced swimming nor the potential for hypothermia secondary to water immersion, factors that could artifactually increase phosphorylated tau levels. The Barnes maze apparatus was purchased from a commercial vendor (Stoelting, Inc., Wood Dale, IL) and consisted of a circular metal platform (91 cm in diameter and elevated to a height of 90 cm from the ground) containing 20 holes, each measuring 5 cm in diameter. With the exception of one of the 20 holes, referred to as the target hole, all of the holes were blocked with a piece of metal, which prevented a means of escape for the mouse. Beneath the target hole there was a small, dark recessed chamber through which the mouse could escape from the open platform. Spatial cues were placed around the maze with bright light, provided by a 150-watt incandescent light bulb (580 lux), as well as white noise (85dB) generated by the Any-maze™ software (Stoelting, Inc., Wood Dale, IL), used as reinforcers. The movements of the mouse in the maze were continuously tracked and recorded using the Any-maze™ computerized video tracking software.
Barnes maze testing consisted of several specific phases of varying duration: an adaptation phase (Day 0), acquisition phase (Days 1-4), probe test I measuring short-term memory (Day 5), and probe test II measuring long-term memory (Day 12). Our Barnes Maze testing protocol was based on previous studies describing the use of this neurobehavioral test in rodents (Barnes, 1979, Chen, et al., 2011, Patil, et al., 2009, Sunyer, et al., 2007). The same investigator performed all phases of the Barnes Maze testing protocol while another investigator, blinded to the treatments, performed the analysis of the computerized behavioral data including the recorded video data. The Barnes Maze testing phases were specifically performed as follows:
Adaptation Phase
Day 0 consisted of an adaptation phase, during which the mouse was acclimated to the testing platform, the target area, and the escape box. Each mouse was placed in the middle of the maze under a dark start box. After 10s, the box was lifted, the white noise then commenced, and the mouse was gently guided towards the target hole. Once in the target hole area, the mouse was passively allowed to enter the target hole under its own volition; however, if the mouse did not enter the hole, they were placed inside the target hole escape box. While in the escape box, the white noise was discontinued, the target hole then covered, and the mouse was allowed to stay in the box for 2 min.
Spatial acquisition Phase (Days 1-4)
In this phase, the mouse was trained to enter the target hole within 180s. During each trial, the mouse was placed under the start box in the center of the testing platform and, after 10s, the box was lifted, the white noise switched on and the mouse was allowed to explore the maze with the goal of reaching the escape box under the target hole. Each trial was terminated when the mouse successfully reached the escape box or at the end of 180s. Once the mouse was in the escape box, the white noise was switched off, the target hole again covered, and the mouse was allowed to stay there for 1 min. If the mouse did not enter the escape box within 180s, it was guided there and left inside for 1 min, with the target hole covered. Three trials, at approximately 15 min intervals, were performed for each mouse, for 4 consecutive days with the goal of reaching asymptotic performance. Before each trial, the maze was cleaned with 40% ethanol to remove olfactory cues. Variables measured during the acquisition phase included total latency, total distance, and total errors.
Probe Test I (Short-term spatial reference memory retention)
On Day 5, probe test I, a test of short-term reference memory retention, was performed. On the morning of this test, the mice were randomly assigned to receive either dexmedetomidine 300 μg/kg (n = 8) or an equivalent volume of saline (n = 8) i.p. Six hours later, probe test I was initiated by placing each mouse in the middle of the Barnes Maze. After 10s, the start box was lifted, the white noise commenced and the mouse was allowed to explore the maze. During the probe test, the target hole was still located in its same position as in the acquisition phase; however, the hole was blocked, the same way as the other 19 holes, such that the mouse could not enter the escape box. Probe test I was terminated after 90s, and the latency to reach the target hole, total errors, primary distance travelled (distance travelled before reaching the target hole) and mean speed (m/s) were measured. In addition, target hole preference was determined by measuring the number of nose pokes for the adjacent and opposite holes and comparing this value to the number of pokes in the target hole region, according to a previously published protocol (Sunyer, et al., 2007).
Probe Test II (Long-term reference memory retention)
On Day 12, probe test II was performed, in the same manner as probe test I, exactly 1 week following the acute administration of dexmedetomidine or saline. Again, the same parameters were measured and compared as in Probe Test I. At the end of probe test II, the mice were sacrificed using cervical dislocation and the cortex and hippocampal tissues dissected for analysis of phosphorylated and total tau levels.
2.9 Statistical Analysis
Group comparisons of immunoblot relative band intensities were performed using a oneway analysis of variance (ANOVA) with Newman-Keuls Multiple Comparison post hoc test applied when appropriate or by means of an unpaired t-test. Group comparisons of rectal temperatures were performed using one-way analysis of variance (ANOVA) or an unpaired t-test. All statistical calculations were performed using Prism® 5 software (GraphPad Software, Inc., San Diego, CA), and all data are reported as mean ± SD with a value of P < 0.05 considered statistically significant.
Barnes Maze performance variables during the acquisition phase were analyzed using a two-way repeated-measures ANOVA with Newman-Keuls Multiple Comparison post hoc test applied when appropriate. Between group analyses of probe test variables were performed using an unpaired t-test. For each probe test, target hole preference (pokes) for each treatment group was determined using ANOVA with Newman-Keuls post hoc test applied when appropriate. All of the Barnes Maze statistical comparisons were performed Prism® 5 software, and these behavioral data are reported as mean ± SEM with P < 0.05 deemed statistically significant.
3. Results
3.1 Dexmedetomidine Increases Tau Phosphorylation Under Normothermic Conditions in the Mouse Hippocampus
We first determined whether the administration of dexmedetomidine, under normothermic conditions, had an effect on levels of phosphorylated tau (p-tau) in the hippocampus of 8 to 10-week-old C57BL/6 male mice. Thirty min (0.5h, n = 6) following administration of 300 μg/kg dexmedetomidine i.p., there was a significant increase (expressed as % of control) in hippocampal tau phosphorylation at the CP13 (170 ± 68%), AT8 (182 ± 36%), and PHF-1 (183 ± 27%) phosphoepitopes (Fig. 1C, D, F, 0.5h), when compared to the saline-treated mice (Ctl, n = 6), while other epitopes did not increase. Interestingly, at 2h (n = 6), dexmedetomidine treatment was still associated with significant increases in hippocampal p-tau levels at the same three phosphoepitopes, despite the return of the righting reflex in all of the mice at this time point (Fig. 1C, D, F, 2h). Dexmedetomidine (Dex) administration had no effect on total tau levels at either time point (Fig. 1H), and rectal temperatures, at the end of the experiment, were similar in all of the study groups: Ctl 37.1 ± 0.6, Dex 0.5h 37.1 ± 0.5, and Dex 2h 37.1 ± 0.7°C.
Figure 1. Dexmedetomidine increases tau phosphorylation in the mouse hippocampus following 2h administration under normothermic conditions.
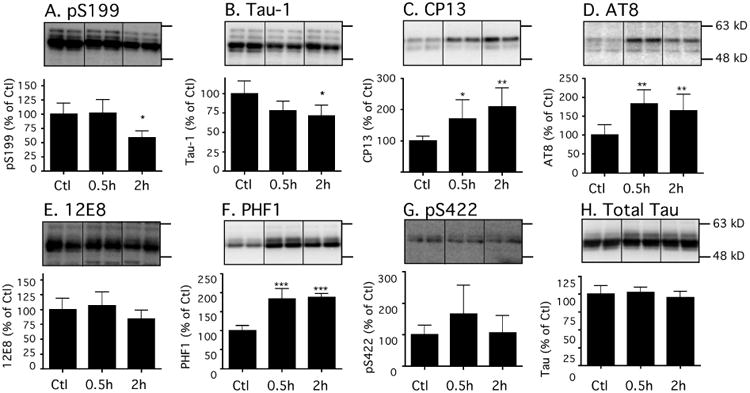
Levels of tau phosphorylation at Ser199, Tau-1 (tau dephosphorylated Ser195/198/199/202), CP13 (Ser202), AT8 (Ser202/Thr205), 12E8 (Ser262/Ser356), PHF-1 (Ser396/Ser404), and Ser422 phosphoepitopes in hippocampal proteins C57BL/6 mice treated with dexmedetomidine 300 μg/kg i.p. and sacrificed 0.5h (n =6) or 2h (n = 6) following treatment. Levels of phosphorylated tau were normalized to total tau (H). Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of Ctl, and are displayed for each phosphoepitope and total tau. For each condition, 2 representative bands are displayed. Data are expressed as mean ± SD, and were analyzed using ANOVA with Newman-Keuls post hoc test. * denotes P < 0.05, ** denotes P < 0.01, and *** denotes P < 0.001 vs. Ctl.
