

Abstract
The last decade of decreasing DNA sequencing costs and proliferating sequencing services in core labs and companies has brought the de-novo genome sequencing and assembly of insect species within reach for many entomologists. However, sequence production alone is not enough to generate a high quality reference genome, and in many cases, poor planning can lead to extremely fragmented genome assemblies preventing high quality gene annotation and other desired analyses. Insect genomes can be problematic to assemble, due to combinations of high polymorphism, inability to breed for genome homozygocity, and small physical sizes limiting the quantity of DNA able to be isolated from a single individual. Recent advances in sequencing technology and assembly strategies are enabling a revolution for insect genome reference sequencing and assembly. Here we review historical and new genome sequencing and assembly strategies, with a particular focus on their application to arthropod genomes. We highlight both the need to design sequencing strategies for the requirements of the assembly software, and new long-read technologies that are enabling a return to traditional assembly approaches. Finally, we compare and contrast very cost effective short read draft genome strategies with the long read approaches that although entailing additional cost, bring a higher likelihood of success and the possibility of archival assembly qualities approaching that of finished genomes.
Sanger Beginnings: The First Insect Genome
The sequencing of the first arthropod genome – Drosophila melanogaster [1] – was planned to generate the ideal dataset for whole genome assembly [2] and the principals employed then are still valid today. An isogenic strain avoided DNA polymorphism assembly issues; milligrams of high quality DNA were isolated from embryonic nuclei avoiding gut and mitochondrial contamination; polytene, genetic and BAC based maps provided long range information for assembly validation; high genome coverage sequence information of different scales (2kb and 10kb inserts, and BAC end sequences) was generated enabling assembly of contigs, and determination of their order and orientation to produce scaffolds. The Celera Assembler [2] was designed with exactly this dataset in mind, and has been improved over the years continuing to be a high quality assembly tool today.
Variations on this approach have been applied to many other species, but in many cases the required inputs - especially isogenic DNA and high long read sequence coverage - could not be provided. Drosophila simulans [3] provided an early warning of polymorphism problems. The initial sequencing plan attempted to collect polymorphism data in addition to a draft reference, generating 1X sequence coverage from multiple strains of D. simulans – a seductive goal. Unfortunately the dataset could not be assembled to high quality, and additional sequence had to be generated from a single inbred strain to rescue the assembly. This same dynamic has played out through successive sequence technologies and insect species, providing a cautionary tale for the designers of de novo reference genome-sequencing projects.
Shorter and shorter (but cheaper and cheaper) sequence reads
New technologies have given us cheaper, but shorter reads, enabling genome sequencing of many more species. De novo sequencing costs for genome assembly fell by a factor of 10 with the introduction of 454 sequencing [4] and another factor of 10 with Illumina short read assembly enabling sequence coverage decisions to be based on assembly strategy rather than cost. The downside has been the increased assembly difficulty leading to lower contig N50 lengths (more assembly false negatives) as short reads cannot straddle as many repeats or polymorphic regions. 454 assembly tools include Newbler ([5] but see [6]) and the CABOG variant of the Celera assembler [7] and have proven adept at assembling reasonable coverage (20X fragment, 30X clone coverage in 3kb and 8kb insert paired end) of inbred insects, but do not address sequence polymorphism. Results can be impressive for inbred Drosophila ([8] Table S1 shows N50’s of 100–400kb except for D. rhopaloa which could not be inbred resulting in a contig N50 of 19kb). More typical results using outbred species include the centipede ([9] 24.7kb contig n50) and the somewhat inbred Heliconious butterfly ([10] 51kb contig N50) that required manual partitioning of haplotypes and re-assembly to improve genome contiguity.
Illumina 100bp reads required higher coverage (as a high proportion of read information is used for overlap determination instead of contig extension) and new de Bruijn kmer graph based assembly tools to efficiently deal with the large numbers of sequence reads [11–14]. Note that storing kmer graph structures in memory for assembly requires large amounts of RAM – often 1Terabyte RAM, 32 core servers are used. Table 1. shows a typical ALLPATHS-LG [12] sequencing strategy of ~150X genome coverage, 100bp paired end (pe) Illumina reads. Short read assembly of arthropod genomes works well (20–50kb contig N50s, 1Mb Scaffold N50) for inbred material (or haploid male hymenoptera), generally requiring ~20ug DNA (1ug of DNA for the 180bp and 500bp libraries, 5ug for the 3kb insert library, and 10ug for the 8kb). However, this and other assemblers are not tuned for polymorphic material, and even mediocre quality assembly for publication (say > 10kb contig N50 enabling annotation of gene models without excessive fragmentation) is not guaranteed. Additionally, despite routine success assembling 3Gb mammalian genomes, it is extremely difficult to assemble polymorphic genomes larger than 2Gb.
