

Abstract
Hotspot mutations in IDH1 and IDH2 cause a differentiation block that can promote tumorigenesis. Two recent papers reported that small molecules targeting mutant IDH1 or mutant IDH2 release this differentiation block and/or impede tumor growth, providing a proof-of-concept that mutant IDHs are therapeutically targetable and that their effects are reversible.
Main Text
The isocitrate dehydrogenases IDH1 and IDH2 are mutated in a variety of cancers, including progressive gliomas and acute myelogenous leukemia (AML). These mutations cause IDH1 or IDH2 to produce R-2-hydroxyglutarate (2HG), an oncometabolite responsible for global changes to DNA methylation, inhibition of histone lysine demethylases, blocks to cellular differentiation, and ultimately, tumorigenesis (Figueroa et al., 2010; Lu et al., 2012; Sasaki et al., 2012; Turcan et al., 2012). The presence of IDH mutations in a wide variety of cancers together with the hotspot nature of these mutations makes them attractive therapeutic target candidates. Recently, two studies published in Science characterized the use of small molecules that specifically target mutant IDH1 or mutant IDH2 (Rohle et al., 2013; Wang et al., 2013).The inhibitors restored 2HG levels to normal physiological levels and reversed several of the biological and epigenetic phenotypes exerted by IDH mutations. These proof-of-concept studies showing IDH mutations are targetable by small molecules highlight a promising therapeutic avenue that necessitates further investigation.
The understanding of the complexities underlying IDH mutations have evolved since their discovery in progressive gliomas (Yan et al., 2009). Pioneering studies in AML and progressive gliomas show that IDH mutations induce a promoter-associated CpG-island methylator phenotype with direct effects on gene expression (Figueroa et al., 2010; Noushmehr et al., 2010). The gene expression profile of mutant cells confers a block to the normal differentiation program, leading to an expansion of progenitor cells that is thought to be a critical step in cancer pathogenesis (Duncan et al., 2012; Turcan et al., 2012). These observations raised the possibility that inhibiting IDH mutants might reverse their tumorigenic effects (Jin et al., 2012) and that design of effective inhibitors would need to take into account the complex downstream effects of IDH mutations.
To assess this therapeutic possibility in the glioma context, Rohle et al. used AGI-5198, a small molecule inhibitor of the most common IDH mutation in gliomas, IDH1-R132H. Treatment of an oligodendroglioma cell line harboring an endogenous IDH1-R132H mutation with this inhibitor reduced growth in soft agar by 40-60% and impeded growth of xenograft tumors derived from that cell line in mice. Analysis of these tumors showed a reduction in proliferative markers but no change in apoptosis, suggesting that the altered tumor growth was due to failure to proliferate as opposed to cell death. Following treatment, several genes involved in glial differentiation were upregulated and found to have lost repressive histone marks H3K9me3 and H3K27me3 at their promoters, implying that the mutant IDH1 inhibitor is capable of erasing histone modifications that influence gene expression. This study therefore demonstrated that, in this model, targeting mutant IDH1 can impair glioma growth in vivo and this growth inhibition is linked to changes in differentiation.
Concurrently, Wang et al. designed AGI-6780, a small molecule that inhibits the most commonly occurring IDH mutation in AML, IDH2-R140Q, by holding the protein in an open conformation (Figure 1). They then used this inhibitor to explore the effects of inhibiting mutant IDH in cells of the hematopoietic system. Treatment with this inhibitor lowered 2HG to normal physiological levels in an erythroleukemia cell line ectopically expressing IDH2-R140Q. The inhibitor also released these cells from the block to differentiation following induction with erythropoietin that was exerted by IDH2-R140Q expression. Complementary studies treating IDH2-mutated primary human AML cells showed a similar reduction in 2HG levels. However, in the mutant primary cells, a burst of proliferation resulted followed by an increase in mature cell types at the expense of progenitor cells. These results imply that mutant IDH2 inhibition can be used to promote differentiation of mutated AML cells. This was similar to the Rohle et al. finding where genes involved in differentiation were expressed following treatment with the inhibitor, suggesting that mutant IDH imparts a block to differentiation that is released upon treatment with inhibitor. The mutant IDH2 inhibitor's ability to reduce 2HG to baseline levels coupled with its differentiating effects on the AML cells opens up avenues to treat AML and to determine the efficacy of this small molecule inhibitor alone or in combination with other therapeutics. The well-characterized nature of the hematopoietic system will permit many eloquent and exciting studies analyzing AGI-6780's effectiveness in an in vivo animal context.