3.2 Tau Hyperphosphorylation in the Mouse Brain 6h after Dexmedetomidine Administration Resolves with the Exception of Hippocampal AT8
As tau hyperphosphorylation was still present in the mouse brain 2h following dexmedetomidine administration, we next examined tau phosphorylation levels in the mouse hippocampus 30 min as well as 6h following dexmedetomidine treatment. The 6h time point was chosen as all mice demonstrated no signs of sedation or hypnosis at 6h; whereas, just 2h following dexmedetomidine anesthesia, some animals still exhibited gait disturbances despite the return of the righting reflex in all of the mice within 1h of drug administration. Furthermore, our previous study with propofol anesthesia in mice demonstrated that 6h following acute propofol administration, hippocampal tau phosphorylation returned to control levels (Whittington, et al., 2011). Therefore, we also focused on this time point to determine whether a similar resolution of hyperphosphorylation would be observed 6h following dexmedetomidine.
In these experiments, we determined the degree of tau phosphorylation at the epitopes shown to be affected at 30 min (Fig. 1), in hippocampal and cortical tissue obtained from male C57BL/6 mice 0.5h (n=6) and 6h (n=5) following normothermic dexmedetomidine administration (300 μg/kg i.p.) as well as 0.5h following saline administration (Ctl, n = 6). Again, 0.5h following dexmedetomidine administration, an increase in phosphorylated tau in the hippocampus was observed at all 3 phosphoepitopes: AT8: 230 ± 19%, CP13: 185 ± 18%, and PHF-1: 210 ± 8% versus the control group (Fig. 2A1-3). Interestingly, tau phosphorylation returned to control levels at PHF-1 and CP13 at 6h, whereas phosphorylation at AT8 (139 ± 14%) remained slightly, but significantly, elevated (P < 0.05) versus the control-treated mice (Fig 2A1, 6h). Levels of total tau at 0.5h remained similar to control mice; however a small, but significant increase in total tau was observed at 6h (Fig 2A4). Of note, rectal temperatures at the end of the study were similar in all groups: Ctl 37.4 ± 0.4, Dex 0.5h 37.2 ± 0.4, and Dex 6h 37.2 ± 0.3°C.
Figure 2. Tau returns to basal levels 6h after dexmedetomidine administration under normothermic conditions in the cortex but not in the hippocampus.
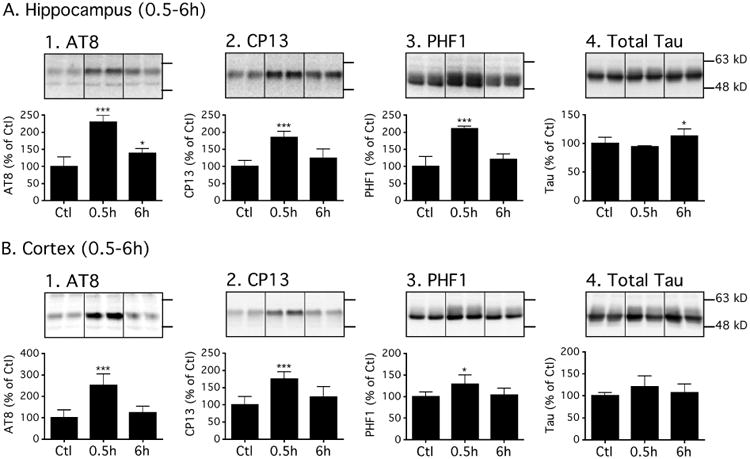
Levels of tau phosphorylation at AT8 (Ser202/Thr205), CP13 (Ser202), and PHF-1 (Ser396/Ser404) phosphoepitopes in hippocampal (A) or cortical (B) proteins of C57BL/6 mice treated with dexmedetomidine 300 μg/kg i.p. and sacrificed 0.5h (n =6) or 6h (n = 5) following treatment. Levels of phosphorylated tau were normalized to total tau (A4 or B4). After 6h, AT8 hyperphosphorylation persisted in the hippocampus while other epitopes returned to basal levels in both hippocampus and cortex. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of Ctl, and are displayed for each phosphoepitope and total tau. For each condition, 2 representative bands are displayed. Data are expressed as mean ± SD, and were analyzed using ANOVA with Newman-Keuls post hoc test. * denotes P < 0.05 and *** denotes P < 0.001 vs. Ctl.
In the cortical tissue, 0.5h following dexmedetomidine administration, significant increases in phosphorylated tau (P < 0.05 and P < 0.001) were also observed at AT8, PHF-1 and CP13 phosphoepitopes (Fig. 2B1-3); moreover, these increases were of the same order of magnitude as those observed in the hippocampus. However, they disappeared by 6h, which was again similar to the pattern observed in the hippocampus, with the exception of the AT8 phosphoepitope. There were no significant changes in total tau at either time point (Fig. 2B4).
Overall, these data indicate that normothermic dexmedetomidine administration acutely induces tau hyperphosphorylation in the mouse hippocampus and cortex. In the hippocampus, this effect persists 2h following drug administration; however, with the exception of a modest increase at the AT8 phosphoepitope, this effect disappears upon complete recovery from the sedative effect of the drug. Similarly, the cortical increase in phosphorylated tau, observed 0.5h following dexmedetomidine administration, resolves by 6h.
3.3 Impact of Dexmedetomidine on Tau Kinases and Phosphatases
We next wanted to determine the mechanism of tau hyperphosphorylation during dexmedetomidine administration. The phosphorylation state of tau is dependent on a balance between the activity of several major tau protein kinases (Planel, et al., 2002) such as glycogen synthase kinase-3β (GSK-3β), c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase or mitogen-activated protein kinase (ERK or MAPK), p38 MAPK, cyclin-dependent kinase 5 (cdk5) and its activators p35/p25, and calmodulin-dependent kinase II (CaMKII), as well as the activity of several protein phosphatases, including protein phosphatase 1 (PP1), protein phosphatase 2B (PP2B), protein phosphatase 2A (PP2A), and PP5 (Tian and Wang, 2002). Furthermore, as we have previously demonstrated that propofol-induced tau hyperphosphorylation, under normothermic conditions, involves a decrease in PP2A activity and expression (Whittington, et al., 2011), we subsequently determined whether a similar mechanism was involved in mediating dexmedetomidine-induced tau phosphorylation.
Thirty minutes and 2h following dexmedetomidine treatment, there was no increase in kinases activation correlating with tau hyperphosphorylation. On the contrary, ERK (Fig. 3C), JNK (Fig. 3E), CaMKII (Fig. 3G), and GSK-3β (Fig. 3N) were inhibited. There was an accumulation of P38 at both time (Fig. 3J), but its activated form remained undetectable (Fig. 3I). There was also accumulation of P35 at 2h (Fig. 3K), but it could not explain the extent of tau phosphorylation at both time points. Collectively, all of these changes indicated an inhibition of some kinases, which was not consistent with the increased tau phosphorylation observed 30 min or 2h following dexmedetomidine administration.
Figure 3. Effects of dexmedetomidine on tau kinases in the mouse hippocampus under normothermic conditions.
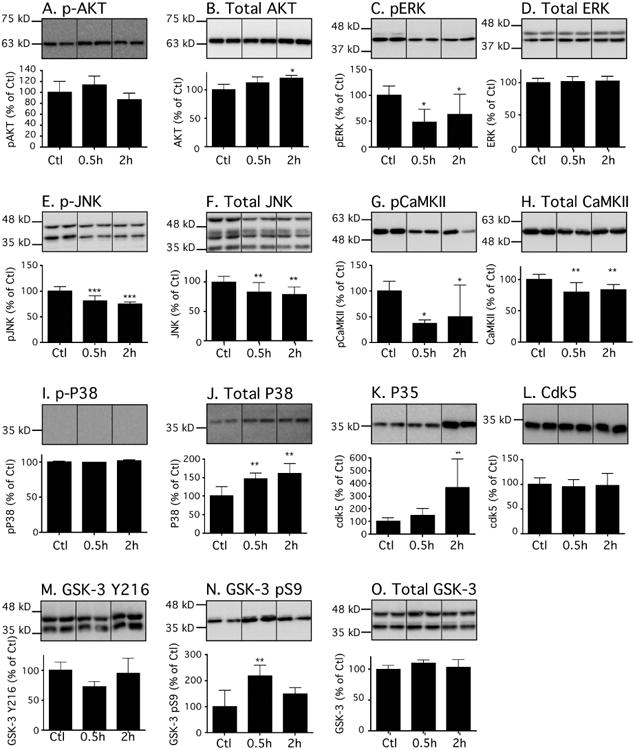
Hippocampal proteins were separated by SDS-PAGE, and levels of tau kinases were determined using antibodies directed at activated or total kinases: (A) phospho-Akt, (B) Akt, (C) phospho-ERK, (D) ERK, (E) phospho-JNK, (F) JNK, (G) phospho-CaMKII, (H) CaMKII, (I) phospho-p38, (J) p38, (K) p35, (L) cdk5, (M) phospho-GSK3β pY216, (N) phospho-GSK3β pS9, and (O) (M) GSK3β. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of saline control (Ctl) and are displayed for each epitope. For each condition, 1 representative band is displayed with Ctl (n = 6), 0.5h (n = 6), and 2h (n=6). Data are expressed as mean ± SD. *, ** and *** denote P < 0.05, P < 0.01 and P < 0.001 vs. Ctl, respectively; ANOVA with Newman-Keuls post hoc test.