Table 1. De novo genome assembly strategies.
Assembly software is designed for a specific sequencing and assembly strategy. Thus sequence must be generated with the assembly software and algorithm in mind, choosing a sequence strategy designed for a different assembly algorithm, or sequencing without thinking about assembly is usually a recipe for poor unpublishable assemblies [38]. Here we survey different assembly strategies, with different sequence and library construction requirements. A typical genome project starts with high quality DNA of as low polymorphism as available, and extends beyond genome assembly to include gene annotation. Relatively inexpensive RNAseq from multiple tissues/or life stages (the authors often chooses adult male, adult female and mixed other life stages) provides transcript data for final genome annotation. For the definition of sex chromosomes, it is often useful to re-sequence at 30X coverage one individual of each sex. Additionally, re-sequencing of individuals at 30X genome coverage followed by alignment to the final reference using standard human analysis tools is the best way to characterize sequence variation within a species.
| Technology | Strategy | Software | DNA | Cost | Notes | |
|---|---|---|---|---|---|---|
| Sanger Paired-end | Overlap-layout-consensus Read length: 700bp Insert sizes, coverage 3kb, 15x 8kbp, 5x |
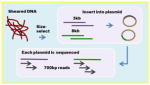
|
Celera assembler | ++++ | +++++ | The original sequencing technology is now obsolete for genome assembly due to cost. |
| 454 Fragment + Paired-end | Overlap-layout-consensus Read length: 600bp Insert sizes, coverage 600bp (fragment), 15x sequence coverage 3kb (pe), 15x clone coverage 8kb (pe), 15x clone coverage |
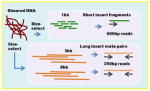
|
Celera assembler Newbler |
+++ | ++++ | Later revisions of the chemistry gave almost Sanger length sequence reads. Systemic homopolymer errors in assemblies can easily be corrected with Illumina sequence. Roche has announced a 2016 end of life for 454 sequencing support. |
| llumina Paired-ends + mate pairs | de Bruijn graph based assembly Read length: 100bp Insert sizes, coverage 180bp, 40x 500bp, 40x 3kb, 40x 8kb, 20x |
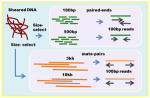
|
AllPaths-LG SOAP de novo SGA Platanus |
+++ | + | Needs large memory machine for assembly. Can assemble large eukaryotic genomes. Not designed for polymorphic genomes (except Platanus) |
| Illumina PCR-free Single library paired-end | de Bruijn graph based assembly Read length: 250bp Insert sizes, coverage 450bp, 60x |
|
DISCOVAR de novo | + | + | Simplified library production. Designed for mammalian levels of sequence polymorphism. DISCOVAR is designed for PCR-free library construction, Platanus is more flexiblegt. Platanus documentation gives no indication of desired sequence coverage or insert size inputs. We would recommend a longer insert size for scaffolding, in addition to shorter insert sizes for the primary sequence data. |
| Multiple k-mer de Bruijn graph Read length: 100bp, 250bp Insert sizes, coverage 450bp, 60x 3kb-40kb (optional) |
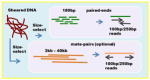
|
Platanus | ||||
| Illumina symthetic long reads (previously Moleculo) | Overlap-layout-consensus Reads: 10kbp sheared to 500–800bp and assembled into 1–18.5kb synthetic reads, 20x |
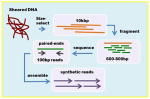
|
Celera assembler | + | +++ | Currently relatively expensive, but has continued potential for cost reduction. Synthetic long reads are very accurate. Possible uneven coverage. |
| PacBio Self-correction | Overlap-layout-consensus Read sizes, coverage 6–15kb reads at 60x |
|
HBAR/Falcon & Celera assembler | ++++ | +++ | All-against-all read alignment for error correction is processing intensive |
| PacBio Circular Consensus Sequencing | Overlap-layout-consensus Reads sizes, coverage 3kb CCS reads, 20X |
|
Celera assembler | ++ | +++ | Trivial error correction to Sanger quality long reads. No possibility of sequence reads error corrected from disparate genomic loci. Assembly of 3kb reads may not be as good as longer reads not be as good as longer reads |
The Problem With Insects (And Many Other Species)
The major goal of high quality genome references is high quality gene model annotation. As average gene loci range in size from 12kb in Drosophila to 25kb and more in larger insect and mammalian genomes, contig N50s of at least 10kb, and scaffold N50s > 300kb are the minimum for high quality gene annotation.