Figure 1. Small molecule inhibitor AGI-6780 binds to mutant IDH2 and releases a block to differentiation.
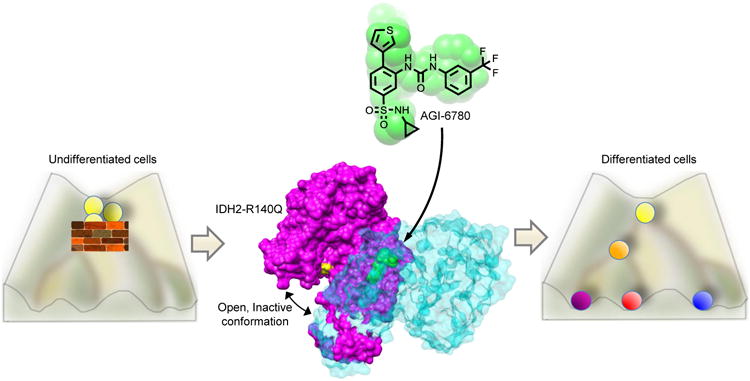
IDH mutations induce a block to normal differentiation programs leading to accumulation of progenitor cells, as shown in the Waddington differentiation diagram on the left. AGI-6780, shown in green, binds allosterically at the mutant IDH2 homodimer interface and holds the enzyme in the catalytically inactive open conformation (middle). Each member of the IDH2 homodimer pair is shown in purple and teal, respectively, with the R140Q mutation in yellow. Inhibition of mutant IDH2 releases the block and induces differentiation (right).
Several hurdles must be overcome before the strategy of targeting IDH mutations can be translated to the clinic. These inhibitors were tested on a select few cell lines, and in the case of the mutant IDH1 inhibitor, only on one glioma cell line. Whether that inhibitor will work similarly in an astrocytic glioma line as it did in an oligodendroglial line, or in a glioma line of a lower or higher tumor grade, will need to be established. Examination of long-term mutant IDH inhibition will be necessary to determine whether growth inhibition can be maintained over time or can be used together with conventional therapeutics. Additionally, all glioma xenografts in the mutant IDH1 inhibitor studies were heterotopic. Though it is promising that the inhibitor was capable of reducing intratumoral 2HG levels in these subcutaneous tumors, its ability to cross the blood-brain-barrier and inhibit glioma growth in the brain is imperative for its success. Though genetically engineered mouse models that express mutant IDH1 in the brain would be an ideal system to assess the efficacy of the IDH1 inhibitor, such models have been unsuccessful in generating tumors. However, orthotopic xenograft models would be a viable option to address this issue. In addition, although the mutant IDH1 inhibitor inhibited glioma cell growth, its inability to induce apoptosis is concerning from a therapeutic standpoint. Determining the long-term fate of those cells exhibiting growth inhibition will be critically important. In the case of the mutant IDH2 inhibitor, these in vitro findings must be verified in animal models of IDH2-mutated AML.
Nevertheless, the proof-of-concept experiments presented by Rohle et al. and Wang et al. contribute greatly to the field by showing the potential for targeting IDH mutations through small molecule inhibitors. Especially encouraging is the fact that cells without IDH mutations were tolerant to treatment with the inhibitors. Intriguingly, promotion of cellular differentiation was a major component of the therapeutic response in all of the above experiments: the in vitro and in vivo ablation of excessive 2HG allows the IDH mutant-mediated differentiation block to be lifted, permitting expression of normal differentiation programs. Given the prevalence of IDH mutations in a variety of cancers, the demonstration that efficient and selective targeting of these mutations can be achieved, and can act as a therapeutic strategy, holds great promise for a large number of patients. Though further analysis is required to generalize the efficacy of these inhibitors in reversing the differentiation block and oncogenic effects imparted by 2HG, the contribution of these tools to understanding the biology of IDH mutations is invaluable.
References
- Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012;22:2339–55. doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 Mutations Result in†a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Pirozzi CJ, Chen LH, Lopez GY, Duncan CG, Feng J, Spasojevic I, Bigner DD, He Y, Yan H. Mutant IDH1 is required for IDH1 mutated tumor cell growth. Oncotarget. 2012;3:774–782. doi: 10.18632/oncotarget.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science. 2013 doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, Delabarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science. 2013 doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]