Therefore, we examined protein phosphatases expression at these same time points (Fig. 4), especially as we have previously observed that intravenous anesthetics can decrease the expression and activity of protein phosphatase (Whittington, et al., 2011). Except for a decrease in demethylated PP2A (Fig. 4E, resulting in PP2A activation), there was no change in any of the tau phosphatases examined. Methylation activates PP2A, while demethylation inhibits it and has been associated with tau hyperphosphorylation (Zhou, et al., 2008).
Figure 4. Effects of dexmedetomidine on tau phosphatases in mouse hippocampal tissue under normothermic conditions.
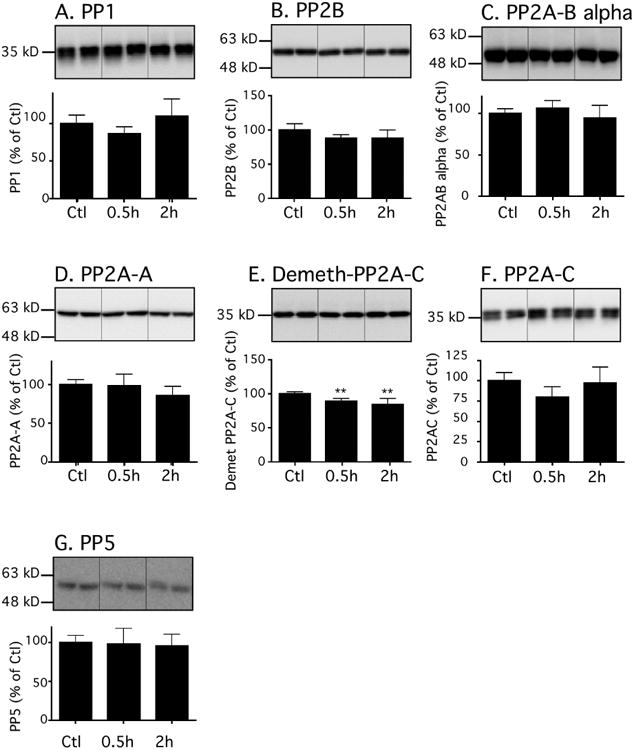
Hippocampal proteins were separated by SDS-PAGE and levels of phosphatases were determined using antibodies directed at the following proteins: (A) PP1 catalytic subunit, (B) PP2B catalytic subunit, (C) PP2A Ba regulatory subunit, (D) PP2A scaffolding subunit (PP2A-A), (E) Demethylated PP2A catalytic subunit, (F) PP2A catalytic subunit, and G. PP5. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of saline control (Ctl) and are displayed for each epitope. For each condition, 1 representative band is displayed. All data are expressed as mean ± SD. ** and *** denote P < 0.01 and P<0.001 vs. Ctl, respectively; Ctl (n = 6), 0.5h (n = 6), and 2h (n = 6); ANOVA with Newman-Keuls post hoc test.
In summary, most of the changes observed indicate the inhibition of several tau kinases (GSK-3β, ERK, JNK, CaMKII) and activation of the phosphatases (PP2A), which fail to mechanistically explain the observed dexmedetomidine-induced tau hyperphosphorylation.
3.4 Hippocampal Tau Hyperphosphorylation at AT8 following In Vivo Dexmedetomidine Administration is Dependent on α2-AR Activation
As the sedative effects of dexmedetomidine are mediated by its agonist effects at α2-ARs in the locus coeruleus (Correa-Sales, et al., 1992), we next examined whether the increases in hippocampal tau phosphorylation observed following dexmedetomidine are dependent on activation of α2-ARs. Establishing the specific role of α2-ARs in mediating dexmedetomidine-induced tau hyperphosphorylation is mechanistically important given that, although dexmedetomidine is a highly specific α2-AR agonist, it is also known to have effects at other receptor types such as imidazoline receptors (Dahmani, et al., 2008). Thus, we measured dexmedetomidine-induced tau phosphorylation, in 8 to 10-week-old C57BL/6 male mice, in the absence or presence of pretreatment with atipamezole, a highly specific α2-AR antagonist (Pertovaara, et al., 2005). We specifically measured phosphorylation at AT8, as our acute dexmedetomidine administration studies demonstrated that this phosphoepitope was associated with the highest and most persistent degree of tau hyperphosphorylation following dexmedetomidine administration.
An initial experiment was performed to determine whether atipamezole had any effect on tau phosphorylation at the maximum dose utilized to antagonize the phosphorylation effects of dexmedetomidine. In this experiment, 8 to 10-week-old C57BL/6 male mice were treated with either atipamezole 2 mg/kg (n = 6) or saline (Ctl; n = 6) i.p. and sacrificed 45 min following each treatment. Normothermia was maintained throughout the study, and there was no significant difference in rectal temperatures between the groups: 37.2 ± 0.4 and 37.0 ± 0.4°C, in the saline and atipamezole groups, respectively. No significant increases in hippocampal tau phosphorylation at AT8 were observed in the atipamezole-treated mice (Fig. 5A), but a minor increase in total tau was observed (Fig. 5B).
Figure 5. Atipamezole abolishes dexmedetomidine-induced tau hyperphosphorylation in the mouse hippocampus under normothermic conditions.
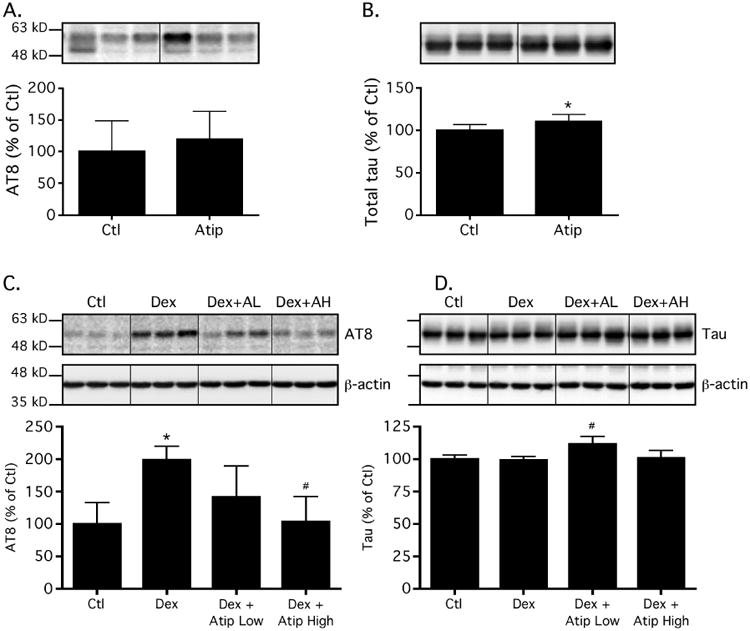
Immunoblot analysis of protein expression levels of tau phosphorylated at the AT8 phosphoepitope (5A) as well as total tau (5B) in the hippocampus of C57BL/6 mice, 45 min following the administration of atipamezole 2 mg/kg (Atip, n = 6) or 0.9% saline (Ctl, n = 6) i.p. These data demonstrate that atipamezole itself had no effect on hippocampal phosphorylated tau levels. In Fig. 5C and Fig. 5D the immunoblot analysis of levels of tau phosphorylated (% Ctl) at the AT8 phosphoepitope (5C) as well as total tau (5D) in the hippocampus of C57BL/6 mice pretreated with 0.9% saline (Dex, n = 5), atipamezole 1 mg/kg (Dex + Atip Low [L], n = 6), or atipamezole 2 mg/kg i.p. (Dex + Atip High [H], n = 6) followed 15 min later by dexmedetomidine 300 mg/kg i.p are shown. Control (Ctl, n = 5) mice received an initial i.p. injection of 0.9% saline, followed 15 min later by a second i.p. saline injection. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. All mice were sacrificed 30 min following the second injection. Both AT8 and total tau were controlled for gel loading using β-actin; however, levels of phosphorylated tau were normalized to total tau. Data are expressed as mean ± SD. * P < 0.05 vs. Ctl and # P < 0.01 vs. Dex group using ANOVA with Newman-Keuls post hoc test.