Unfortunately, insects and other invertebrates have a particularly bad list of attributes for genome assembly that compromise contig N50 sizes:
Often they cannot be reared in the lab – which precludes any breeding for genome homozygocity - and instead must be collected on field trips necessitating the use of some material for species identification. Even if research colonies are available, annual and longer lifecycles can make inbreeding unrealistic.
Insects are often physically small, such that very little DNA (nanograms) can be obtained from a single individual, necessitating pooled polymorphic individuals to make libraries. In cases with intermediate sized individuals, we prioritize a single individual for the majority of sequence, and pooled individuals for larger insert libraries, where significant material is lost in agarose gel size selection.
Due to the large species diversity within the arthropods, there are generally no high quality genome assemblies of phylogentically close species to aid in assembly (with the possible exception of the Lepidoptera [15])
DNA preps often have to be optimized for a new insect species, as entomologists are not trained in molecular methods and standard protocols have not been determined. From our experience, Qiagen spin columns do not produce DNA of appropriate quality, but Qiagen midi sized drip column kits often give good results.
Although holometabola often have small (~500Mb) genomes, outside the holometabola, arthropods can have large genomes (1.5Gb spiders, 3Gb cockroaches, 5Gb mantis, and bristletails, 7Gb grasshoppers [16]), thus costs are variable (compared to the relative stability of the 3Gb mammals) and larger than the 175Mb Drosophila experience would indicate.
New Technologies are Revolutionizing Genome Assembly
The above describes the reality that high quality insect genome assembly is not guaranteed. Optimization of assembly parameters by empirical testing is critical, and can perhaps improve a current genome assembly 2X or more for a specific dataset and software combination, but the limitations of current data and techniques remain. New sequencing technologies and assembly strategies are overcoming the traditional assembly problems of data polymorphism and repetitiveness with the goal of creating near ‘finished’ archival quality genome assemblies.
New assembly software
New assembly software attempts to account for polymorphism and take advantage of longer 250bp Illumina reads. DISCOVAR [17] from David Jaffe’s group who wrote the excellent ALLPATHS-LG is designed around 250bp paired end reads from a single PCR free library - an extremely low cost strategy. However, as it was designed with relatively low human polymorphism in mind, additional testing will be required to assess the extent of polymorphism it can handle, despite excellent results in mammalian assemblies. Platanus [18] has been used extensively for assemblies by the BGI, and is designed with multiple approaches to dealing with polymorphism. Its use of multiple kmer lengths in the de Bruijn graph, enables effective use of Illumina reads from 100bp to 250bp.
Long reads and their assembly
Long reads offer inherent advantages for genome assembly: they span polymorphic regions, repeats and transposable elements, and provide long-range information for contig scaffolding intrinsically avoiding many assembly problems. Additionally, fewer reads for the same coverage reduces computational demands. Read lengths are often longer than contigs from mediocre short read assemblies.
The TruSeq synthetic long read technology from Illumina [19] is a library construction kit and cloud software solution producing high-quality sub assemblies of barcoded 10kb fragments (synthetic long reads) from shorter Illumina reads. Resulting synthetic long reads are then easily assembled into the full genome using the Celera Assembler or other overlap layout consensus assembler. This strategy was designed and successfully used for a highly polymorphic colonial tunicate genome [20]. There can be some issues from un-even genome representation [19], so a combination of this technique and a less biased short read assembly might be ideal. Synthetic long reads are especially exciting due to the continued potential for cost reduction from this ever more massively parallel sequencing technology.