Next, the tau phosphorylation response to acute dexmedetomidine administration was examined in the absence and presence of atipamezole (Fig. 5C) and normalized to total tau (Fig. 5D). As expected with dexmedetomidine administration, 15 min after pre-treatment with saline (control for atipamezole, n = 5), resulted in a significant increase (P < 0.001) in hippocampal phosphorylated tau levels at AT8, 30 min following dexmedetomidine administration (n = 5, Dex). However, pretreatment with either atipamezole 1 mg/kg (Dex + Atip Low [L], n =6) or 2 mg/kg (Dex + Atip High [H], n = 6) 15 min prior to dexmedetomidine, abolished the significant increase in phosphorylated tau levels observed at AT8 in Sal + Dex treated mice (Fig. 4C, Dex + AL/AH, respectively), such that levels of phosphorylated tau in atipamezole-pretreated groups were not significantly different from saline-treated controls (Ctl, n = 5). Moreover, mice pretreated with either dose of atipamezole did not become sedated following the administration of dexmedetomidine, as expected. Again, rectal temperatures at the end of the study were similar in all groups: Ctl 37.2 ± 0.7, Dex 37.2 ± 0.7, Dex+ Atip Low 37.4 ± 0.6, and Dex + Atip High 37.1 ± 0.5 °C.
3.5 Acute Dexmedetomidine Administration Impairs Spatial Memory
Barnes Maze testing was performed in a separate group of 8 to 10-week-old C57BL/6 male mice in order to determine whether dexmedetomidine, at a dose demonstrated to produce a transient increase in hippocampal and cortical tau phosphorylation, impacts short-term and long-term spatial reference memory. Data from the acquisition phase of testing (Days 1-4) demonstrate that the mice quickly learned how to reach the target hole, as asymptotic performance in terms of latency, distance travelled, and number of errors was reached (Fig. 6A).
Figure 6. Barnes Maze testing demonstrates that dexmedetomidine-increased tau phosphorylation is associated with the impairment of spatial reference memory.
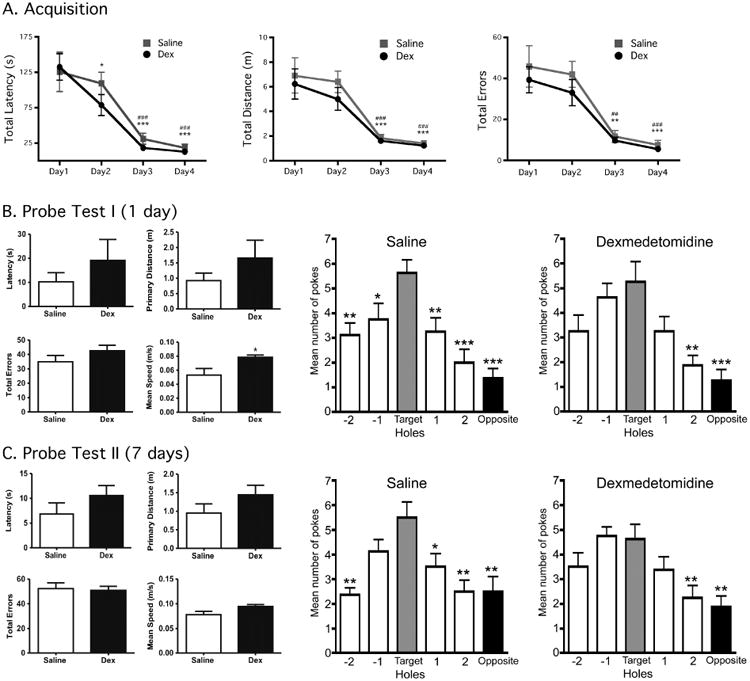
Acquisition phase testing data (mean ± SEM) in C57BL/6 mice demonstrate that asymptotic performance is readily achieved in the Barnes Maze with 4 days of training (6A), prior to treatment. By day 3, total latency (s), total distance (m) and total errors in the saline (n = 8) and dexmedetomidine (Dex, n = 8) groups was significantly different vs. their respective day 1 performance for each variable, denoting good learning performance. *, **, *** denote P < 0.05, P < 0.01, and P < 0.001 vs. Day 1 (Saline), respectively and ##, ### denote P < 0.01, and P < 0.001 vs. Day 1 (Dex) using repeated measures ANOVA with Newman-Keuls post hoc test. Probe Test I (6B) data (mean ± SEM) include latency (s), primary distance (m), total errors, mean speed (m/s) in C57BL/6 mice, and were obtained 6h following treatment with dexmedetomidine 300 μg/kg or saline i.p. In saline-treated mice, a clear preference for the target hole (Target vs. Hole -1 and Hole 1) can be observed demonstrating good short-term spatial memory retention. Dexmedetomidine-treated mice had decreased preference for the target hole, suggesting impaired short-term spatial memory retention. Probe Test II (6C) data (mean ± SEM) were obtained 1 week following dexmedetomidine or saline treatment and demonstrate no changes in latency, primary distance, total errors and mean speed. For saline-treated mice a preference for the target hole could still be observed (Target vs. Hole 1 but not Hole -1), while there was no significant target hole preference in dexmedetomidine-injected mice, suggesting that the treatment impaired long-term spatial reference memory retention. For the pokes data, analysis was done using ANOVA with Newman-Keuls post hoc test. *, **, and ***, denote P < 0.05, P < 0.01, and P < 0.001, vs. the target hole.
Probe Test I (Day 5), designed to measure short-term memory retention following the administration of dexmedetomidine 300 μg/kg (n = 8) or saline (n = 8) i.p., revealed that dexmedetomidine treatment had no impact on latency, primary distance travelled, and the total number of errors committed in the Barnes Maze when compared to saline (control) mice (Fig. 6B). It should be noted that although Probe Test I was performed 6h following the administration of dexmedetomidine or saline, the dexmedetomidine mice actually moved at a speed that was significantly higher than that of the saline mice, suggesting that at that time point there was no evidence of dexmedetomidine-induced motor retardation (Fig. 6B, mean speed). In both the dexmedetomidine and saline-treated groups, the number of visits (“nose pokes”) to adjacent and target holes were measured and compared to the target hole. These data reveal that on the day of Probe Test I, the dexmedetomidine-treated group had impaired target hole preference compared to saline-treated animals; hence indicating impaired short-term spatial reference memory (Fig. 6B).
Probe Test II (Day 12) was performed 1 week following dexmedetomidine or saline administration to determine the effect of anesthesia treatment on long-term memory. Again, there were no significant differences observed in latency, primary distance, and total errors committed between the dexmedetomidine and saline-treated groups (Fig. 6C). For saline-treated mice, a preference for the target hole could still be observed (Target vs. Hole 1 but not Hole -1), while there was no significant target hole preference in dexmedetomidine-injected mice, suggesting that the treatment impaired long-term spatial reference memory retention as well. Hippocampal tissue samples were obtained at the end of Probe Test II and revealed no significant increase in tau phosphorylation at this time point (1 week after dexmedetomidine or saline administration, data not shown).
In summary, these data reveal that acute administration of dexmedetomidine can impair both short-term and long-term spatial reference memory. At this juncture, it remains to be determined whether the transient increase in tau phosphorylation during treatment has any causative role in this impairment.
3.6 Dexmedetomidine Induces Tau Hyperphosphorylation In Vitro
Although our mouse studies eliminated hypothermia as a possible cause of dexmedetomidine-induced tau hyperphosphorylation, the possibility that changes in tau phosphorylation following dexmedetomidine were secondary to another systemic physiological change (e.g., changes in mean arterial pressure, cerebral blood flow, or the cerebral metabolic rate of oxygen) could not be completely excluded using this in vivo approach. Thus, we performed in vitro studies to determine whether dexmedetomidine-induced tau hyperphosphorylation is the result of a direct pharmacologic effect and not the secondary to an anesthetic-induced systemic physiological change.
To determine the effect of dexmedetomidine on tau phosphorylation in vitro, experiments were performed in SH-SY5Y human neuroblastoma cells stably transfected to constitutively express a human tau isoform with 3 microtubule-binding domains (Tau-SH-SY5Y cells). We previously have demonstrated that this transfected cell culture model is well suited for examining the tau phosphorylation effects of anesthetics (Whittington, et al., 2011), particularly as the basal levels of tau in native SH-SY5Y cells is very low. In these cell culture experiments, following a 4h incubation at 37°C with either dexmedetomidine 0.1 μM (n = 5) or 10 μM (n = 5) in growth medium or, as a control, in growth medium alone (n = 4), a significant increase in phosphorylated tau levels at AT8 was observed solely following treatment with dexmedetomidine 10 μM (Fig. 7A1). This increase in phosphorylated tau, following treatment with dexmedetomidine 10 μM, was not associated with an increase in total tau (Fig. 7A2).
Figure 7. Exposure to dexmedetomidine increases tau phosphorylation in Tau-SH-SY5Y cells in a dose-dependent manner.
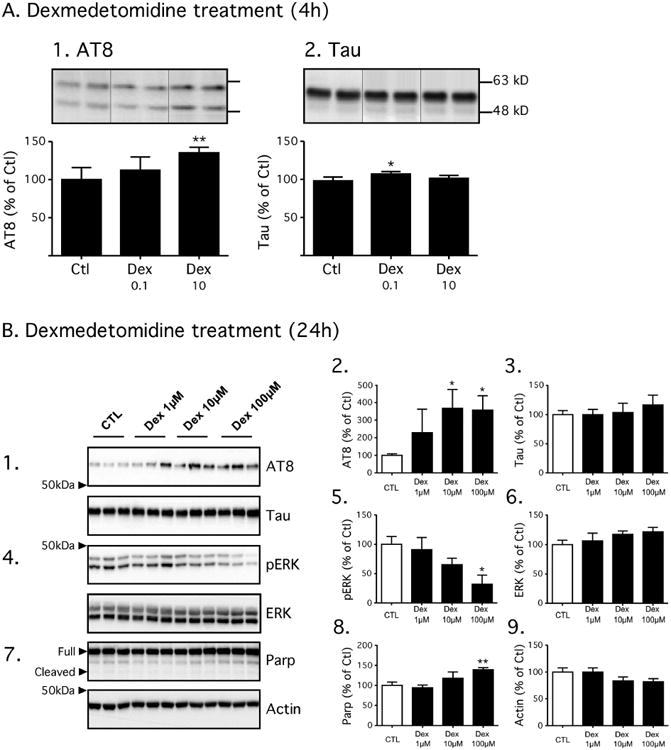
In Fig. 7A, Cells were harvested following a 4h exposure to dexmedetomidine 0.1 μM (Dex 0.1, n = 5), dexmedetomidine 10 μM (Dex 10, n = 5) growth medium or growth medium alone (Ctl, n = 4) at 37°C. The level of tau phosphorylation was determined using antibodies directed at AT8 (7A1) or total tau (7A2). In Fig. 7B, cells were harvested following a 24h exposure to dexmedetomidine (Dex) 1 μM, 10 μM, or 100 μM in growth medium or growth medium alone (Ctl) at 37°C, and n =3 for each condition. The level of tau phosphorylation was determined using antibodies directed at AT8 and demonstrated a dose-dependent increase when normalized to total tau (7B1-3). Following Dex treatment, a progressive decrease in phospho-ERK was observed with no change in total ERK (7B4-B6), and this mirrored what was observed in the mouse hippocampus. A slight increase in full-length Poly(ADP-ribose) polymerase (PARP) was seen with Dex 100 μM (7B7-B8) when normalized to β-actin (7B9); however, no increase in PARP cleavage was observed suggestion that apoptosis did not impact the observed phosphorylation changes. All data are expressed as mean ± SD. * and ** denote P < 0.05 and P<0.01 vs. Ctl; ANOVA with Newman-Keuls post hoc test.
Given this increase in tau phosphorylation following a brief exposure to dexmedetomidine 10 μM, additional experiments were performed in the Tau-SH-SY5Y cells in which the cells were incubated for 24h with either dexmedetomidine (1, 10, or 100 μM) in growth medium or growth medium alone (n = 3 for each condition) at 37°C to determine the impact of time of exposure on dexmedetomidine-induced tau phosphorylation. Significant increases in tau phosphorylation were observed at AT8 following incubation with Dex 10 μM and 100 μM doses with no significant change in total tau levels (Fig. 7B1-3). Consistent with what was seen in the hippocampal tissue, a significant decrease in p-ERK levels was observed with Dex 100 μM with no change in total ERK (Fig. 7B4-6). A slight increase in full length PARP levels (full) was also observed with Dex 100 μM (Fig 7B7-8); however, no changes in cleaved PARP were seen suggesting that apoptosis did not significantly affect the capacity of dexmedetomidine to increase tau phosphorylation in vitro. In summary, dexmedetomidine increases tau phosphorylation in Tau-SH-SY5Y cells in a dose dependent manner suggesting that this anesthetic can directly increase tau phosphorylation in the absence of a change in a major systemic physiological variable.
3.7 Dexmedetomidine Induces Tau Hyperphosphorylation and Accumulation of Insoluble Tau in hTau Mice
We next wanted to assess whether dexmedetomidine could affect tau phosphorylation and pathology in a mouse model of tauopathy. We chose to examine the effect of the drug in hTau mice, which overexpress human tau on a tau knockout background. These mice develop tau pathology as characterized by accumulation of insoluble tau and formation of neurofibrillary tangles (Andorfer, et al., 2005, Andorfer, et al., 2003). This model is more relevant to study AD-like tau pathogenesis, as it expresses non-mutated human tau (tau mutations result in fronto-temporal dementia, not AD). Thirty minutes after injection of 300 μg/kg of dexmedetomidine i.p. to 3-month-old male hTau mice, there was significant tau hyperphosphorylation at the AT8 and PHF-1 epitopes (Fig. 8 panels 1 & 3), and accumulation of insoluble tau (Fig. 8 panel 5), but no change in total or soluble tau (Fig. 8, Panels 4 and 6). These results demonstrate that dexmedetomidine can induce rapid acceleration of tau pathology in a mouse model of tauopathy.
Figure 8. Immunoblot analysis of tau solubility after dexmedetomidine treatment in hTau mice.
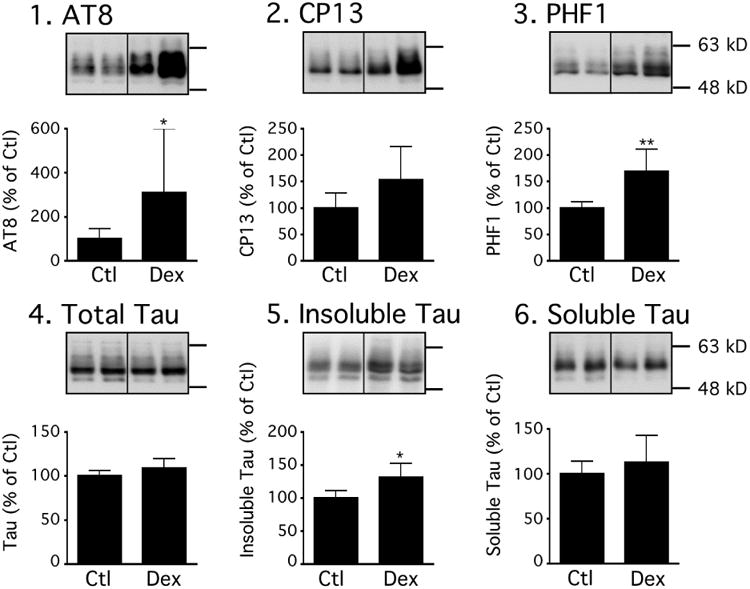
Proteins from cortices of control (Ctl; n=6), and dexmedetomidine-treated (Dex; n=6) hTau mice were extracted and analyzed by Western blot (Total fraction) with AT8 (Panel 1), CP13 (Panel 2), PHF-1 (Panel 3) and Total Tau (Panel 4) antibodies. Tau from sarkosyl-insoluble (Panel 5) and heat-stable soluble fractions (Panel 6) was evaluated by immunoblot analysis with a phospho-independent tau antibody. There was increased accumulation of insoluble tau. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of saline control (Ctl) and are displayed for each epitope. For each condition, 2 representative bands are displayed. All data are expressed as mean ± SD. * denote P < 0.05.
4. Discussion
The findings of the present study demonstrate that the sedative-analgesic agent, dexmedetomidine, possesses the capacity to directly induce tau phosphorylation in vivo as well as in vitro. Specifically, we observed that the acute administration of dexmedetomidine, under normothermic conditions, resulted in tau hyperphosphorylation in the mouse hippocampus and cortex. This hippocampal tau hyperphosphorylation persisted for 2h to 6h following dexmedetomidine administration and was dependent on activation of α2-ARs. Treatment with dexmedetomidine induced spatial memory impairment in nontransgenic mice and increased tau pathology in hTau mice. Furthermore, in vitro studies revealed that the hyperphosphorylation observed following dexmedetomidine exposure, is a direct, dose-dependent phenomenon.