Pacific Biosciences (PacBio) RSII reads have matured, and now routinely generate read lengths >20kb with averages in the 10kb range. Unfortunately, techniques for Illumina error correction of PacBio reads such as the pacbioToCA utility [21] within the Celera Assembler, and PBcR [21] have proved computationally inefficient in our hands due to the difficulties of aligning high coverage 100bp Illumina data to high coverage 15% error rate long reads (although a promising new algorithmic approach was recently released [22]). A different approach, PBJelly [23] enables gap filling in draft assemblies by alignment to an existing draft genome, avoiding the error correction step except when generating consensus sequence in gap regions. However, pure PacBio error correction strategies appear to give the best results. HGAP/HBAR/Falcon (Here called HGAP) [24–26] assembly trades high sequence coverage (50–70X) to overcome the 15% error rate, by using shorter reads to correct the longest 15–20X of reads enabling traditional assembly using the Celera assembler. This has produced effectively finished “archival” quality genome assemblies with contig N50s 5Mb and above for isogenic melanogaster and Arabidopsis [27,28] genomes, although haploid human assembly, whilst equally successful, was computationally demanding for the error correction step [29]. The effectiveness of HGAP on polymorphic genomes has yet to be tested, but the length of the error corrected reads suggests a significant improvement over current methods. The megabase sized contigs of these assemblies simplifies and almost obviates finishing, and prevents future re-visiting of sequencing to make these assemblies truly archival – of high enough quality to be placed in a database and used for many decades to come.
Pacific Biosciences Circular Consensus Sequencing (CCS) [30] is an alternate, often neglected method of error correcting PacBio sequence reads. Circularized shorter templates are sequenced multiple times by a single long read enabling error correction on a single molecule. This avoids the possibility of independent, but similar, genomic loci erroneously error correcting each other. The advantage is computationally trivial error correction, as no all-against-all read comparison is required, but the disadvantage of shorter error corrected read lengths (current read lengths enable ~3kb CCS reads with at least 3 sequence passes of the molecule), and slightly less efficient production of error corrected data. This can be an effective approach for genomes with significant highly repetitive content and larger genomes to avoid the computational difficulties of all against all alignment for error correction.
Finally, Oxford Nanopore’s infamous single molecule sequencing is in beta testing at the time of writing. Anecdotal information suggests the technology enables extremely long reads up to 100kb in length, but quality issues remain significant. Despite these high error rates, an initial assembly of a eukaryote has been reported [31]. Given the revolutionary assembly potential of such read lengths, we are watching this technology with interest.
New assembly validation and scaffolding technologies
Genome assembly validation has fallen out of favor, as the costs associated are often greater than that of the Illumina assembly itself. Genetic maps, physical maps and other long-range independent data are expensive to generate, and taxonomically close insect species with high quality genome sequences are rare. Minimally, BUSCO analysis should be performed (see Waterhouse’s review in this issue [32]), indicating the completeness of the assembled gene set –an item of critical importance for most users. For comprehensive analysis of assembly quality, additional data is required. Two new technologies are described here, but it should be noted that long-range sequence information requires long pieces of DNA, either from intact nuclei or extremely long DNA molecules, which may require additional material.
Optical mapping (commercially available from OpGen) developed in 1990s [33] directly images restriction digests of confined long linear DNA molecules, generating large scale restriction maps. A similar, but more scalable technology from BioNano Genomics [34] uses semiconductor fabricated nanogrooves, and sequence motif fluorescent labeling to enable high throughput data collection of similar motif maps. The high throughput data collection method has reduced costs to enable routine validation of genome assemblies, and has been used for assembly super scaffolding to increase scaffold N50 lengths.
An orthogonal data source for genome assembly validation is chromatin interaction sequencing (Hi-C) [35]. This sequencing protocol captures pairs of DNA fragments physically close to each other in three dimensional chromatin structures. The proximity of the paired sequences is inversely related to the distance between them, and this fact is used to produce ‘ultra-long-range’ genome assembly scaffolding for entire chromosome arms [35]. To date, this has been used on high quality genomes including Drosophila, but the technique is rapidly gaining acceptance.