While it has been well established that both inhalational and intravenous anesthetics can increase tau phosphorylation by inducing hypothermia (Planel, et al., 2007), this study further supports the notion that certain anesthetics can, nevertheless, induce tau hyperphosphorylation in the absence of hypothermia, as previously reported (Run, et al., 2009, Whittington, et al., 2011). The use of dexmedetomidine as a sedative agent in the intensive care unit setting has steadily increased in recent years (Wunsch, et al., 2010), particularly as the drug has been associated with a lower incidence of delirium when compared with the benzodiazepine midazolam (Riker, et al., 2009). The hyperphosphorylation of tau at several phosphoepitopes and the increase tau pathology in vivo, suggests that, despite the lower incidence of delirium (Riker, et al., 2009) and improved cognitive performance (Mirski, et al., 2010) associated with clinical use of the drug when compared to other sedative agents, dexmedetomidine still possesses the capacity to initiate tau hyperphosphorylation and aggregation, which are changes linked to the potential development of neurofibrillary pathology (Grundke-Iqbal, et al., 1986a, Grundke-Iqbal, et al., 1986b, Iqbal, et al., 2005).
Interestingly, the level of tau phosphorylation observed with dexmedetomidine, under normothermic conditions, was of the same order of magnitude of tau phosphorylation previously reported with propofol (Whittington, et al., 2011). However, unlike propofol where hippocampal tau hyperphosphorylation was solely present at AT8 at 2h, following dexmedetomidine administration hippocampal tau hyperphosphorylation was still present at AT8, PHF-1, and CP13 at this same time point. This seems to suggest that the phosphorylation effects of dexmedetomidine persist longer than that of propofol for a given similar sedation endpoint (i.e., loss of righting reflex lasting approximately 1h or less), which may be partly the result of pharmacokinetic differences between the two drugs. Specifically, in contrast to dexmedetomidine, following acute administration, propofol displays a relatively faster distribution and metabolism primarily in two exponential distribution phases (Kanto and Gepts, 1989). Indeed, the cognitive effects of acute dexmedetomidine administration have been demonstrated to persist longer clinically when compared to propofol (Bustillo, et al., 2002). Nevertheless, similar to what was observed with propofol (Whittington, et al., 2011), within 6h of dexmedetomidine administration, the hyperphosphorylation mostly resolved, suggesting that in nontransgenic mice, this response is also short-lived.
While we could not identify the kinases and/or phosphatases mediating tau hyperphosphorylation, our results revealed that alpha-2 adrenergic receptors (α2-ARs) play a key role in the process following dexmedetomidine administration. Interestingly, although our study is the first demonstration of the regulation of tau phosphorylation by α2-ARs, a recent article has shown that β2-ARs can mediate tau hyperphosphorylation by activating JNK and ERK (Wang, et al., 2013). At this juncture, we cannot exclude the mechanistic possibility that our observation is being mediated through a response downstream to α2-ARs activation, but we can rule out a mechanism similar to β2-ARs as both JNK and ERK where inhibited in parallel with tau hyperphosphorylation following dexmedetomidine. Nevertheless, our study lends further support to the hypothesis that drugs that modulate the adrenergic system can potentially impact tau pathology.
The results of the Barnes Maze testing indicate that the transient phosphorylation response following acute dexmedetomidine administration paralleled the impairment of short-term spatial reference memory. As tau hyperphosphorylation has been suggested to detach tau from MT, resulting in the destabilization of MT structure and the disruption of axonal transport ultimately leading to synaptic changes and memory impairment (Avila, et al., 2004, Trojanowski and Lee, 1994), we examined the effect of acute dexmedetomidine administration on synaptic markers and the ability of tau to bind to preformed, taxol-stabilized MT according to our published protocol (Planel, et al., 2009, Planel, et al., 2008). There was no change in tau binding to MT (Figure S1) and on pre- (Septin 3, SNAP25, synaptophysin) or post- (drebrin, PSD95) synaptic markers (Figure S2), indicating they were not the source of the memory impairment. As recent studies (Le Freche, et al., 2012) have shown that repeated exposure to inhalational anesthesia may result in a more persistent state of tau phosphorylation correlating with memory impairment, determining whether tau phosphorylation is just a co-occurrence or has a role in the drug-induced memory impairment will be important for future studies. In this context, investigating the effects of prolonged dexmedetomidine exposure will be particularly relevant as a recent study demonstrated that 31.5% of patients in an ICU setting are receiving dexmedetomidine for more than 1 day (Wunsch, et al., 2010).
The exposure of Tau-SH-SY5Y cells to dexmedetomidine in vitro was also associated with increased tau phosphorylation. Although a modest increase in phosphorylated tau was observed following a 4h exposure to 10 μM dexmedetomidine, the most significant increases in phosphorylation were observed following exposure to 10 and 100 μM dexmedetomidine for 24h, suggesting that dexmedetomidine-induced tau phosphorylation is a dose- and, perhaps even, an exposure time-dependent phenomenon. It is important to note that we examined tau phosphorylation in these cells using concentrations of dexmedetomidine that have been shown to prevent apoptosis in cortical neurons (Sanders, et al., 2010) and to possess a neuroprotective effect in an organotypic hippocampal slice culture model of traumatic brain injury (Schoeler, et al., 2012). However, we solely observed tau hyperphosphorylation at concentrations of dexmedetomidine 10 and 100 μM, concentrations that would be considered to be significantly higher than the maximal therapeutic plasma concentration in previously reported in humans, 0.05 μM (Bloor, et al., 1992, Ebert, et al., 2000, Narimatsu, et al., 2007, Talke, et al., 1999). Nevertheless, these in vitro studies still demonstrate that the tau phosphorylation observed following dexmedetomidine administration, at concentrations previously demonstrated to provide cortical neuroprotection in vitro (Sanders, et al., 2010), is a direct effect of the drug on the cells and occurs independently of changes in systemic physiological variables such as temperature, arterial blood pressure, and cerebral blood flow. Moreover, these findings suggest that a certain critical concentration needs to be achieved before changes in tau phosphorylation become manifest.
Although tau hyperphosphorylation and aggregation were observed with dexmedetomidine, it would be extremely premature and unwarranted to extrapolate these findings to the clinical sphere at this juncture. First of all, in terms of negative cognitive effects such as delirium, dexmedetomidine use has been associated with a lower incidence of delirium (Riker, et al., 2009). Moreover, dexmedetomidine was not compared directly with equipotent sedative doses of other commonly used sedatives such as midazolam, propofol, or opioid agents; therefore, it currently is unknown whether dexmedetomidine has less or more of an impact on tau phosphorylation and pathology than any of the aforementioned agents. Indeed, these current studies demonstrate that, similar to what we observed with propofol (Whittington, et al., 2011), the effects of acute dexmedetomidine administration on tau phosphorylation are primarily transient.
5. Conclusions
In conclusion, the present study clearly demonstrates that dexmedetomidine increases tau phosphorylation and aggregation in vivo under normothermic conditions with the potential to impact spatial reference memory, and that, at least in vitro, the hyperphosphorylation of tau is a dose-dependent phenomenon. While hyperphosphorylation is a critical step in the development of neurofibrillary pathology (Alonso, et al., 2001), further studies are warranted in transgenic mouse models of AD to determine whether the degree of hyperphosphorylation observed following prolonged dexmedetomidine administration is associated increased tau pathology and neurocognitive impairment. This is particularly important, given that, in the absence of more effective treatments for these neurodegenerative conditions, as life expectancy continues to increase the number of patients with an underlying tauopathy (Alzheimer's disease, Parkinson's disease, traumatic brain injury etc.) will continue to rise, thus increasing the likelihood that these patients will encounter the need for surgery and anesthesia.
Supplementary Material
Figure S1. No alteration in tau binding to microtubules is observed after dexmedetomidine administration in hTau mice. To determine whether tau hyperphosphorylation induced by dexmedetomidine could impair tau binding to microtubules, a microtubule (MT) binding assay was performed using a previously reported procedure (Planel, et al., 2008). There was no effect of dexmedetomidine treatment on the amount of free or bound tau (Panel A, n=6), or tubulin (Panel B, n=6), indicating no impairment of tau binding to MT. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of Ctl.
Figure S2. No alteration of pre- or post-synaptic markers after dexmedetomidine administration in hTau mice. To assess whether dexmedetomidine treatment could affect synaptic proteins, proteins from cortices of control (Ctl; n=6), and dexmedetomidine-treated (Dex; n=6) hTau mice were extracted and analyzed by Western blot. There were no changes in post-synaptic markers (A. drebrin, B. PSD95) or pre-synaptic markers (C. Septin 3, D. SNAP25, E. synaptophysin). Relative immunoreactive band intensities are expressed as a percent of Ctl.
Highlights.
Dexmedetomidine increases tau phosphorylation in the mouse hippocampus and cortex.
Dexmedetomidine also increases tau phosphorylation in SH-SY5Y cells in a dose dependent manner.
The effects of dexmedetomidine on tau are mediated by the activation of the α2-AR.
This tau hyperphosphorylation is associated with spatial memory impairment.
Acknowledgments
The authors thank Dr. Peter Davies (Hofstra North Shore-LIJ School of Medicine, Manhasset, NY, USA) for the gift of the PHF-1 and CP13 antibodies, Dr. Peter Seubert (Neotope Biosciences, San Francisco, CA) for the gift of the 12E8 antibody, and Dr. Luc Buée (Research Director, Unité 422, INSERM, Lille, France) for the gift of the Tau-SH-SY5Y cells.
Supported by grant R01GM101698 (RAW) from the National Institutes of Health, and grants to E.P. from the Canadian Institute of Health Research (MOP-106423, PCN-102993), Fonds de Recherche en Santé du Québec (16205, 20048), the Natural Sciences and Engineering Research Council of Canada (354722), and the Alzheimer Society of Canada. A.N., F.R.P., M.G. and N.E.K. are recipients of an Alzheimer Society of Canada Biomedical Award. G.T. is recipient of a Didier Mouginot Doctoral Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Robert A. Whittington, Email: raw9@cumc.columbia.edu.
László Virág, Email: lv10@columbia.edu.
Maud Gratuze, Email: maud.gratuze.1@ulaval.ca.
Franck R. Petry, Email: franck.petry.1@ulaval.ca.
Anastasia Noël, Email: anastasia.noel@hotmail.fr.
Isabelle Poitras, Email: isabelle.poitras.2@ulaval.ca.
Geoffrey Truchetti, Email: geoffrey.truchetti.1@ulaval.ca.
François Marcouiller, Email: Francois.Marcouiller@crchul.ulaval.ca.
Marie-Amélie Papon, Email: amelie_pap@hotmail.com.
Noura El Khoury, Email: noura.elkhoury@utoronto.ca.
Kevin Wong, Email: kevin.wong712@gmail.com.
Alexis Bretteville, Email: alexisbretteville.pro@gmail.com.
Françoise Morin, Email: Francoise.Morin@crchul.ulaval.ca.
Emmanuel Planel, Email: Emmanuel.Planel@fmed.ulaval.ca.
References
- Alonso AC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments / straight filaments. Proc Natl Acad Sci U S A. 2001;98(12):6923–8. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25(22):5446–54. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86(3):582–90. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42(3 Pt 1):631–9. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Augustinack J, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103(1):26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84(2):361–84. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- Baranov D, Bickler PE, Crosby GJ, Culley DJ, Eckenhoff MF, Eckenhoff RG, Hogan KJ, Jevtovic-Todorovic V, Palotas A, Perouansky M, Planel E, Silverstein JH, Wei H, Whittington RA, Xie Z, Zuo Z. Consensus statement: First International Workshop on Anesthetics and Alzheimer's disease. Anesth Analg. 2009;108(5):1627–30. doi: 10.1213/ane.0b013e318199dc72. doi:108/5/1627[pii]10.1213/ane.0b013e318199dc72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. Journal of comparative and physiological psychology. 1979;93(1):74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Bianchi SL, Tran T, Liu C, Lin S, Li Y, Keller JM, Eckenhoff RG, Eckenhoff MF. Brain and behavior changes in 12-month-old Tg2576 and nontransgenic mice exposed to anesthetics. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloor BC, Ward DS, Belleville JP, Maze M. Effects of intravenous dexmedetomidine in humans. II. Hemodynamic changes. Anesthesiology. 1992;77(6):1134–42. doi: 10.1097/00000542-199212000-00014. [DOI] [PubMed] [Google Scholar]
- Bretteville A, Planel E. Tau aggregates: toxic, inert, or protective species? J Alzheimers Dis. 2008;14(4):431–6. doi: 10.3233/jad-2008-14411. [DOI] [PubMed] [Google Scholar]
- Brunden KR, Yao Y, Potuzak JS, Ferrer NI, Ballatore C, James MJ, Hogan AM, Trojanowski JQ, Smith AB, 3rd, Lee VM. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer's disease and related tauopathies. Pharmacol Res. 2011;63(4):341–51. doi: 10.1016/j.phrs.2010.12.002. doi:S1043-6618(10)00227-6[pii]10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere T, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Buee L, Delacourte A. Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol (Berl) 1999;97(3):221–30. doi: 10.1007/s004010050978. [DOI] [PubMed] [Google Scholar]
- Bustillo MA, Lazar RM, Finck AD, Fitzsimmons B, Berman MF, Pile-Spellman J, Heyer EJ. Dexmedetomidine may impair cognitive testing during endovascular embolization of cerebral arteriovenous malformations: a retrospective case report series. J Neurosurg Anesthesiol. 2002;14(3):209–12. doi: 10.1097/01.ANA.0000017492.93942.DE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Prior M, Dargusch R, Roberts A, Riek R, Eichmann C, Chiruta C, Akaishi T, Abe K, Maher P, Schubert D. A novel neurotrophic drug for cognitive enhancement and Alzheimer's disease. PLoS ONE. 2011;6(12):e27865. doi: 10.1371/journal.pone.0027865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Sales C, Rabin BC, Maze M. A hypnotic response to dexmedetomidine, an alpha 2 agonist, is mediated in the locus coeruleus in rats. Anesthesiology. 1992;76(6):948–52. doi: 10.1097/00000542-199206000-00013. [DOI] [PubMed] [Google Scholar]
- Dahmani S, Paris A, Jannier V, Hein L, Rouelle D, Scholz J, Gressens P, Mantz J. Dexmedetomidine increases hippocampal phosphorylated extracellular signal-regulated protein kinase 1 and 2 content by an alpha 2-adrenoceptor-independent mechanism: evidence for the involvement of imidazoline I1 receptors. Anesthesiology. 2008;108(3):457–66. doi: 10.1097/ALN.0b013e318164ca81. [DOI] [PubMed] [Google Scholar]
- Delobel P, Flament S, Hamdane M, Mailliot C, Sambo AV, Begard S, Sergeant N, Delacourte A, Vilain JP, Buee L. Abnormal Tau phosphorylation of the Alzheimer-type also occurs during mitosis. J Neurochem. 2002;83(2):412–20. doi: 10.1046/j.1471-4159.2002.01143.x. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology. 2000;93(2):382–94. doi: 10.1097/00000542-200008000-00016. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301(Pt 3):871–7. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci U S A. 1993;90(11):5066–70. doi: 10.1073/pnas.90.11.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189(3):167–9. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87(15):5827–31. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986a;261(13):6084–9. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986b;83(13):4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60(5):729–36. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- Hamdane M, Sambo AV, Delobel P, Begard S, Violleau A, Delacourte A, Bertrand P, Benavides J, Buee L. Mitotic-like tau phosphorylation by p25-Cdk5 kinase complex. J Biol Chem. 2003;278(36):34026–34. doi: 10.1074/jbc.M302872200. [DOI] [PubMed] [Google Scholar]
- Hocker J, Paris A, Scholz J, Tonner PH, Nielsen M, Bein B. Differential effects of alpha 2-adrenoceptors in the modulation of the thermoregulatory response in mice induced by meperidine. Anesthesiology. 2008;109(1):95–100. doi: 10.1097/ALN.0b013e31817c02fc. [DOI] [PubMed] [Google Scholar]
- Hunter JC, Fontana DJ, Hedley LR, Jasper JR, Lewis R, Link RE, Secchi R, Sutton J, Eglen RM. Assessment of the role of alpha2-adrenoceptor subtypes in the antinociceptive, sedative and hypothermic action of dexmedetomidine in transgenic mice. British journal of pharmacology. 1997;122(7):1339–44. doi: 10.1038/sj.bjp.0701520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739(2-3):198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Julien C, Bretteville A, Planel E. Biochemical isolation of insoluble tau in transgenic mouse models of tauopathies. Methods Mol Biol. 2012;849:473–91. doi: 10.1007/978-1-61779-551-0_32. [DOI] [PubMed] [Google Scholar]
- Kanto J, Gepts E. Pharmacokinetic implications for the clinical use of propofol. Clin Pharmacokinet. 1989;17(5):308–26. doi: 10.2165/00003088-198917050-00002. [DOI] [PubMed] [Google Scholar]
- Le Freche H, Brouillette J, Fernandez-Gomez FJ, Patin P, Caillierez R, Zommer N, Sergeant N, Buee-Scherrer V, Lebuffe G, Blum D, Buee L. Tau Phosphorylation and Sevoflurane Anesthesia: An Association to Postoperative Cognitive Impairment. Anesthesiology. 2012 doi: 10.1097/ALN.0b013e31824be8c7. [DOI] [PubMed] [Google Scholar]
- Mailliot C, Podevin-Dimster V, Rosenthal RE, Sergeant N, Delacourte A, Fiskum G, Buee L. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2000;20(3):543–9. doi: 10.1097/00004647-200003000-00013. [DOI] [PubMed] [Google Scholar]
- Maurage CA, Sergeant N, Ruchoux MM, Hauw JJ, Delacourte A. Phosphorylated serine 199 of microtubule-associated protein tau is a neuronal epitope abundantly expressed in youth and an early marker of tau pathology. Acta Neuropathol (Berl) 2003;105(2):89–97. doi: 10.1007/s00401-002-0608-7. [DOI] [PubMed] [Google Scholar]