Low input DNA and large genome sizes are still problematic
As described above, in many cases input DNA quantity from a single individual can be very small, and pooled individuals increase the polymorphism and chances of a poor assembly product. But some of the above strategies require relatively small amounts of DNA (although the DNA must still not be degraded). Illumina Synthetic reads require only 500ng of DNA, depending on the amount of synthetic long-read sequence desired. The DISCOVAR or Platanus assembly of a 250bp read length Illumina data can be made from as little as 10ng of DNA using low input DNA library protocols. Unfortunately this is not a PCR free library technique, but ignoring this requirement may be a lessor of evils. In our hands Platanus assembly of an outbred 400Mb diplura genome from 250bp paired reads with a 400bp insert generated 11kb contig N50 from approximately 50ng of DNA, although the scaffolds are small due to an absence of long-range sequence information.
Very large genomes are also problematic, with the definition of very large decreasing with increased polymorphism in the dataset. With ALLPATHS-LG, many groups have routinely assembled 3Gb mammalian genomes to high quality, but more polymorphic insect genome assemblies often fail with genomes greater than 2Gb. The ALLPATHS-LG software is not designed for genomes larger than 4Gb, and run times grow into multiple months. MaSuRCA [36] is the only assembler designed for very large genomes having been successfully applied to the 22Gb loblolly pine genome [37], however, we have had difficulty applying this software to polymorphic insect genomes from 2.5 to 5Gb. Additional research is required to address this problem.
Summary
New long read based assembly strategies, such as Pacific Biosciences and Illumina synthetic long-reads, are highly recommended due to a higher probability of assembly completion, longer contiguous sequences and potentially archival “finished” quality assembly products. Cost effective draft assembly of longer 250bp Illumina reads is also improved due to new software, although the success rate in significantly polymorphic datasets is unknown and expectations of archival quality genome references for little investment are unrealistic. New validation technologies enable quality assurance and improved super-scaffolding of genome assemblies. Genome assembly from low DNA quantity and of large and polymorphic genomes is still a significant challenge.
Highlights.
Insect genome assembly is difficult, unreliable and often low quality
Obstacles include DNA polymorphism, inability to inbreed and limited DNA quantities
New short read assembly tools are more cost effective and higher quality than ever
Assembly of long sequence reads is robust and can produce archival quality genomes
New assembly validation tools are now cost effective at genome scale
Acknowledgments
SR and SCM were supported by NHGRI grant U54 HG003273, Richard Gibbs PI, funding the Baylor College of Medicine Human Genome Sequencing Center. We would like to acknowledge Dr. Kim C. Worley for editing and intellectual advice in the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest.
- 1.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- *2.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. The initial Drosophila melanogaster assembly paper is perhaps the original eukaryotic assembly paper and still a good introduction to the concepts. [DOI] [PubMed] [Google Scholar]
- 3.Drosophila 12 Genomes C. Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 4.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knight J. 454 Life Sciences, editor. Newbler. 2005. p. 454. sequence read assembly software. [Google Scholar]
- 6.Nederbragt AJ. On the middle ground between open source and commercial software -the case of the Newbler program. Genome Biol. 2014;15:113. doi: 10.1186/gb4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen ZX, Sturgill D, Qu J, Jiang H, Park S, Boley N, Suzuki AM, Fletcher AR, Plachetzki DC, FitzGerald PC, et al. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res. 2014;24:1209–1223. doi: 10.1101/gr.159384.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chipman AD, Ferrier DE, Brena C, Qu J, Hughes DS, Schroder R, Torres-Oliva M, Znassi N, Jiang H, Almeida FC, et al. The First Myriapod Genome Sequence Reveals Conservative Arthropod Gene Content and Genome Organisation in the Centipede Strigamia maritima. PLoS Biol. 2014;12:e1002005. doi: 10.1371/journal.pbio.1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heliconius-Genome-Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 14.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.d’Alencon E, Sezutsu H, Legeai F, Permal E, Bernard-Samain S, Gimenez S, Gagneur C, Cousserans F, Shimomura M, Brun-Barale A, et al. Extensive synteny conservation of holocentric chromosomes in Lepidoptera despite high rates of local genome rearrangements. Proc Natl Acad Sci U S A. 2010;107:7680–7685. doi: 10.1073/pnas.0910413107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregory TR. Animal Genome Size Database. 2014. [Google Scholar]