- Mirski MA, Lewin JJ, 3rd, Ledroux S, Thompson C, Murakami P, Zink EK, Griswold M. Cognitive improvement during continuous sedation in critically ill, awake and responsive patients: the Acute Neurological ICU Sedation Trial (ANIST) Intensive care medicine. 2010;36(9):1505–13. doi: 10.1007/s00134-010-1874-9. [DOI] [PubMed] [Google Scholar]
- Narimatsu E, Niiya T, Kawamata M, Namiki A. Lack in effects of therapeutic concentrations of dexmedetomidine and clonidine on the neuromuscular blocking action of rocuronium in isolated rat diaphragms. Anesth Analg. 2007;104(5):1116–20. doi: 10.1213/01.ane.0000260317.02748.83. tables of contents. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Lu J, Guo T, Saper CB, Franks NP, Maze M. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98(2):428–36. doi: 10.1097/00000542-200302000-00024. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39(6):669–73. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Papon MA, Whittington RA, El-Khoury NB, Planel E. Alzheimer's disease and anesthesia. Front Neurosci. 2011;4:272. doi: 10.3389/fnins.2010.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil SS, Sunyer B, Hoger H, Lubec G. Evaluation of spatial memory of C57BL/6J and CD1 mice in the Barnes maze, the Multiple T-maze and in the Morris water maze. Behav Brain Res. 2009;198(1):58–68. doi: 10.1016/j.bbr.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Haapalinna A, Sirvio J, Virtanen R. Pharmacological properties, central nervous system effects, and potential therapeutic applications of atipamezole, a selective alpha2-adrenoceptor antagonist. CNS Drug Rev. 2005;11(3):273–88. doi: 10.1111/j.1527-3458.2005.tb00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Bretteville A, Liu L, Virag L, Du AL, Yu WH, Dickson DW, Whittington RA, Duff KE. Acceleration and persistence of neurofibrillary pathology in a mouse model of tauopathy following anesthesia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009 doi: 10.1096/fj.08-122424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Krishnamurthy P, Miyasaka T, Liu L, Herman M, Kumar A, Bretteville A, Figueroa HY, Yu WH, Whittington RA, Davies P, Takashima A, Nixon RA, Duff KE. Anesthesia-induced hyperphosphorylation detaches 3-repeat tau from microtubules without affecting their stability in vivo. J Neurosci. 2008;28(48):12798–807. doi: 10.1523/JNEUROSCI.4101-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Richter KEG, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27(12):3090–7. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Sun X, Takashima A. Role of GSK-3 beta in Alzheimer's disease pathology. Drug Development Research. 2002;56(3):491–510. [Google Scholar]
- Riker RR, Shehabi Y, Bokesch PM, Ceraso D, Wisemandle W, Koura F, Whitten P, Margolis BD, Byrne DW, Ely EW, Rocha MG. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA: the journal of the American Medical Association. 2009;301(5):489–99. doi: 10.1001/jama.2009.56. [DOI] [PubMed] [Google Scholar]
- Rissman RA. Stress-induced tau phosphorylation: functional neuroplasticity or neuronal vulnerability? Journal of Alzheimer's disease: JAD. 2009;18(2):453–7. doi: 10.3233/JAD-2009-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong CX. Anesthesia induces phosphorylation of tau. J Alzheimers Dis. 2009;16(3):619–26. doi: 10.3233/JAD-2009-1003. doi:549M633456804L08[pii]10.3233/JAD-2009-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RD, Sun P, Patel S, Li M, Maze M, Ma D. Dexmedetomidine provides cortical neuroprotection: impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta anaesthesiologica Scandinavica. 2010;54(6):710–6. doi: 10.1111/j.1399-6576.2009.02177.x. [DOI] [PubMed] [Google Scholar]
- Schoeler M, Loetscher PD, Rossaint R, Fahlenkamp AV, Eberhardt G, Rex S, Weis J, Coburn M. Dexmedetomidine is neuroprotective in an in vitro model for traumatic brain injury. BMC neurology. 2012;12:20. doi: 10.1186/1471-2377-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, et al. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270(32):18917–22. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Analytical biochemistry. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Sunyer B, Patil S, Höger H, Lubec G. Barnes maze, a useful task to assess spatial reference memory in the mice. Nature Protocol Exchange. 2007 doi: 10.1038/nprot.2007.390. [DOI] [Google Scholar]
- Szendrei GI, Lee VM, Otvos L., Jr Recognition of the minimal epitope of monoclonal antibody Tau-1 depends upon the presence of a phosphate group but not its location. J Neurosci Res. 1993;34(2):243–9. doi: 10.1002/jnr.490340212. [DOI] [PubMed] [Google Scholar]
- Talke PO, Caldwell JE, Richardson CA, Kirkegaard-Nielsen H, Stafford M. The effects of dexmedetomidine on neuromuscular blockade in human volunteers. Anesth Analg. 1999;88(3):633–9. doi: 10.1097/00000539-199903000-00031. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Planel E, Chui DH, Sato S, Miyasaka T, Sahara N, Murayama M, Kikuchi N, Yoshioka K, Rivka R, Takashima A. c-jun N-terminal kinase hyperphosphorylates R406W tau at the PHF-1 site during mitosis. Faseb J. 2006;20(6):762–4. doi: 10.1096/fj.05-4362fje. [DOI] [PubMed] [Google Scholar]
- Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer's disease. Neurosignals. 2002;11(5):262–9. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Paired helical filament tau in Alzheimer's disease. The kinase connection. Am J Pathol. 1994;144(3):449–53. [PMC free article] [PubMed] [Google Scholar]
- Vulliemoz Y, Virag L, Whittington RA. Interaction of the alpha-2 adrenergic- and opioid receptor with the cGMP system in the mouse cerebellum. Brain Res. 1998;813(1):26–31. doi: 10.1016/s0006-8993(98)00967-6. [DOI] [PubMed] [Google Scholar]
- Wang D, Fu Q, Zhou Y, Xu B, Shi Q, Igwe B, Matt L, Hell JW, Wisely EV, Oddo S, Xiang YK. beta2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem. 2013;288(15):10298–307. doi: 10.1074/jbc.M112.415141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21(5):719–27. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Whittington RA, Bretteville A, Dickler MF, Planel E. Anesthesia and tau pathology. Progress in neuro-psychopharmacology & biological psychiatry. 2013;47:147–55. doi: 10.1016/j.pnpbp.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington RA, Virag L, Marcouiller F, Papon MA, El Khoury NB, Julien C, Morin F, Emala CW, Planel E. Propofol directly increases tau phosphorylation. PLoS ONE. 2011;6(1):e16648. doi: 10.1371/journal.pone.0016648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56(2-3):343–56. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- Wunsch H, Kahn JM, Kramer AA, Wagener G, Li G, Sladen RN, Rubenfeld GD. Dexmedetomidine in the care of critically ill patients from 2001 to 2007: an observational cohort study. Anesthesiology. 2010;113(2):386–94. doi: 10.1097/ALN.0b013e3181e74116. [DOI] [PubMed] [Google Scholar]
- Zhou XW, Gustafsson JA, Tanila H, Bjorkdahl C, Liu R, Winblad B, Pei JJ. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol Dis. 2008;31(3):386–94. doi: 10.1016/j.nbd.2008.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. No alteration in tau binding to microtubules is observed after dexmedetomidine administration in hTau mice. To determine whether tau hyperphosphorylation induced by dexmedetomidine could impair tau binding to microtubules, a microtubule (MT) binding assay was performed using a previously reported procedure (Planel, et al., 2008). There was no effect of dexmedetomidine treatment on the amount of free or bound tau (Panel A, n=6), or tubulin (Panel B, n=6), indicating no impairment of tau binding to MT. Dividing lines represent areas where lanes from the same blot were removed and the remaining lanes were spliced together. Relative immunoreactive band intensities are expressed as a percent of Ctl.
Figure S2. No alteration of pre- or post-synaptic markers after dexmedetomidine administration in hTau mice. To assess whether dexmedetomidine treatment could affect synaptic proteins, proteins from cortices of control (Ctl; n=6), and dexmedetomidine-treated (Dex; n=6) hTau mice were extracted and analyzed by Western blot. There were no changes in post-synaptic markers (A. drebrin, B. PSD95) or pre-synaptic markers (C. Septin 3, D. SNAP25, E. synaptophysin). Relative immunoreactive band intensities are expressed as a percent of Ctl.