- *17.Weisenfeld NI, Yin S, Sharpe T, Lau B, Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, et al. Comprehensive variation discovery in single human genomes. Nat Genet. 2014;46:1350–1355. doi: 10.1038/ng.3121. Discovar requires 250bp Illumina sequence reads and enables cost effective draft (but not archival quality) genomes for low polymorphism species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *18.Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24:1384–1395. doi: 10.1101/gr.170720.113. The Platanus assembler described in this manuscript is the only de-novo genome assembler designed especially for polymorphic genomes, but no advice is given on ideal coverage or insert sizes, or the extent of polymorphism that it can accommodate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **19.McCoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, Pushkarev D, Petrov DA, Fiston-Lavier AS. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS One. 2014;9:e106689. doi: 10.1371/journal.pone.0106689. McCoy et al further explore the use of Illumina synthetic long reads for repeat analysis, and importantly describe some of the limits of the current protocols. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **20.Voskoboynik A, Neff NF, Sahoo D, Newman AM, Pushkarev D, Koh W, Passarelli B, Fan HC, Mantalas GL, Palmeri KJ, et al. The genome sequence of the colonial chordate, Botryllus schlosseri. Elife. 2013;2:e00569. doi: 10.7554/eLife.00569. This paper describes the use of Illumina synthetic long reads (then Moleculo) Illumina synthetic long reads on an extremely polymorphic colonial tunicate. Previous sanger projects sequencing sea urchins, and Ciona seq squirt with similarly large polymorphism rates (3–8%) were problematic, and the approach here immediately gave acceptable results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye C, Hill C, Koren S, Ruan J, Ma ZS, Yorke JA, Zimin A. DBG2OLC: Efficient Assembly of Large Genomes Using the Compressed Overlap Graph. 2015. arXiv. [Google Scholar]
- 23.English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One. 2012;7:e47768. doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 25.Chin J. HBAR-DTK. 2013. [Google Scholar]
- 26.Chin J. FALCON. 2013. [Google Scholar]
- **27.Pacific-Biosciences. Data Release: Preliminary de novo Haploid and Diploid Assemblies of Drosophila melanogaster. PacBio Blog. 2014;2014 Datasets and description of a de-novo Pacific biosciences de-novo assembly of Drosophila melanogaster generating assemblies with 5Mb contig N50’s. [Google Scholar]
- 28.Pacific-Biosciences. New Data Release: Arabidopsis Assembly Offers Glimpse of De Novo SMRT Sequencing for Larger Genomes. PacBio Blog. 2013;2014 [Google Scholar]
- 29.Pacific-Biosciences. Data Release: ~54x Long-Read Coverage for PacBio-only De Novo Human Genome Assembly. PacBio Blog. 2014;2014 [Google Scholar]
- 30.Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010;38:e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodwin S, Gurtowski J, Ethe-Sayers S, Deshpande P, Schatz M, McCombie WR. BioRxiv pre-print server. 2015. Oxford Nanopore Sequencing and de novo Assembly of a Eukaryotic Genome. [Google Scholar]
- 32.Waterhouse RM. A maturing understanding of the composition of the insect gene repertoire. Current Opinion in Insect Science. 2015 doi: 10.1016/j.cois.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz DC, Li X, Hernandez LI, Ramnarain SP, Huff EJ, Wang YK. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science. 1993;262:110–114. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- **34.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol. 2012;30:771–776. doi: 10.1038/nbt.2303. Bionano’s genome mapping technology is a cost effective method of assembly validation generating sequence motif maps of DNA molecules upto 1Mb in length. This paper describes the use of the technology for the complex human HLA region. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **35.Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. This paper describes the use of chromatin sequencing to scaffold draft assemblies to the chromosome arm level. The library construction protocol requires intact cells with intact three dimensional DNA structure, but sequencing does not require extensive coverage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zimin AV, Marcais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. The MaSuRCA genome assembler. Bioinformatics. 2013;29:2669–2677. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37.Neale DB, Wegrzyn JL, Stevens KA, Zimin AV, Puiu D, Crepeau MW, Cardeno C, Koriabine M, Holtz-Morris AE, Liechty JD, et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014;15:R59. doi: 10.1186/gb-2014-15-3-r59. A description of the assembly of the 22Gb loblolly pine genome the largest genome assembled to date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richards S. Please note that this advice, often ignored by otherwise brilliant researchers, is the main reason we agreed to write this review. [Google Scholar]